 Han Daicheng1†
Han Daicheng1† Zhang Jingxuan
Zhang Jingxuan Wang Bo
Wang Bo- 1Department of Neonatology, Maternal and Child Health Hospital of Hubei Province, Wuhan, China
- 2Hubei Key Laboratory of Embryonic Stem Cell Research School of Basic Medical Sciences, Hubei University of Medicine, Shiyan, China
- 3Department of Pathology, Maternal and Child Health Hospital of Hubei Province, Wuhan, China
- 4Department of Clinical Laboratory, Maternal and Child Health Hospital of Hubei Province, Wuhan, China
TARP syndrome is a rare X-linked genetic condition caused by mutations in the RBM10 gene. Primary clinical characteristics of TARP syndrome include Talipes equinovarus, Atrial septal defect, Robin sequence and Persistent left superior vena cava. Newly reported cases identified a few novel RBM10 variants and atypical manifestations associated with TARP syndrome, thus expanding the genetic and clinical spectrum of TARP syndrome. Here we report a molecularly confirmed TARP syndrome with distinctive clinical features including pulmonary arteriovenous malformation, single umbilical artery, and coagulopathy. We identified a frameshift RBM10 variant that might be associated with his distinctive clinical features.
Introduction
TARP syndrome is a rare X-linked genetic disorder that causes several birth defects (Niceta et al., 2019). This syndrome was first described in 1970 as Robin’s syndrome (Gorlin et al., 1970). TARP stands for Talipes equinovarus, Atrial septal defect, Persistence of the left superior vena cava and Robin sequence (a set of abnormalities including small lower jaw (micrognathia), displacement of the tongue toward the back of the oral cavity (glossoptosis), dyspnea and an abnormal opening in the roof of the mouth (cleft palate)) (Gorlin et al., 1970). These are the most common clinical features in the early reported cases (Kurpinski et al., 2003).
However, as more patients have been identified by genetic testing, the original TARP acronym doesn’t describe the full phenotypic spectrum of this syndrome. In some cases, patients have only one or two of these features (Powis et al., 2017). Meanwhile, additional clinical features including central nervous system dysfunction, renal abnormalities, cardiac lesions, and distal limb defects have been reported, which demonstrates broad phenotypic heterogeneity among patients with TARP syndrome (Johnston et al., 2010).
TARP syndrome is caused by loss-of-function mutations in the RBM10 gene (Johnston et al., 2014). This gene maps to chromosome Xp11.23-q13.3 in human, which encodes an RNA binding motif protein that is involved in alternative splicing (Coleman et al., 1996; Thiselton et al., 2002). The function of RBM10 is not fully understood. RBM10 has been shown to regulate alternative splicing of pre-mRNA of NUMB, FAS, Dlg4, SMN2 and Even its own pre-mRNA resulting in alternative splicing-coupled nonsense-mediated mRNA decay (AS-NMD) (Loiselle and Sutherland, 2018). Disease mutations include missense and frameshift mutations that lead to abnormal or truncated proteins (Højland et al., 2018; Imagawa et al., 2020). The identification of RMB10 variants could help to clarify its clinical variabilities and shed light on the pathogenesis of TARP syndrome.
Here we report a molecularly confirmed TARPS with distinctive clinical features including pulmonary arteriovenous malformation, single umbilical artery, and coagulopathy. We identified a frameshift RBM10 variant in this patient that might be associated with his distinctive clinical features.
Methods
Patients and Samples
A male newborn was delivered by cesarean section because of oligoamnios at a gestational age of 35 weeks and 5 days with a birth weight of 1535 g. The karyotype of his mother is 47, XXX. The parents are Chinese and Han nationality, they had no family history of genetic disease. The mother and the father were 35 and 42 years old respectively at conception. This pregnancy was achieved by in vitro fertilization (IVF) due to primary infertility. During the pregnancy, the fetus was diagnosed with small mandible, single umbilical artery and permanent left superior vena cava by ultrasonic testing in 25th week of pregnancy, the mother was diagnosed with oligoamnios and asthma in her 35th week of pregnancy and treated accordingly. Immediately after birth, the newborn was admitted to the neonatal intensive care unit (NICU) because of severe respiratory distress and asphyxia. The baby looked pale and abnormally blue (cyanosis), an indication of poor circulation or insufficient oxygenation of the blood. Because it was difficult to perform endotracheal intubation in the newborn, we gave him maximal ventilator support, with a FiO2 of 100%. However, due to the severe coagulopathy and pulmonary hemorrhage, 8 h after birth, the patient died.
