 Maria Luce Genovesi1
Maria Luce Genovesi1 Barbara Torres2Marina Goldoni2Eliana Salvo3Claudia Cesario4Massimo Majolo5
Barbara Torres2Marina Goldoni2Eliana Salvo3Claudia Cesario4Massimo Majolo5 Tommaso Mazza6Carmelo Piscopo7
Tommaso Mazza6Carmelo Piscopo7 Laura Bernardini2*
Laura Bernardini2*- 1Department of Experimental Medicine, Sapienza University of Rome, Rome, Italy
- 2Medical Genetics Division, IRCCS Casa Sollievo Della Sofferenza Foundation, San Giovanni Rotondo, Italy
- 3Medical Genetics, Department of Biomedical and Biotechnological Sciences, University of Catania, Catania, Italy
- 4Translational Cytogenomics Research Unit, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy
- 5Hospital Directorate, National Hospital A.O.R.N. “Antonio Cardarelli”, Naples, Italy
- 6Laboratory of Bioinformatics, IRCCs Casa Sollievo Della Sofferenza Foundation, San Giovanni Rotondo, Italy
- 7Medical and Laboratory Genetics Unit, National Hospital A.O.R.N. “Antonio Cardarelli”, Naples, Italy
Fibrillin proteins are extracellular matrix glycoproteins assembling into microfibrils. FBN1, FBN2, and FBN3 encode the human fibrillins and mutations in FBN1 and FBN2 cause connective tissue disorders called fibrillinopathies, affecting cardiovascular, dermal, skeletal, and ocular tissues. Recently, mutations of the less characterized fibrillin family member, FBN3, have been associated in a single family with Bardet–Biedl syndrome (BBS). Here, we report on a patient born from two first cousins and affected by developmental delay, cognitive impairment, obesity, dental and genital anomalies, and brachydactyly/syndactyly. His phenotype was very similar to that reported in the previous FBN3-mutated family and fulfilled BBS clinical diagnostic criteria, although lacking polydactyly, the most recurrent clinical feature, as the previous siblings described. A familial SNP-array and proband’s WES were performed prioritizing candidate variants on the sole patient’s runs of homozygosity. This analysis disclosed a novel homozygous missense variant in FBN3 (NM_032447:c.5434A>G; NP_115823:p.Ile1812Val; rs115948457), inherited from the heterozygous parents. This study further supports that FBN3 is a candidate gene for a BBS-like syndrome characterized by developmental delay, cognitive impairment, obesity, dental, genital, and skeletal anomalies. Anyway, additional studies are necessary to investigate the exact role of the gene and possible interactions between FBN3 and BBS proteins.
Introduction
Fibrillin proteins are extracellular matrix glycoproteins that assemble into microfibrils with architectural functions in many connective tissues (Corson et al., 2004; Sabatier et al., 2011). These 10–12 nm extracellular calcium-binding microfibrils occur either in association with elastin or in elastin-free bundles and provide long-term force bearing structural support (Corson et al., 2004). Fibrillins and microfibrils are also involved in matrix deposition and activation of growth factors belonging to the transforming growth factor β (TGF-β) superfamily, including TGF-β and bone morphogenetic proteins (BMPs), which regulate a broad array of developmental and homeostatic processes (Reinhardt, 2014).
FBN1 (fibrillin 1; MIM * 134797), FBN2 (fibrillin 2; MIM * 612570), and FBN3 (fibrillin 3; MIM * 608529) encode the three human fibrillins that show a highly conserved modular domain organization and about 61–69% of homology at the amino acid level and are also conserved among species (Hubmacher et al., 2006).
FBN1 is expressed throughout life, whereas FBN2 and FBN3 are primarily expressed during embryonic development. Indeed, the developing embryo seems to require all the three fibrillins, while the adult organism only fibrillin-1 (Corson et al., 2004; Sabatier et al., 2011). Mutations in FBN1 and FBN2 cause connective tissue disorders called fibrillinopathies. FBN1 has been associated with several autosomal dominant (AD) disorders: acromicric dysplasia (MIM # 102370), ectopia lentis familial (MIM # 129600), geleophysic dysplasia 2 (MIM # 614185), Marfan lipodystrophy syndrome (MIM # 616914), Marfan syndrome (MIM # 154700), MASS syndrome (MIM # 604308), stiff skin syndrome (MIM # 184900), and Weill–Marchesani syndrome 2 (MIM # 608328). FBN2 is causative of AD congenital contractural arachnodactyly (MIM # 121050) and AD macular degeneration early-onset (MIM # 616118). These fibrillinopathies share some common features and are all characterized by clinical manifestations in cardiovascular, dermal, skeletal, and ocular tissues. Sometimes also palate malformations and neurologic features are reported (Robinson et al., 2006). FBN3 seems to play a role in the pathogenesis of polycystic ovary syndrome 1 (PCOS1; MIM % 184700) (Urbanek et al., 2005; Stewart et al., 2006; Ewens et al., 2010; Jordan et al., 2010). Recently, the homozygous missense variant of FBN3 (NM_032447) c.2804G>A (p.Arg935Gln) has been associated with Klippel–Trenaunay–Weber syndrome (MIM % 149000) (Liu et al., 2019), while the compound heterozygous variants c.3616G>A (p.Val1206Ile) and c.6037C>T (p.Arg2013Trp) have been associated with Bardet–Biedl syndrome 1 (BBS1; MIM # 209900) (Wang et al., 2017), a multisystemic ciliopathy characterized by retinal dystrophy, obesity, post-axial polydactyly, renal dysfunction, learning difficulties, and hypogonadism (Forsythe and Beales, 2013; Niederlova et al., 2019; Forsyth and Gunay-Aygun, 2003).
