
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Genet. , 04 August 2022
Sec. Human and Medical Genomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.921910
 Zimeng Gao1
Zimeng Gao1 Stephanie M. Rice2
Stephanie M. Rice2 Sascha Wodoslawsky3Sara C. Long3
Sascha Wodoslawsky3Sara C. Long3 Zi-Xuan Wang4Mehnoosh Torkzaban5Ana Milena Angarita Africano2
Zi-Xuan Wang4Mehnoosh Torkzaban5Ana Milena Angarita Africano2 Jinglan Liu4Huda B. Al-Kouatly2*
Jinglan Liu4Huda B. Al-Kouatly2*We aim to determine the spectrum of cytogenetic abnormalities and outcomes in unbalanced offspring of asymptomatic constitutional balanced t(9;22) carriers through a systematic literature review. We also include a case of a constitutional balanced t(9;22) carrier from our institution. Among the 16 balanced t(9;22) carriers in our review, 13 were maternal and 3 were paternal. Of the 15 unbalanced translocation cases identified, 13 were live births, one was a missed abortion, and one resulted in pregnancy termination. The spectrum of established syndromes reported among the live births was the following: trisomy 9p syndrome (6/13), dual trisomy 9p and DiGeorge syndrome (3/13), dual 9q subtelomere deletion syndrome and DiGeorge syndrome (1/13), 9q subtelomere deletion syndrome (1/13), and DiGeorge syndrome (1/13). One unbalanced case did not have a reported syndrome. The phenotype of the unbalanced cases included cardiac abnormalities (5/13), neurological findings (7/13), intellectual disability (6/10), urogenital anomalies (3/13), respiratory or immune dysfunction (3/13), and facial or skeletal dysmorphias (13/13). Any constitutional balanced reciprocal t(9;22) carrier should be counseled regarding the increased risk of having a child with an unbalanced translocation, the spectrum of possible cytogenetic abnormalities, and predicted clinical phenotype for the unbalanced derivative.
Chromosomal translocations are structural abnormalities characterized by the exchange of chromosomal segments between nonhomologous and homologous chromosomes. Balanced translocations refer to instances of chromosomal rearrangement without apparent loss in genetic material and full functionality, while unbalanced translocations result in an overall genetic gain or loss (Morin et al., 2017). Balanced or unbalanced structural translocations have a frequency of 1.7% in the general population (Paththinige et al., 2019). Apparently balanced chromosomal arrangements have an estimated frequency of 0.5% in newborns, of which 14% are de novo (Jacobs et al., 1992).
Apparently constitutional balanced translocations associated with normal phenotypes can be transmitted through generations without major adverse effects, except for an increased risk of reproductive loss or abnormal phenotypes in offspring (Gribble et al., 2005). Phenotypically normal carriers of balanced translocations are at a higher risk of producing offspring with unbalanced translocations with potential outcomes of developmental delay, congenital cardiac defects, urogenital anomalies, respiratory or immune dysfunctions, or facial or skeletal dysmorphias (Ganguly et al., 2011). These carriers of balanced translocations are frequently identified only after recurrences of unbalanced offspring within families. If an abnormal phenotype is identified in a balanced translocation carrier, chromosomal microarray analysis (CMA) or next-generation sequencing (NGS) may be performed to detect possible copy-number variation (CNV) at breakpoints of the translocation (Weckselblatt and Rudd, 2015). Submicroscopic deletions or duplications can be seen in 46% of individuals with an abnormal phenotype who carry a de novo apparently balanced chromosomal rearrangement, and in 25% of affected individuals with an inherited apparently balanced chromosomal rearrangement (Tabet et al., 2015).
Several uncommon constitutional balanced translocations have been reported, which include: t(8;22)(q24.13;q11.21), t(4;8)(p16;p23), t(4;11)(p16.2;p15.4), t(8;12)(p23.1;p13.31), and t(9;22)(q34;q11.2). These translocations result from nonallelic homologous recombination through breaks in adenine and thymine residue-rich (AT-rich) repeats repaired by nonhomologous end-joining (Czuchlewski et al., 2011; Ou et al., 2011). Some balanced translocations such as the Philadelphia chromosome, t(9;22)(q34;q11.2), may be postnatally acquired. This acquired translocation is characterized by a BCR-ABL1 gene fusion responsible for chronic myeloid leukemia (CML), certain subtypes of acute myeloid leukemia (AML), and acute lymphoblastic leukemia (ALL).