Molecular Analysis
We performed whole-exome sequencing (WES) on the family. The Novaseq6000 platform (Illumina, San Diego, United States), with 150 bp pair-end sequencing mode, was used for sequencing the genomic DNA of the family. The sequencing reads were aligned to the human reference genome (hg38/GRCh38) using the Burrows-Wheeler Aligner tool.
Autopsy Examination
Autopsy examination were performed including gross anatomy, histologic examinations of the lung biopsy specimen, hematoxylin and eosin (HE) staining, cranial magnetic resonance imaging (MRI) and so on.
Results
This baby had hypotonia and a set of abnormalities including a small lower jaw (micrognathia), displacement of the tongue toward the back of the oral cavity (glossoptosis), and an abnormal opening in the roof of the mouth (cleft palate), which is known as Pierre Robin sequence suggesting a diagnosis of TARP syndrome. He had low-set ears which is consistent with previously reported TARP cases (Figure 1). Further identification of the mutation in the RBM10 gene established the diagnosis. The patient presented with massive pulmonary pleural effusions and pulmonary hemorrhage (Figure 2) and coagulation disorder (Table 1).
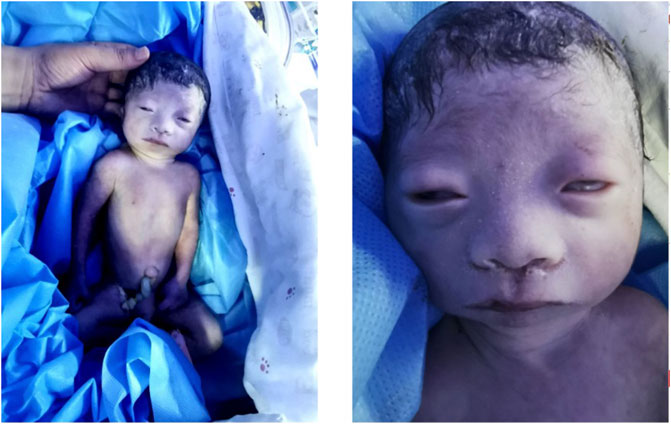
FIGURE 1. Clinical features. The baby had hypotonia, widely spaced eyes, sparse eyelashes, small lower jaw, wide nasal bridge, wide mouth with downturned corners and low-set ears.
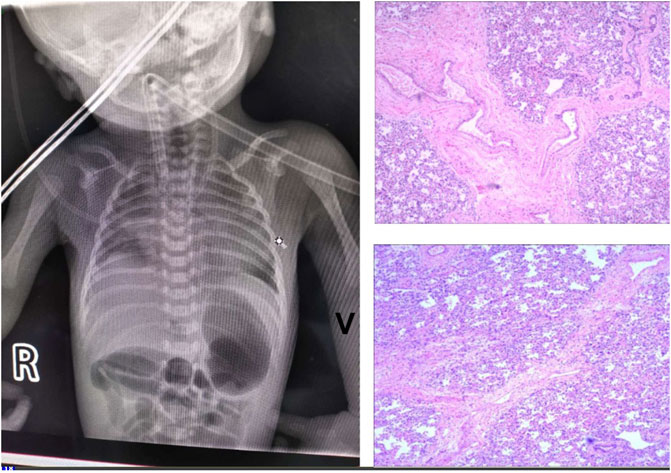
FIGURE 2. Lung imaging. (A) Chest X-Ray showed massive pulmonary pleural effusions and hemorrhage. (B) HE staining showed diffusive interstitial fibrosis (*). magnification of ×100. (C) Histologic examinations of the lung biopsy specimen demonstrated pulmonary arteriovenous malformations. There was increased number of blood vessels, some developing sac-like dilation and thickening walls (arrows).
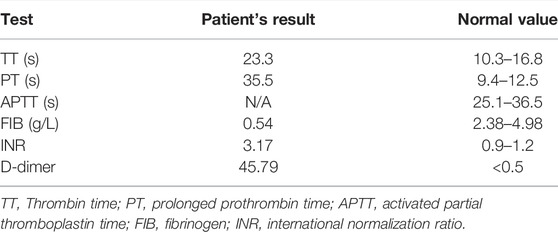
TABLE 1. Coagulation tests.
The diagnostic whole-exome sequencing showed the patient was hemizygous for a c.1113_1119del variant in RBM10. This mutation creates a frameshift at Ile372. We detected the same mutation in the mother and the parents had no family history of genetic disease (Figure 3).