In this work, we report on a patient with a homozygous FBN3 missense variant identified by a combined SNP-array and whole-exome sequencing approach and a phenotype characterized by cognitive impairment, developmental delay, learning difficulties, strabismus, arched palate, dental and genital anomalies, brachydactyly/syndactyly, and obesity, supporting that FBN3 alteration could be associated with a phenotype resembling BBS.
Materials and Methods
Case Report
Second of four siblings, the proband was born from consanguineous non-affected parents (first cousins) of Moroccan origin at full term (39W) of a physiological pregnancy and after natural delivery, with a birth weight of 2,500 kg (5–10 percentile). He was evaluated at the age of 8 years for delayed psychomotor development, learning difficulties, impaired verbal fluency and attention capacity, speech delay, and gross and fine motor abnormalities. Clinical examination revealed various facial dysmorphisms: hypotelorism, eyelid fissures tilted down, beaked nose, arched palate, and large ears. He also showed microdontia with periodontal disease and numerous dental caries, with the removal of three teeth. He had a mild strabismus with hyperopia, bilateral brachydactyly and syndactyly between II and III toes fingers, micropenis with right cryptorchidism, and central obesity (Figure 1). He suffered from frequent respiratory infections and mild obstructive apnea. Audiological tests revealed bilateral conductive hearing loss. The last ophthalmological examination, performed at age 9, documented optic discs with fuzzy, hyperemic margins, and marked vascular tortuosity. He regularly followed a rehabilitation therapy for several years. No abnormalities were found after cardiological, gastroenterological, and neurological investigations, including brain MRI and E.E.G. At the last evaluation (age 11), he had normal head circumference and stature parameters (respectively 51.5 and 137 cm) but a significant overweight, with +2DS of both weight and BMI (respectively, 52 kg and 27.7). FRAXA and methylation for Prader–Willi syndrome were normal.
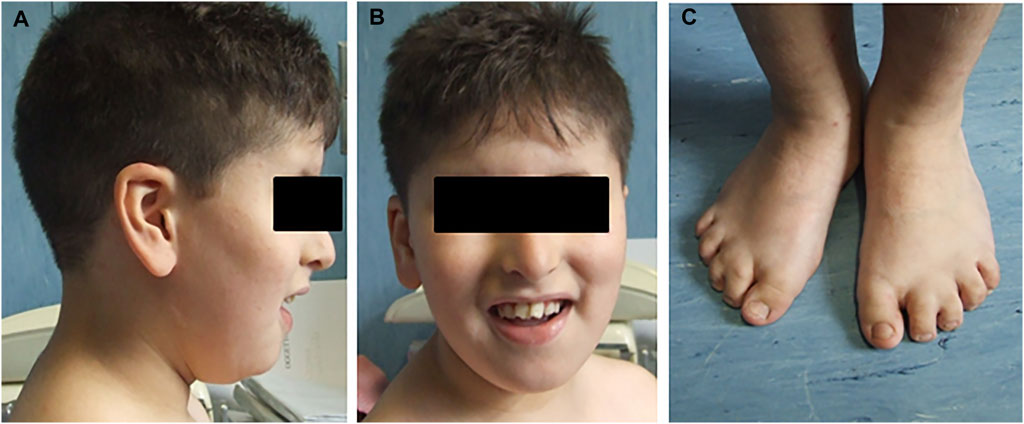
FIGURE 1. Pictures report some clinical characteristics of the proband. (A) Facial dysmorphisms, beaked nose, and large ears. (B) Dental abnormalities. (C) Bilateral brachydactyly and syndactyly between II and III toes fingers.
SNP-Array
Genomic screening for copy number variations (CNVs) was performed on the proband and, successively, on his three healthy siblings’ DNA using a SNP-array platform (GeneChip 6.0, Affymetrix, Santa Clara, CA, United States), carried out following the manufacturer’s recommendations and analyzed using ChAS software (v3.1; Affymetrix). A total of 270 healthy controls belonging to the International HapMap Project were used as a reference sample in data analysis (Affymetrix). Called CNVs were represented by at least 25 contiguous probes and 75 kb as a minimum size and were classified according to the American College of Medical Genetics (ACMG) recommendations (Riggs et al., 2020). Moreover, all CNVs represented by at least five contiguous probes and laying within a disease-gene (OMIM, Online Mendelian Inheritance in Man; https://www.omim.org/) were considered. Runs of homozygosity (ROH) analysis of autosome chromosomes was performed using the SNPs and filtered considering 3 Mb of length as a minimum size (Kearney et al., 2011).