In our systematic literature review of constitutional reciprocal t(9;22) carriers, we describe the genetic and phenotypic characteristics of their unbalanced offspring. Our review was inspired by a case at our institution, in which a patient with a history of recurrent pregnancy loss carried a constitutional balanced t(9,22)(q34;q11.2). This uncommon translocation, which mimics the somatic balanced translocation seen in CML, AML, and ALL subtypes, has only been reported twice in prior literature (McGoey and Lacassie, 2009; Czuchlewski et al., 2011). In this review, we investigate how this translocation pertains to her pregnancies.
A 31-year-old G3P0020 woman of Indian descent presented for prenatal genetic counseling with her partner at 13w5d gestation. The patient has a past medical history of pulmonic stenosis and hidradenitis suppurativa. Her partner is also of Indian descent and a self-reported heterozygote for Factor V Leiden identified through direct-to-consumer genetic testing. The couple had previously experienced two early first trimester pregnancy losses, and genetic testing was not performed on the couple or the products of conception in either pregnancy loss. The couple underwent a workup for recurrent pregnancy loss at a local reproductive endocrinology and infertility clinic.
The patient’s partner had a normal male karyogram (46, XY), while the patient was found to have an apparently balanced reciprocal translocation between chromosomes 9 and 22 [46,XX,t(9;22)(q34;q11.2)] (Figure 1C; Appendix B). The breakpoints of her constitutional translocation are similar to those of the somatic balanced translocation t(9,22)(q34;q11.2) observed in CML and certain AML and ALL subtypes. Given these break point similarities, fluorescent in situ hybridization (FISH) was performed on both interphase nuclei and metaphase cells (Appendix B). The interphase FISH study showed no BCR-ABL1 fusion (Figure 1A), while metaphase FISH demonstrated a translocation between the long arms of chromosomes 9 and 22 at 9q34 and 22q11.2, respectively, without involvement of the ABL1 and BCR genes (Figures 1B, D). Despite having seemingly identical chromosomal alterations by conventional cytogenetic analysis, the negative FISH results confirmed that our patient did not have a pathogenic t(9;22)(9q34;22q11.2) that results in a BCR-ABL1 chimeric product.
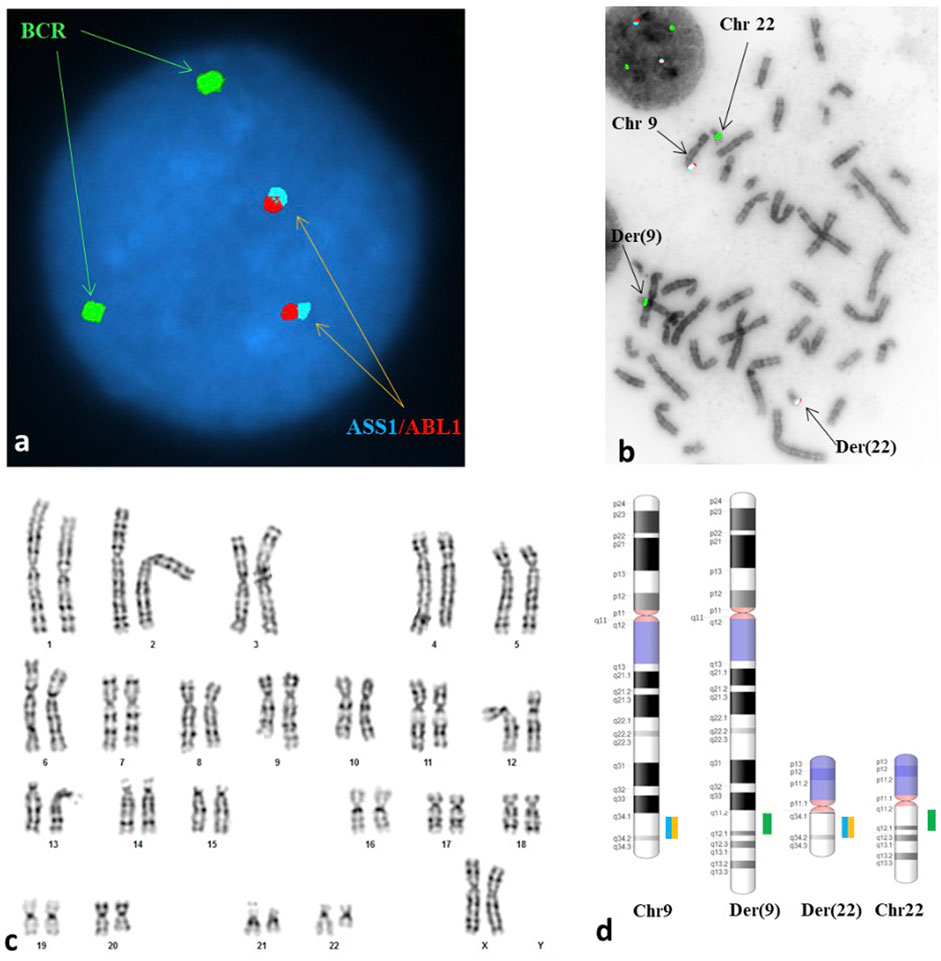
FIGURE 1. A reciprocal translocation of t(9;22)(q34;q11.2) involving neither the ABL1 locus at 9q34 nor the BCR at 22q11.2 was seen in each of the metaphases analyzed. (A) Interphase FISH study showed a normal signal pattern; probes for the ASS1*, ABL1 and BCR were labeled in aqua, red, and green, respectively. (B) Metaphase FISH study showed an abnormal hybridization pattern with no translocation of BCR/ABL1 observed. The arrows are pointing to the derivative chromosome 9 [der(9)] carrying a signal for BCR but no signals for the ASS1 and ABL1, the