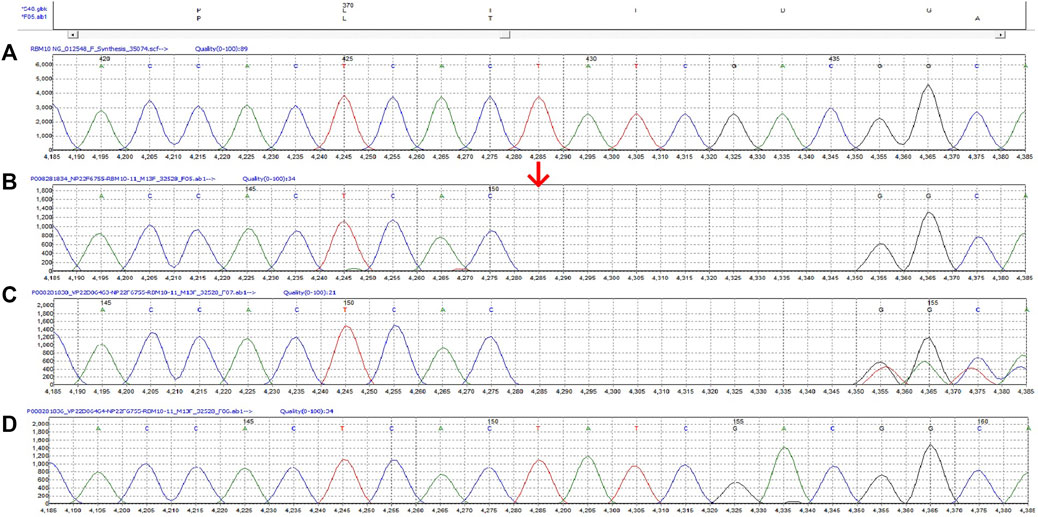
FIGURE 3. The c.1113_1119del variant in RBM10 of this family. (A) The reference sequence (B) Proband (hemizygote) (C) Mother (heterozygote) (D) Father.
Autopsy examination were performed 48 h after death and showed persistent left superior vena cava and pulmonary arteriovenous malformation, which were responsible for his death. The autopsy also revealed massive bilateral pleural effusions. Histologic examinations of the lung biopsy specimen demonstrated pulmonary arteriovenous malformations. There was increased number of blood vessels, some developing sac-like dilation with thickening walls. HE staining showed diffusive interstitial fibrosis (Figure 2).
Cranial MRI during autopsy detected small lower jaw, displacement of the tough toward the back of the oral cavity and cleft palate and lissencephaly (Figure 4). There was no gyri/sulci clearly seen for both cerebral and cerebellum cortex which are structural brain abnormalities frequently reported in TARP patients (Figure 4).
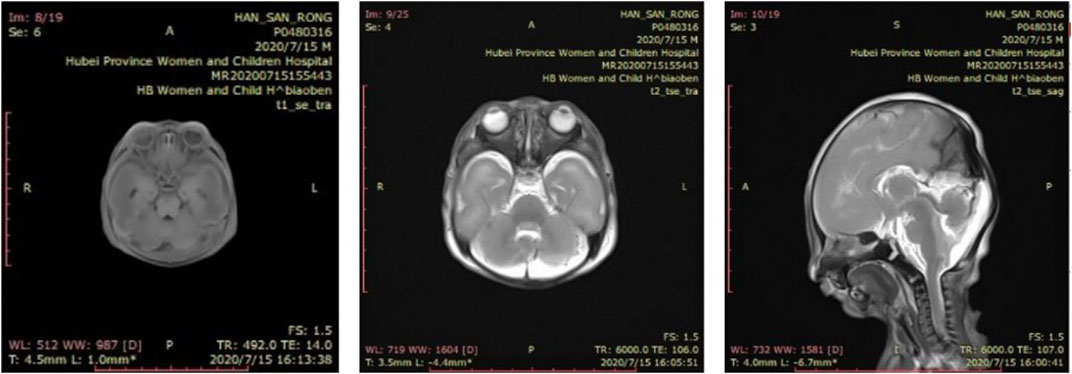
FIGURE 4. Brain imaging. Cranial MRI showed small lower jaw, displacement of the tough toward the back of the oral cavity and cleft palate and lissencephaly. There was no gyri/sulci clearly seen for both cerebral and cerebellum cortex. (A) T1-weighted coronal image (B) T2-weighted coronal image (C) T2-weighted sagittal image.