Whole-Exome Sequencing (WES)
WES on the proband’s genomic DNA was performed at CRIBI (Centro di Ricerca Interdipartimentale per le Biotecnologie Innovative) Genomics (Padua, Italy), using the Ion TargetSeq™ Exome Enrichment Kit (Applied Biosystems, Foster City, CA, United States) as an exome capture kit and the Ion Proton™ System (Applied Biosystems) as a sequencing platform. Reads alignment to the human genome (UCSC GRCh37/hg19) and variant calling were performed through the Torrent Suite Software v5.2.1. Variants and gene annotation was performed using ANNOVAR (Wang et al., 2010; July 2017 release) and resorting to the dbNSFP v3.5 gathered annotations (Liu et al., 2016). Single nucleotide variants (SNVs) and small insertions and deletions (Indels) were filtered based on quality criteria and their effect, only high-quality variants and those with an effect on the coding sequence and splice site regions (±10 bp) were retained. The resulting variants were further filtered to retain only those with unknown frequency or a minor allele frequency (MAF) ≤1%, according to the gnomAD database (v2.1.1), and prioritized based on their predicted functional impact, evaluated through the Combined Annotation Dependent Depletion (CADD) score (v1.6), using a value of 10 as a threshold (Kircher et al., 2014). Subsequently, the variants were first prioritized on the basis of SNP-array results, considering all the ROH identified in the patient and not shared with his healthy siblings, and then considering different modes of inheritance, in order to exclude damaging variants in heterozygosity or compound heterozygosity. The selected candidate variant was validated in the proband and analyzed in the other family members through Sanger sequencing, using the ABI BigDye Terminator Sequencing Kit (v3.1) (Applied Biosystems, Foster City, CA, United States) and a 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, United States). Sequence electropherograms were analyzed using Chromas LITE (v2.01; Technelysium Pty Ltd., Brisbane, Australia).
Results
SNP-array analysis did not disclose any non-polymorphic CNV, whereas identified a list of widespread ROH, with a total percentage of 4.7% calculated on autosome chromosomes and consistent with the degree of parental relationship (Kearney et al., 2011). This analysis extended to the healthy siblings allowed to refine the sole patient’s ROH, likely including the disease-gene. WES resulted in adequate coverage and depth. A total of 48,015 variants were identified and filtered using several criteria, obtaining 9,776 high-quality variants with an effect on the coding and splice site regions; 956 variants with frequency ≤ 1% in gnomAD or with unknown frequency; 670 variants with a CADD score ≥ 10, predicted to have a high functional impact on the protein. Variants prioritized on the patient’s exclusive ROH revealed a homozygous missense variant in FBN3 (NM_032447:c.5434A>G; NP_115823:p.Ile1812Val; rs115948457) mapping within a 6.6 Mb ROH as the most promising variant, while not likely damaging variants were found hypothesizing different transmission patterns. The FBN3 variant had a frequency of 0.002596 in dbSNP 151, a total frequency of 0.0007317 (0.007421 in the African population) in gnomAD and a CADD score of 19.82. The DANN score was 0.9957. This variant was reported in the homozygous state only once in gnomAD. It is predicted as “probably damaging” by PolyPhen-2, as “damaging” by FATHMM, FATHMM-MKL, and FATHMM-XF, and as “disease causing” by MutationTaster. According to the American College of Medical Genetics (ACMG) guidelines, this variant was classified as likely benign considering its frequency in Africans (VarSome, https://varsome.com/), although it is much rarer in the other populations, or as a variant of uncertain significance (VUS) (InterVar, https://wintervar.wglab.org/; Franklin, https://franklin.genoox.com/). Sanger sequencing confirmed the presence of the homozygous variant in the proband (II:2); both the parents (I:1, I:2), one sister (II:3) and the youngest brother (II:4), all non-affected, resulted heterozygous, while the other non-affected sister (II:1) homozygous wild-type, corroborating that the variant co-segregated with the disease. Two missense variants in the same gene, NM_032447:c.3616G>A; NP_115823:p.Val1206Ile; rs747241812 and NM_032447:c.6037C>T; NP_115823:p.Arg2013Trp; rs183278638, were described in compound heterozygosity in two Chinese siblings with clinical diagnosis of BBS and showed developmental delay/cognitive impairment and speech delay, obesity, diabetes mellitus, dental anomalies, and retinitis pigmentosa. Comparison of those clinical features with the clinical characteristics of the patient herein reported showed high similarity, supporting the pathogenicity of the homozygous missense variant detected (Table 1).