derivative chromosome 22 [der(22)] carrying signals for the ASS1 and ABL1 but no signal for the BCR, the normal chromosome 9 [chr 9] and the normal chromosome 22 [chr 22]. (C) Karyogram exhibited an apparently balanced reciprocal translocation between the long arm of chromosome 9 at 9q34 and the short arm of chromosome 22 at 22q11.2, with morphology indistinguishable from the Philadelphia translocation. (D) A schematic illustration of the normal copies of chromosomes 9 and 22 as well as the derivative chromosomes 9 and 22 resulted from the reciprocal translocation. The breakpoints at each derivative chromosome involved were located at the vicinity but proximal to the loci involved in the Philadelphia translocation. *ASS1 is a control probe at 9q34 overlapping the ABL1 probe.
Additionally, no evidence of leukemic process was identified on complete blood count (CBC). Further workup with reverse transcription-polymerase chain reaction (RT-PCR) was recommended to quantitatively detect t(9;22) BCR-ABL1 transcripts. The absence of transcripts would rule out the possibility of a cryptic translocation that could result in leukemia, though unlikely given the patient’s normal CBC. The patient declined further workup with RT-PCR.
The patient’s nuchal translucency ultrasound and first trimester sequential screen were normal. After reviewing her karyotype and discussing the increased risk of an unbalanced translocation in her fetus, the patient elected amniocentesis. The results of the amniocentesis revealed a normal male fetal karyotype (46,XY) with no clinically significant numerical or structural chromosome abnormalities. Chromosome 9 and 22 homologs appeared normal at the 450-band resolution of karyotyping. Additionally, the CMA was consistent with a normal male fetus without clinically significant CNV or copy neutral regions detected at a resolution of 50 Kb or higher (Appendix B). Finally, both her amniotic fluid alpha-fetoprotein (AF-AFP) testing and limited fetal anatomy ultrasound completed at the time of amniocentesis were normal.
An echocardiogram was performed to evaluate the patient’s pulmonic stenosis at 18 weeks gestation, which indicated an increase in trans-pulmonic gradient as compared to her cardiac MRI images from two years prior. This increase, though still in the mild range, was attributed to the high-flow state in pregnancy. A repeat echocardiogram was completed at 30 weeks gestation, which indicated no significant changes since the prior study at 18 weeks.
The patient underwent parental CMA to determine the etiology of her pulmonic valve stenosis by evaluating sub-microscopic CNV(s) not detectable by standard karyotyping. Her CMA results were normal, making the translocation an unlikely cause of her cardiac finding. However, the normal result does not exclude the presence of a CNV <50 Kb or the possibility of a monogenic disorder as the cause of the stenosis. The patient declined single gene testing.