Discussion
TARP syndrome is a rare development defect during embryogenesis. Most affected males have died before or shortly after birth, usually due to various heart conditions associated with the disease (Kurpinski et al., 2003). There have been a few exceptional cases of long-term survival (Gripp et al., 2011). In our case, the patient died 8 h after birth, probably due to severe coagulopathy and massive pulmonary hemorrhage. The classic features of TARP syndrome described in the early reported cases include club foot, atrial septal defect, Robin sequence, and persistent left superior vena cava. However, newly reported cases demonstrate significant variability in clinical manifestations (Kaeppler et al., 2018; Imagawa et al., 2020). In some cases, cortical visual impairment, profound intellectual disability, and chronic lung disease are observed. Our patient showed recognizable Robin sequence and persistent left superior vena cava but lacks other features of TARP syndrome. Brain abnormalities including cerebellar hypoplasia and mega cisterna magna, which are frequent in TARP syndrome (Johnston et al., 2014) were absent in our patient. Instead, our patient had pulmonary arteriovenous malformation, single umbilical artery, and coagulopathy, which have not been reported before.
RBM10 mutations are the cause of TARP syndrome (Johnston et al., 2014). Phenotypic diversity of this condition is thought to be the result of genetic variations of the RBM10 gene including frameshift, nonsense, and deletion mutations (Wang et al., 2013). We identified a frameshift mutation (c.1113_1119del, p. Ile372fs) in exon 11 of the RBM10 gene. Although frameshift mutations are in general cause the reading of the codons after the mutation to code for different amino acid resulting in loss-of-function protein, different frameshift mutants contribute to different phenotypes in patients with TAPR syndrome. Unlike other studies showing various heart conditions and brain abnormalities associated with TARP syndrome, we found this RBM10 variant was associated with severe coagulopathy and pulmonary arteriovenous malformation.
Previous functional analysis of six RBM10 mutations that predicted to be pathogenic from the COSMIC database including one nonsense mutation, four missense mutation, and one frameshift mutation F227fs*39 (c.678delC)) found that when expressed in HEK293, the expressions of nonsense and frameshift mutation were absent demonstrating they lead to non-sense-mediated decay (Zhao et al., 2017). Another study using mouse cells has shown that RBM10 deletion could lead to splicing changes of multiple target genes that affect normal palate development and cause human disease (Rodor et al., 2017). To clarify the role of this mutant in the pathogenesis of the disease, it would be interesting the perform the functional analysis of this mutant in vitro or in animal models of lung injury or alveolar hemorrhage.
To conclude, we identified a RBM10 variant associated with TARP syndrome. Awareness of the expanded phenotypic spectrum will improve the diagnosis and genetic counselling of TARP syndrome.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
HD and XS are responsible for clinical diagnosis and treatment. ZJ and HJ are responsible for pathological examination. WB is responsible for genetic testing and thesis writing.
Funding
This work was supported by the Public Health Leading Talents Training Program of Hubei Province under Grant No. 1020013003.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Coleman, M. P., Ambrose, H. J., Carrel, L., Németh, A. H., Willard, H. F., and Davies, K. E. (1996). A Novel Gene, DXS8237E, Lies within 20 Kb Upstream of UBE1 in Xp11.23 and Has a Different X Inactivation Status. Genomics 31 (1), 135–138. doi:10.1006/geno.1996.0022
Gorlin, R. J., Červenka, J., Anderson, R. C., Sauk, J. J., and Bevis, W. D. (1970). Robin's Syndrome. Am. J. Dis. Child. 119 (2), 176–178. doi:10.1001/archpedi.1970.02100050178020