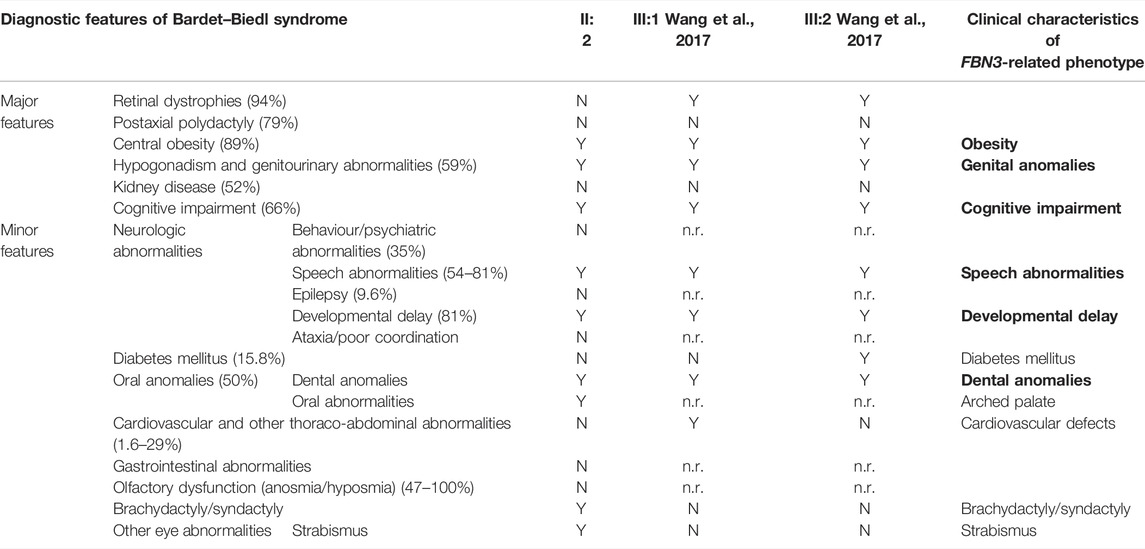
TABLE 1. Left: diagnostic features of Bardet–Biedl syndrome with their relative prevalence (Forsyth and Gunay-Aygun, 2003; Forsythe and Beales, 2013) and (center) clinical characteristics of our (II:2) and Wang’s patients (III:1 and III:2; Wang et al., 2017). Four primary features or three primary features and two secondary features establish the diagnosis of Bardet–Biedl syndrome (Forsyth and Gunay-Aygun, 2003). Right: clinical characteristics (in bold those showed by all reported patients) of the BBS-like phenotype herein described. N: absent; n.r.: not reported; Y: present.
Discussion
Autozygosity, or the inheritance of two copies of an ancestral allele, facilitates the mapping of mutations by flagging the surrounding haplotypes as tractable runs of homozygosity, a process known as autozygosity mapping (Alkuraya, 2013). This approach has been successfully used to discover new disease-genes in consanguineous families (Vahidnezhad et al., 2018) and the application of SNP-array and WES analyses together with advanced bioinformatic filtering approaches, focused on likely damaging homozygous variants, accelerated and made more cost-effective this process with an exponential increase of diagnostic yield (Dharmadhikari et al., 2019). We reported a north-African patient born from two first cousins with a novel homozygous variant of FBN3, the less characterized member of fibrillin protein family, detected by an integrated analysis of familial SNP-array and proband’s WES, by prioritizing candidate variants within the sole patient’s ROH. Compound heterozygous variants of FBN3 have been recently described in two siblings with a clinical diagnosis of Bardet–Biedl syndrome (BBS) (Wang et al., 2017). BBS is a rare autosomal recessive syndrome (with an incidence of 1/160,000 and 1/13,500 in some Arab populations), characterized by major symptoms with very variable penetrance, such as retinal dystrophy, obesity, post-axial polydactyly, renal dysfunction, learning difficulties, hypogonadism, and other minor clinical characteristics (Forsythe and Beales, 2013; Niederlova et al., 2019; Forsyth and Gunay-Aygun, 2003). Seen the extremely variable clinical spectrum, four primary features or three primary features and two secondary features are necessary to establish the diagnosis (Forsyth and Gunay-Aygun, 2003) reported in Table 1. The two previously reported patients presented both with intellectual disability, developmental delay, learning disorders, obesity, and genital anomalies, associated with ocular defects, such as retinitis pigmentosa, nystagmus and blepharophimosis, diabetes mellitus, and dental anomalies, all criteria sufficient to diagnose BBS (Wang et al., 2017). Interestingly, also the patient herein reported fulfills the diagnostic criteria of BBS, having three primary (obesity, genital anomalies, and cognitive impairment) and three secondary BBS features (developmental and speech delay, oral/dental anomalies, and brachydactyly/syndactyly). It is noteworthy that in all these cases the polydactyly, one of the most common clinical characteristics of the syndrome, is absent.
BBS is caused by mutations in numerous BBS genes, all encoding proteins involved in cilia biogenesis and function, and is consequently considered a multisystemic ciliopathy (Forsythe and Beales, 2013). Most of these genes encode for the subunits of the BBSome, an octameric protein complex; while others encode for chaperonin assisting the BBSome assembly. Last, the third most common group of genes encodes a GTPase assisting the BBSome function (Forsythe and Beales, 2013). Therefore, BBS is considered a disease resulting from dysfunction of primary cilia, involved in cellular trafficking and transport, although the pathogenesis of the majority of BBS clinical defects has yet to be elucidated.
Overall, pathogenic variants detected in BBS genes account for about 80% of BBS patients (Forsythe and Beales, 2013), suggesting that other genes not yet discovered are responsible for a subset of cases.
The variant herein reported is suggested to cause a distinct phenotype milder than BBS with an estimated frequency, calculated based on its frequency reported in the international database, of 1/20,000 in the African population and of 1/1,800,000 in general.
FBN3, also known as KIAA1776, encodes the less characterized member of the human fibrillin family. The FBN3 gene has 64 exons and maps on chromosome 19p13.2. The protein is composed of 2,809 amino acids and shows a N-terminal signal sequence, numerous epidermal calcium-binding growth factor (EGF) domains, and some EGF or EGF-like domains, all including six cysteine residues, and several TGF β-binding protein-like (TB) domains, made of eight cysteine modules (Hubmacher et al., 2006; Piha-Gossack et al., 2012) (Figure 2). The C-terminal half of FBN3 multimerizes and strongly interacts with fibronectin, similar to the C-terminal halves of FBN1 and FBN2 (Sabatier et al., 2009).