The remainder of the pregnancy was uneventful except for the identification of a fetal right pelvic kidney with normal renal parenchyma diagnosed at 20 weeks gestation by anatomy ultrasound. The patient had a spontaneous vaginal delivery of a live male infant weighing 3.37 kg at 40w3d. Apgar scores were 8 and 9 at 1 and 5 minutes, respectively. The neonate was discharged on the second day of life with no complications. He is currently in good health at 17 months of age without any health consequences from his right pelvic kidney.
The literature review was conducted using PubMed and Scopus from database inception to 10 October 2021. Articles discussing constitutional translocations of chromosomes 9 and 22 were identified without limitations on study date or type. Only literature in English were included (Figure 2). The following search terms were used to identify extant literature: 9;22, (9;22)(q34;q11.2), t(9;22)(q34;11.2), translocation, congenital, constitutional, and inherited (Appendix A). The reference list of each article identified was reviewed for additional articles. Data extraction from the articles was performed according to PRISMA guidelines (Figure 2) (Page et al., 2021). We refer to unbalanced t(9;22) offspring of balanced t(9;22) carriers as “cases” in our review.
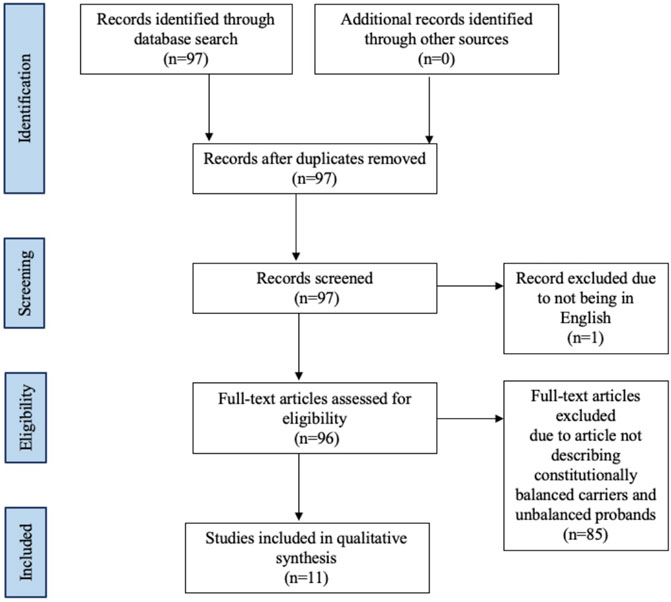
FIGURE 2. Flow diagram of studies identified in the systematic review.
A total of 11 studies including 15 cases of unbalanced translocations from a balanced t(9;22) carrier parent met our inclusion criteria. Table 1 summarizes the cytogenetics of these unbalanced cases. One balanced translocation carrier had primary ovarian insufficiency and thus no pregnancies reported (Peterson et al., 2019). Twelve cases were maternal in origin, while three were paternal. Of the 15 pregnancies included in our literature review, 13 resulted in live births, one resulted in a missed abortion, and one pregnancy was terminated (Tables 2, 3). Of the 13 live births, three cases passed away from cardiopulmonary complications within six months of birth and 10 cases survived to adulthood or the time of publication with ages ranging from 22 months to 35 years. Pregnancy complications were reported for five of the 13 live births (Table 2). Two neonates were small for gestational age, one of whom also had polyhydramnios and required cesarean delivery secondary to fetal distress. One neonate had an irregular fetal heart rate detected at 33 weeks gestation. Two neonates had uneventful prenatal courses.

TABLE 1. Summary of cytogenetic findings of unbalanced t(9;22) cases.