Gripp, K. W., Hopkins, E., Johnston, J. J., Krause, C., Dobyns, W. B., and Biesecker, L. G. (2011). Long-Term Survival in TARP Syndrome and Confirmation of RBM10 as the Disease-Causing Gene. Am. J. Med. Genet. 155 (10), 2516–2520. doi:10.1002/ajmg.a.34190
Højland, A. T., Lolas, I., Okkels, H., Lautrup, C. K., Diness, B. R., Petersen, M. B., et al. (2018). First Reported Adult Patient with TARP Syndrome: A Case Report. Am. J. Med. Genet. 176 (12), 2915–2918. doi:10.1002/ajmg.a.40638
Imagawa, E., Konuma, T., Cork, E. E., Diaz, G. A., and Oishi, K. (2020). A Novel Missense Variant in RBM10 Can Cause a Mild Form of TARP Syndrome with Developmental Delay and Dysmorphic Features. Clin. Genet. 98 (6), 606–612. doi:10.1111/cge.13835
Johnston, J. J., Sapp, J. C., Curry, C., Horton, M., Leon, E., Cusmano-Ozog, K., et al. (2014). Expansion of the TARP Syndrome Phenotype Associated with De Novo Mutations and Mosaicism. Am. J. Med. Genet. 164 (1), 120–128. doi:10.1002/ajmg.a.36212
Johnston, J. J., Teer, J. K., Cherukuri, P. F., Hansen, N. F., Loftus, S. K., Chong, K., et al. (2010). Massively Parallel Sequencing of Exons on the X Chromosome Identifies RBM10 as the Gene that Causes a Syndromic Form of Cleft Palate. Am. J. Hum. Genet. 86 (5), 743–748. doi:10.1016/j.ajhg.2010.04.007
Kaeppler, K. E., Stetson, R. C., Lanpher, B. C., and Collura, C. A. (2018). Infant Male with TARP Syndrome: Review of Clinical Features, Prognosis, and Commonalities with Previously Reported Patients. Am. J. Med. Genet. 176 (12), 2911–2914. doi:10.1002/ajmg.a.40645
Kurpinski, K. T., Magyari, P. A., Gorlin, R. J., Ng, D., and Biesecker, L. G. (2003). Designation of the TARP Syndrome and Linkage to Xp11.23-q13.3 without Samples from Affected Patients. Am. J. Med. Genet. 120A (1), 1–4. doi:10.1002/ajmg.a.10201
Loiselle, J. J., and Sutherland, L. C. (2018). RBM10: Harmful or Helpful‐Many Factors to Consider. J. Cell. Biochem. 119 (5), 3809–3818. doi:10.1002/jcb.26644
Niceta, M., Barresi, S., Pantaleoni, F., Capolino, R., Dentici, M. L., Ciolfi, A., et al. (2019). TARP Syndrome: Long-Term Survival, Anatomic Patterns of Congenital Heart Defects, Differential Diagnosis and Pathogenetic Considerations. Eur. J. Med. Genet. 62 (6), 103534. doi:10.1016/j.ejmg.2018.09.001
Powis, Z., Hart, A., Cherny, S., Petrik, I., Palmaer, E., Tang, S., et al. (2017). Clinical Diagnostic Exome Evaluation for an Infant with a Lethal Disorder: Genetic Diagnosis of TARP Syndrome and Expansion of the Phenotype in a Patient with a Newly Reported RBM10 Alteration. BMC Med. Genet. 18 (1), 1–5. doi:10.1186/s12881-017-0426-3
Rodor, J., FitzPatrick, D. R., Eyras, E., and Cáceres, J. F. (2017). The RNA-Binding Landscape of RBM10 and its Role in Alternative Splicing Regulation in Models of Mouse Early Development. RNA Biol. 14 (1), 45–57. doi:10.1080/15476286.2016.1247148
Thiselton, D. L., McDowall, J., Brandau, O., Ramser, J., d'Esposito, F., Bhattacharya, S. S., et al. (2002). An Integrated, Functionally Annotated Gene Map of the DXS8026-ELK1 Interval on Human Xp11.3-Xp11.23: Potential Hotspot for Neurogenetic Disorders. Genomics 79 (4), 560–572. doi:10.1006/geno.2002.6733
Wang, Y., Gogol‐Döring, A., Hu, H., Fröhler, S., Ma, Y., Jens, M., et al. (2013). Integrative Analysis Revealed the Molecular Mechanism Underlying RBM10‐Mediated Splicing Regulation. EMBO Mol. Med. 5 (9), 1431–1442. doi:10.1002/emmm.201302663
Keywords: TARP, RBM10, X-linked, pulmonary arteriovenous malformation, coagulopathy
Citation: Daicheng H, Shiwen X, Jingxuan Z, Junbo H and Bo W (2022) A Frameshift RBM10 Variant Associated With TARP Syndrome. Front. Genet. 13:922048. doi: 10.3389/fgene.2022.922048
Received: 17 April 2022; Accepted: 22 June 2022;
Published: 04 August 2022.
Edited by:
Marco Tartaglia, Bambino Gesù Children’s Hospital (IRCCS), ItalyCopyright © 2022 Daicheng, Shiwen, Jingxuan, Junbo and Bo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wang Bo, d2FuZ2JvMTAwNUAxNjMuY29t
†These authors have contributed equally to this work