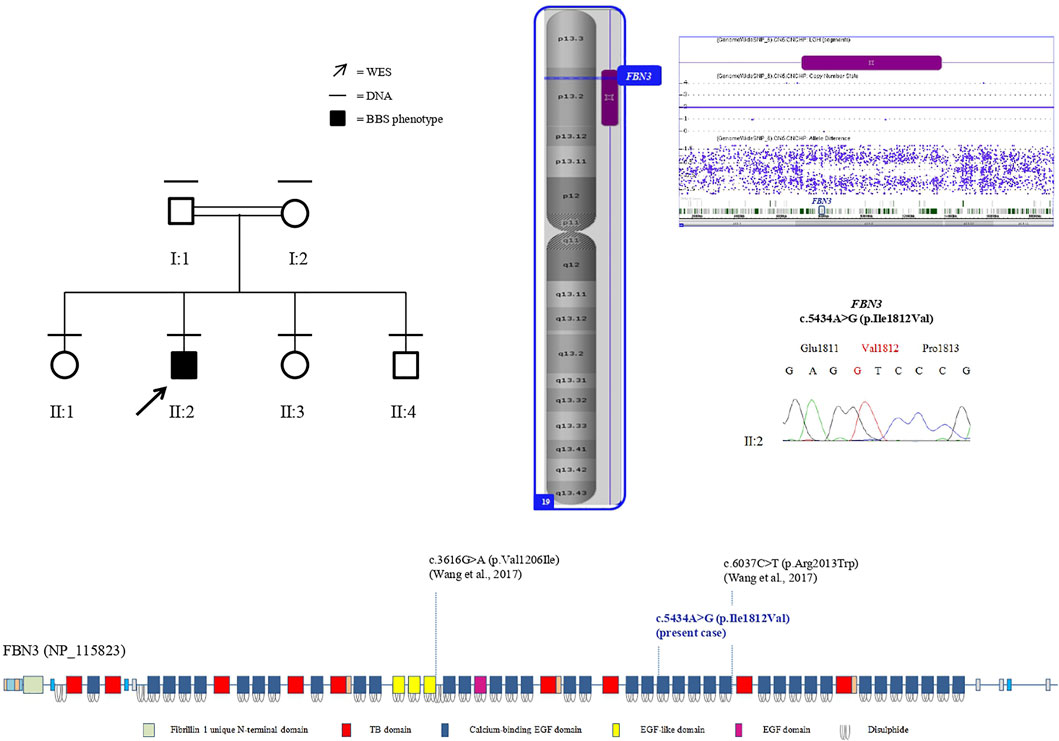
FIGURE 2. Left: pedigree of the family. Black lines indicate individuals for whom DNA was collected; the arrow indicates the proband that underwent WES. Right: the sole patient’s ROH (purple bar) on chromosome 19, including FBN3, assessed by SNP-array analysis (imaged by ChAS software, v3.1). Electropherogram shows the proband’s homozygous genotype of FBN3 (c.5434A>G) variant. Below: protein domains scheme of FBN3 reconstructed from Pfam (https://pfam.xfam.org/protein/Q75N90) and localization of variants reported so far.
An EGF domain contains six characteristic cysteine residues, which form three disulfide bonds in a 1–3, 2–4, and 5–6 arrangement, and frequently a consensus sequence for calcium-binding at its N-terminus, representing an essential structural and functional domain of the protein and conceivably mediating protein–protein interactions (Piha-Gossack et al., 2012). The missense variant p.Ile1812Val herein identified falls in a conserved amino acid within one of the calcium-binding EGF domains (https://pfam.xfam.org/protein/Q75N90) and a difference in the properties between the wild-type isoleucine and the mutant valine might disturb the EGF domain structure. It is not possible to assess the functional consequences of this variant on the protein. Anyway, it is noteworthy that the reciprocal substitution, a valine replaced by an isoleucine, in a EGF-like domain has been detected in the previously reported patients (Wang et al., 2017) (Figure 2) and the same amino acid change, an isoleucine replaced by a valine, has been also described in FBN1 in two patients affected by FBN1-related disease: p.Ile724Val (Tjeldhorn et al., 2006), and p.Ile2616Val (Howarth et al., 2007), both falling in a calcium-binding EGF-like domain and classified as VUS and likely pathogenic, respectively, following the most recent classification criteria (https://franklin.genoox.com/).
FBN3 is highly expressed in numerous tissues in the early stages of embryo development, from the 6th to the 12th gestational week: perichondrium, perineurium, perimysium, skin, developing bronchi, glomeruli, pancreas, kidney, heart, and testis and at the prospective basement membranes in developing epithelia and endothelia (Corson et al., 2004; Sabatier et al., 2011).
FBN3 is also found in the connective tissue of both the central nervous system and the peripheral nervous system (Sabatier et al., 2011). Moreover, expression studies detected high levels of FBN3 in the developing brain (Corson et al., 2004), but also in the adult brain (Nagase et al., 2000) and this expression pattern distinguishes FBN3 from the other fibrillins.