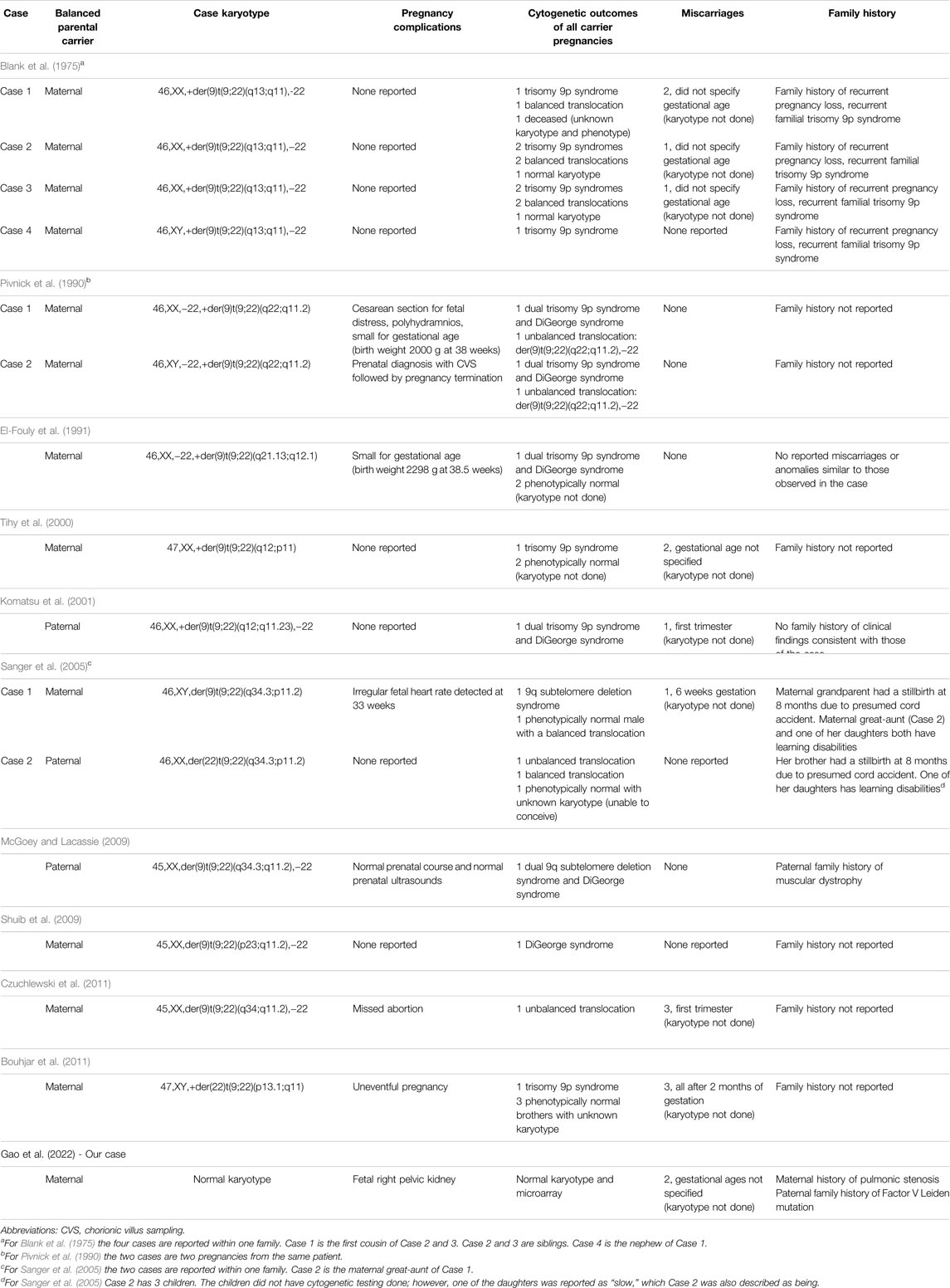
TABLE 2. Pregnancy outcomes of constitutional reciprocal t(9;22) carriers.
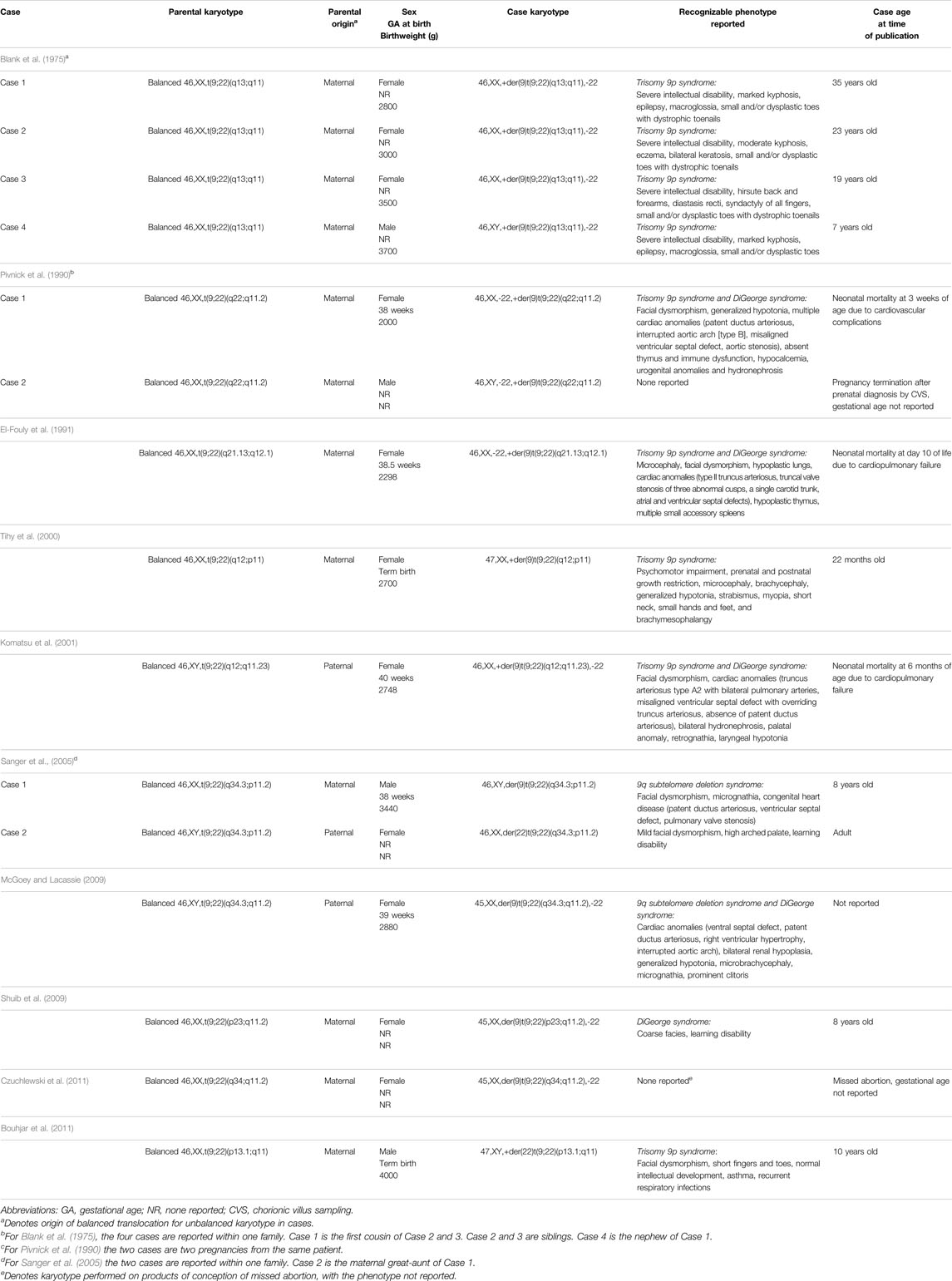
TABLE 3. Unbalanced translocation cases resulting from constitutional reciprocal t(9;22) carriers.
The phenotypes of the unbalanced cases include an array of features such as cardiac abnormalities, neurological findings, intellectual disability, urogenital abnormalities, respiratory and immune dysfunctions, and facial or skeletal dysmorphias (Table 3, Table 4). Only a few of these findings were found in isolation.

TABLE 4. Summary of phenotypes of unbalanced t(9;22) cases.
Congenital heart disease was found in five of 13 live births. Of these five cases, three passed away from cardiopulmonary complications within six months of birth, one case was eight years old at time of report, and one case did not have a reported clinical outcome. Of these same five cases, three had a dual diagnosis of trisomy 9p syndrome and DiGeorge syndrome (22q11.2 deletion syndrome), one had 9q subtelomere deletion syndrome, and one had a dual diagnosis of 9q subtelomere deletion syndrome and DiGeorge syndrome (Table 3). All three neonatal deaths from cardiac abnormalities had dual diagnoses of trisomy 9p and DiGeorge syndrome. All eight unbalanced cases without significant cardiac abnormalities survived to adulthood or to the time of publication.
Seven of 13 live births presented with neurological findings. Two cases had epilepsy (Blank et al., 1975), three had generalized hypotonia (Pivnick et al., 1990; Tihy et al., 2000; McGoey and Lacassie, 2009), and one had laryngeal hypotonia (Komatsu et al., 2001). El-Fouly, Tihy, and McGoey also describe microcephaly, brachycephaly, or a combination of both conditions in their publications (McGoey and Lacassie, 2009; Tihy et al., 2000; El-Fouly et al., 1991).