How deleterious variants in FBN3 could lead to a BBS phenotype has yet to be clarified. Seen the high rate of homology between fibrillins and taking into account that FBN3 is globally expressed in the same organs of FBN1 and FBN2, with some differences suggestive of FBN3 unique functions in certain tissues (Corson et al., 2004; Sabatier et al., 2011), insights inferred from the knowledge of other fibrillin family members can be used to deduct the possible clinical consequences of FBN3 impairment.
Fibrillins play both a well-known structural and an instructive role reflecting their ability to sequester TGF-β and BMP complexes in the extracellular matrix. In particular, the fibrillin-rich microfibrils concentrate the ligands locally and regulate cell differentiation within a spatial context during organ formation (positive regulation) and restrict their bioavailability and modulate cell performance in a timely fashion during tissue remodeling/repair (negative regulation). Therefore, fibrillins are integral components of a broader biological network orchestrating morphogenetic and homeostatic programs in multiple organ systems (Ramirez and Sakai, 2010).
Known mutations in the fibrillin genes cause a spectrum of phenotypes reflecting their role in the cell, and ranging from the Marfan syndrome to patients displaying only tall stature and mild marfanoid skeletal features (Loeys et al., 2003). Moreover, it is noteworthy that mice lacking Fbn2 display a limb-patterning defect (syndactyly), due to the impaired balance between chondrogenic outgrowth and interdigital cell death, which are under the control of several signaling molecules, including BMPs (Arteaga-Solis et al., 2001). Interestingly, syndactyly is one of the digit anomalies shown by our patients together with brachydactyly, which is considered another digit defect due to perturbation of BMP trafficking (Mundlos, 2009).
A characteristic of FBN1-related diseases is the alteration of subcutaneous adipose tissue (Muthu and Reinhardt, 2020). There is much evidence that abnormal adipose tissue metabolism strongly correlates with elevated TGF-β levels in humans and mouse (Samad et al., 1997; Fain et al., 2005; Yadav et al., 2011). It has also been demonstrated that FBN1 pathogenic variants lead to an upregulation of TGF-β signaling (Andelfinger et al., 2016; Lin et al., 2020). All these data support the hypothesis that obesity, recorded in all the patients herein described, is caused by an increased TGF-β signaling determined by damaging FBN3 variants alternating an EGF-like domain.
Overall, type I fibrillinopathies are characterized by clinical symptoms in skeletal, cardiovascular, ocular, and adipose tissues, showing a close overlap with clinical features recorded in the FBN3-mutated subjects.
FBN3 variants could be, therefore, associated with a distinct BBS phenotype and partially overlapping with fibrillin-related disorders and characterized by developmental delay and cognitive impairment, obesity, dental, and genital anomalies, in association with other more variable clinical abnormalities including ocular defects, abnormal development of the extremities, cardiovascular, and palate anomalies (Table 1). The FBN3 expression pattern in the developing and adult central nervous system could explain the neurodevelopmental disorder shown by the current and previous cases, a clinical feature that is vice versa absent in the other fibrillin-related diseases. Conversely, the absence in all cases described so far of polydactyly, one of the most common clinical characteristics of BBS, could be due to the distinct molecular and cellular pathways altered in FBN3-mutated and BBS patients.
In conclusion this is the second study associating FBN3 with a BBS phenotype and it supports FBN3 as a candidate gene for a BBS-like syndrome.
Up to date, there is no evidence that FBN3 and BBS proteins are structurally or functionally related and further studies are necessary to investigate the molecular basis of this new clinical condition.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
This study was conducted in accordance with the 1984 Declaration of Helsinki and its subsequent revisions. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article. Molecular testing carried out in this report is based on the routine clinical care of our institution.
Author Contributions
MLG performed part of experiments, analyzed and interpreted data and wrote and reviewed the manuscript; BT and MG performed part of experiments, analyzed and interpreted data and participated to the manuscript writing; ES and CC analyzed and interpreted data and participated to the manuscript writing; MM provided clinical data of the family; TM performed bioinformatic data analysis and curation and manuscript editing; CP provided clinical data of the family and participated to the conceptualization of the work and manuscript writing and editing; LB conceptualized and supervised the work, participated in the manuscript writing and editing. All authors contributed to the manuscript revision, read the manuscript, and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the family participating to this study. This study was partially funded by the Italian Ministry of Health (Ricerca Corrente to IRCCS Casa Sollievo della Sofferenza Foundation).
References
Alkuraya, F. S. (2013). The Application of Next-Generation Sequencing in the Autozygosity Mapping of Human Recessive Diseases. Hum. Genet. 132, 1197–1211. doi:10.1007/s00439-013-1344-x
Andelfinger, G., Loeys, B., and Dietz, H. (2016). A Decade of Discovery in the Genetic Understanding of Thoracic Aortic Disease. Can. J. Cardiol. 32, 13–25. doi:10.1016/j.cjca.2015.10.017
Arteaga-Solis, E., Gayraud, B., Lee, S. Y., Shum, L., Sakai, L., and Ramirez, F. (2001). Regulation of Limb Patterning by Extracellular Microfibrils. J. Cell Biol. 154, 275–282. doi:10.1083/jcb.200105046
Corson, G. M., Charbonneau, N. L., Keene, D. R., and Sakai, L. Y. (2004). Differential Expression of Fibrillin-3 Adds to Microfibril Variety in Human and Avian, but Not Rodent, Connective Tissues. Genomics 83, 461–472. doi:10.1016/j.ygeno.2003.08.023
Dharmadhikari, A. V., Ghosh, R., Yuan, B., Liu, P., Dai, H., Al Masri, S., et al. (2019). Copy Number Variant and Runs of Homozygosity Detection by Microarrays Enabled More Precise Molecular Diagnoses in 11,020 Clinical Exome Cases. Genome Med. 11, 30. doi:10.1186/s13073-019-0639-5
Ewens, K. G., Stewart, D. R., Ankener, W., Urbanek, M., McAllister, J. M., Chen, C., et al. (2010). Family-based Analysis of Candidate Genes for Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 95, 2306–2315. doi:10.1210/jc.2009-2703
Fain, J. N., Tichansky, D. S., and Madan, A. K. (2005). Transforming Growth Factor β1 Release by Human Adipose Tissue Is Enhanced in Obesity. Metabolism 54, 1546–1551. doi:10.1016/j.metabol.2005.05.024
Forsyth, R., and Gunay-Aygun, M. (2003). “Bardet-Biedl Syndrome Overview,” in GeneReviews® [Internet]. Editors M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, G. Mirzaaet al. (Seattle (WA): University of Washington, Seattle). Available from: https://www.ncbi.nlm.nih.gov/books/NBK1363/(updated 2020 Jul 23).
Forsythe, E., and Beales, P. L. (2013). Bardet-Biedl Syndrome. Eur. J. Hum. Genet. 21, 8–13. doi:10.1038/ejhg.2012.115
Howarth, R., Yearwood, C., and Harvey, J. F. (2007). Application of dHPLC for Mutation Detection of the Fibrillin-1 Gene for the Diagnosis of Marfan Syndrome in a National Health Service Laboratory. Genet. Test. 11 (2), 146–152. doi:10.1089/gte.2006.0514
Hubmacher, D., Tiedemann, K., and Reinhardt, D. P. (2006). Fibrillins: from Biogenesis of Microfibrils to Signaling Functions. Curr. Top. Dev. Biol. 75, 93–123. doi:10.1016/S0070-2153(06)75004-9
Jordan, C. D., Bohling, S. D., Charbonneau, N. L., and Sakai, L. Y. (2010). Fibrillins in Adult Human Ovary and Polycystic Ovary Syndrome: Is Fibrillin-3 Affected in PCOS? J. Histochem Cytochem. 58, 903–915. doi:10.1369/jhc.2010.956615
Kearney, H. M., Kearney, J. B., and Conlin, L. K. (2011). Diagnostic Implications of Excessive Homozygosity Detected by SNP-Based Microarrays: Consanguinity, Uniparental Disomy, and Recessive Single-Gene Mutations. Clin. Laboratory Med. 31, 595–613. doi:10.1016/j.cll.2011.08.003
Kircher, M., Witten, D. M., Jain, P., O'Roak, B. J., Cooper, G. M., and Shendure, J. (2014). A General Framework for Estimating the Relative Pathogenicity of Human Genetic Variants. Nat. Genet. 46, 310–315. doi:10.1038/ng.2892
Lin, M., Liu, Z., Liu, G., Zhao, S., Li, C., Chen, W., et al. (2020). Deciphering Disorders Involving Scoliosis and COmorbidities (DISCO) studyGenetic and Molecular Mechanism for Distinct Clinical Phenotypes Conveyed by Allelic Truncating Mutations Implicated in FBN1. Mol. Genet. Genomic Med. 8, e1023. doi:10.1002/mgg3.1023
Liu, H.-Y., Zhou, L., Zheng, M.-Y., Huang, J., Wan, S., Zhu, A., et al. (2019). Diagnostic and Clinical Utility of Whole Genome Sequencing in a Cohort of Undiagnosed Chinese Families with Rare Diseases. Sci. Rep. 9 (1), 19365. doi:10.1038/s41598-019-55832-1
Liu, X., Wu, C., Li, C., and Boerwinkle, E. (2016). dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 37, 235–241. doi:10.1002/humu.22932
Loeys, B. L., Matthys, D. M., and de Paepe, A. M. (2003). Genetic Fibrillinopathies : New Insights in Molecular Diagnosis and Clinical Management. Acta Clin. Belg. 58, 3–11. doi:10.1179/acb.2003.58.1.001
Mundlos, S. (2009). The Brachydactylies: a Molecular Disease Family. Clin. Genet. 76, 123–136. doi:10.1111/j.1399-0004.2009.01238.x
Muthu, M. L., and Reinhardt, D. P. (2020). Fibrillin-1 and Fibrillin-1-Derived Asprosin in Adipose Tissue Function and Metabolic Disorders. J. Cell Commun. Signal. 14, 159–173. doi:10.1007/s12079-020-00566-3