At least six of the 10 live births (60%) who were able to be cognitively assessed have some degree of intellectual or learning disability. Blank et al. (1975) reports four cases with severe intellectual disability. Sanger and Shuib each report one case with a learning disability (Sanger et al., 2005; Shuib et al., 2009).
Urogenital findings were reported in three of the 13 cases. Pivnick et al. (1990) described one case of hydronephrosis and hypoplastic and posteriorly fused flat labia majora, while Komatsu et al. (2001) reported bilateral hydronephrosis in one case. McGoey and Lacassie (2009) reported one case with bilateral renal hypoplasia, unilateral simple renal cysts, and a prominent clitoris.
Three of the 15 cases had pulmonary or immune dysfunction. One case reported in Pivnick et al. (1990) had an absent thymus with subsequent immune dysfunction. El-Fouly et al. (1991) reported one case with hypoplastic lungs and thymus, and Bouhjar et al. (2011) reported one case with asthma with recurrent respiratory infections.
All unbalanced cases with reported phenotypes had some form of facial or skeletal dysmorphia. Seven of the 13 live births had facial dysmorphia, and four had malformations such as microbrachycephaly, microcephaly, micrognathia, retrognathia, and macroglossia (McGoey and Lacassie, 2009; Blank et al., 1975; Pivnick et al., 1990; Komatsu et al., 2001; El-Fouly et al., 1991; Sanger et al., 2005; Shuib et al., 2009; Bouhjar et al., 2011). Six cases had reported skeletal anomalies, all of which include small bones in the hands and feet. Blank et al. (1975) reports four cases with small and/or dysplastic toes, Tihy et al. (2000) reports one case of small hands and feet, and Bouhjar et al. (2011) reports one case of short fingers and toes.
Nine of the 15 balanced carriers (60%) had at least one prior miscarriage (Table 2). Recurrent pregnancy loss, defined as ≥2 pregnancy losses, occurred in five (33%) pregnancies. All but one miscarriage involved balanced translocations of maternal origin (Komatsu et al., 2001). Four of the balanced carriers, three of which were maternal and one of which was paternal, did not have any miscarriages. Additionally, the four family members in Blank et al. (1975) with trisomy 9p syndrome, including the parents, maternal grandparents, and first cousins of Case 1, reported several miscarriages. Three balanced carriers did not specify a history of miscarriage.
Eight of the 15 unbalanced cases included information on their siblings. Case 1 in Blank et al. (1975) had two siblings, one of whom had a balanced translocation, and the other resulted in an infant death without a reported cause or genetic workup. Case 2 and 3 in the same paper were siblings with three other siblings in the household, two of whom had the balanced translocation, while the other had a normal karyotype. The unbalanced cases described by El-Fouly et al. (1991) and Tihy et al. (2000) each have two siblings who are phenotypically normal with an unknown karyotype. Case 1 in Sanger et al. (2005) has one brother with a balanced translocation. Case 2 has two brothers, one with a balanced translocation and the other phenotypically normal with an unknown karyotype, but reported infertility. The cases in Bouhjar et al. (2011) had three total siblings, all of whom were phenotypically normal but had unknown karyotypes.
The apparently balanced translocations of healthy individuals can remain undetected until pregnancy when patients present with recurrent pregnancy loss or fetal congenital abnormalities (Gardner, 2004). Our patient, who only had a past medical history of mild pulmonic stenosis, sought reproductive counseling due to recurrent pregnancy loss and was diagnosed with a constitutional balanced t(9;22)(q34;q11.2). The unusual cytogenetics of our patient inspired this literature search, which found no previously published systematic reviews describing the cytogenetics and phenotypic characteristics of unbalanced t(9;22) cases.
Though balanced translocations are common, occurring in at least 1 in 500 individuals, our patient’s constitutional balanced t(9;22)(q34;q11.2) is rare (Gardner, 2004). Her distinct translocation and abnormal metaphase FISH finding, albeit without gene fusion of BCR-ABL1, could have led to a misdiagnosis of CML if she had also presented with leukocytosis. Czuchlewski et al. (2011) reported a patient with constitutional t(9;22)(q34;11.2) and leukocytosis, which was differentiated from CML using FISH and the identification of the constitutional translocation in all cells examined. FISH demonstrated a juxtaposition of two intact probes rather than the fusion seen in the Philadelphia chromosome. Our patient’s translocation was also observed in all cells examined, indicating a similar germline translocation that mimics the Philadelphia chromosome.