Nagase, T., Kikuno, R., Hattori, A., Kondo, Y., Okumura, K., and Ohara, O. (2000). Prediction of the Coding Sequences of Unidentified Human Genes. XIX. The Complete Sequences of 100 New cDNA Clones from Brain Which Code for Large Proteins In Vitro. DNA Res. 7 (6), 347–355. doi:10.1093/dnares/7.6.347
Niederlova, V., Modrak, M., Tsyklauri, O., Huranova, M., and Stepanek, O. (2019). Meta‐analysis of Genotype‐phenotype Associations in Bardet‐Biedl Syndrome Uncovers Differences Among Causative Genes. Hum. Mutat. 40, 2068–2087. doi:10.1002/humu.23862
Piha-Gossack, A., Sossin, W., and Reinhardt, D. P. (2012). The Evolution of Extracellular Fibrillins and Their Functional Domains. PLoS One 7, e33560. doi:10.1371/journal.pone.0033560
Ramirez, F., and Sakai, L. Y. (2010). Biogenesis and Function of Fibrillin Assemblies. Cell Tissue Res. 339, 71–82. doi:10.1007/s00441-009-0822-x
Reinhardt, D. P. (2014). Microfibril-associated Disorders. J. Glaucoma 23 (8 Suppl. 1), S34–S35. doi:10.1097/IJG.0000000000000114
Riggs, E. R., Andersen, E. F., Cherry, A. M., Kantarci, S., Kearney, H., Patel, A., et al. (2020). Technical Standards for the Interpretation and Reporting of Constitutional Copy-Number Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 22 (2), 245–257. doi:10.1038/s41436-019-0686-8
Robinson, P. N., Arteaga-Solis, E., Baldock, C., Collod-Beroud, G., Booms, P., De Paepe, A., et al. (2006). The Molecular Genetics of Marfan Syndrome and Related Disorders. J. Med. Genet. 43, 769–787. doi:10.1136/jmg.2005.039669
Sabatier, L., Chen, D., Fagotto-Kaufmann, C., Hubmacher, D., McKee, M. D., Annis, D. S., et al. (2009). Fibrillin Assembly Requires Fibronectin. MBoC 20, 846–858. doi:10.1091/mbc.e08-08-0830
Sabatier, L., Miosge, N., Hubmacher, D., Lin, G., Davis, E. C., and Reinhardt, D. P. (2011). Fibrillin-3 Expression in Human Development. Matrix Biol. 30, 43–52. doi:10.1016/j.matbio.2010.10.003
Samad, F., Yamamoto, K., Pandey, M., and Loskutoff, D. J. (1997). Elevated Expression of Transforming Growth Factor-β in Adipose Tissue from Obese Mice. Mol. Med. 3, 37–48. doi:10.1007/bf03401666
Stewart, D. R., Dombroski, B. A., Urbanek, M., Ankener, W., Ewens, K. G., Wood, J. R., et al. (2006). Fine Mapping of Genetic Susceptibility to Polycystic Ovary Syndrome on Chromosome 19p13.2 and Tests for Regulatory Activity. J. Clin. Endocrinol. Metab. 91, 4112–4117. doi:10.1210/jc.2006-0951
Tjeldhorn, L., Rand-Hendriksen, S., Gervin, K., Brandal, K., Inderhaug, E., Geiran, O., et al. (2006). Rapid and Efficient FBN1 Mutation Detection Using Automated Sample Preparation and Direct Sequencing as the Primary Strategy. Genet. Test. 10 (4), 258–264. doi:10.1089/gte.2006.258-264
Urbanek, M., Woodroffe, A., Ewens, K. G., Diamanti-Kandarakis, E., Legro, R. S., Strauss, J. F., et al. (2005). Candidate Gene Region for Polycystic Ovary Syndrome on Chromosome 19p13.2. J. Clin. Endocrinol. Metabolism 90, 6623–6629. doi:10.1210/jc.2005-0622
Vahidnezhad, H., Youssefian, L., Jazayeri, A., and Uitto, J. (2018). Research Techniques Made Simple: Genome-wide Homozygosity/Autozygosity Mapping Is a Powerful Tool for Identifying Candidate Genes in Autosomal Recessive Genetic Diseases. J. Investigative Dermatology 138, 1893–1900. doi:10.1016/j.jid.2018.06.170
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 38, e164. doi:10.1093/nar/gkq603
Wang, Y., Garraoui, A., Zeng, L., Lai, M., He, F., Wang, H., et al. (2017). FBN3 Gene Involved in Pathogenesis of a Chinese Family with Bardet-Biedl Syndrome. Oncotarget 8, 86718–86725. doi:10.18632/oncotarget.21415
Keywords: Bardet–Biedl syndrome, FBN3, fibrillinopathies, SNP-array, whole-exome sequencing
Citation: Genovesi ML, Torres B, Goldoni M, Salvo E, Cesario C, Majolo M, Mazza T, Piscopo C and Bernardini L (2022) Case Report: A Novel Homozygous Missense Variant of FBN3 Supporting It Is a New Candidate Gene Causative of a Bardet–Biedl Syndrome–Like Phenotype. Front. Genet. 13:924362. doi: 10.3389/fgene.2022.924362
Received: 20 April 2022; Accepted: 07 June 2022;
Published: 15 July 2022.
Edited by:
Antonio Percesepe, University of Parma, ItalyReviewed by:
Diana Valverde, University of Vigo, SpainYiyang Wu, Vanderbilt University Medical Center, United States
Copyright © 2022 Genovesi, Torres, Goldoni, Salvo, Cesario, Majolo, Mazza, Piscopo and Bernardini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Bernardini, bC5iZXJuYXJkaW5pQGNzcy1tZW5kZWwuaXQ=