Balanced t(9;22) carriers and their families should be counseled on the fertility implications and the reproductive risks of t(9;22) to help guide parents in their reproductive decision-making. Firstly, parents with a balanced t(9;22) have an increased risk of pregnancy loss. Though it is unclear if our patient’s previous pregnancy losses were related to her balanced translocation, we found in our review that most balanced t(9;22) carriers had at least one miscarriage, many of whom had recurrent pregnancy loss. Translocations of maternal origin carry an increased risk for recurrent pregnancy loss, while translocations of paternal origin increase the risk of male infertility (Griffin and Finch, 2005). In a cohort study of 132 Sri Lankan balanced translocation carriers, 63.6% experienced subfertility or recurrent pregnancy loss, likely due to an unbalanced translocation (Paththinige et al., 2019). These manifestations carry profound consequences for patients attempting family planning.
Secondly, balanced t(9;22) carriers should be counseled on their risk of having a child with an unbalanced translocation. Based on the breakpoints of our patient’s translocation, she has an increased likelihood of having a child with DiGeorge syndrome, 9q subtelomeric deletion syndrome, dual genetic diagnosis of both syndromes, or other unbalanced genomic outcomes (McGoey and Lacassie, 2009). A prospective trial in 2012 analyzed the reproductive outcomes of 54 balanced translocation carriers over two years and found that 12.5% had a subsequent pregnancy with the same or a different unbalanced translocation cytogenetically or phenotypically from their first affected pregnancy (Kochhar and Ghosh, 2013). Children who carry their parents’ balanced translocation also have a theoretical risk of transmitting an unbalanced translocation to their future offspring (Sanger et al., 2005). Balanced t(9;22) carriers are also at an increased risk of having a child with a spectrum of phenotypes associated with unbalanced t(9;22) cases, including cardiac abnormalities, neurological findings, intellectual disability, urogenital anomalies, respiratory or immune dysfunction, and facial or skeletal dysmorphias. However, our findings do suggest that unbalanced cases born to parents with balanced t(9;22) have a high rate of survival into adulthood in the absence of major cardiac abnormalities.
This systematic literature review is the only comprehensive assessment of constitutional t(9;22) carriers and the outcomes and phenotypes of their unbalanced offspring. We also incorporated a case from our institution of a balanced t(9;22)(q34;q11.2), an analog of the somatic balanced translocation seen in the Philadelphia chromosome of CML. There are only two other published cases of a patient with balanced t(9;22)(q34;q11.2) (McGoey and Lacassie, 2009; Czuchlewski et al., 2011). The rarity of such a translocation underscores the importance of publishing similar cases.
A limitation of this review is the inclusion of clinical cases of t(9;22) with various chromosomal breakpoints, each of which are associated with a range of possible phenotypic outcomes. There is a relatively small number of accrued cases and each paper reported their cases with a varying level of detail. Additionally, five of the 15 balanced translocation carriers included in the review did not have the karyotype results for each of their offspring reported, limiting insight into the cytogenetic variability amongst siblings (Blank et al., 1975; Tihy et al., 2000; El-Fouly et al., 1991; Sanger et al., 2005; Bouhjar et al., 2011). Reported family histories of balanced translocation carriers and history of prior miscarriages were also sparse. Given the known propensity of balanced t(9;22) for familial recurrence and recurrent pregnancy losses, a lack of reporting limits our study’s ability to review the significance of these correlations in this population.
Our review provides a cytogenetic breakpoint-based genotype-phenotype correlation for unbalanced cases of parental constitutional reciprocal translocation (9;22) carriers. This literature review helps providers give anticipatory guidance to balanced t(9;22) carriers on potential pregnancy outcomes. Balanced t(9;22) carriers should be counseled on the increased risk of recurrent pregnancy loss, subfertility, and having a child with an unbalanced translocation that could lead to an array of phenotypic manifestations. Phenotypic findings in unbalanced cases include cardiac abnormalities, neurological findings, intellectual disability, urogenital anomalies, respiratory or immune dysfunction, and facial or skeletal dysmorphias. Genetic counseling is recommended for any balanced translocation carrier or breakpoints regardless of the chromosomes that are involved.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Study conception and design: HA-K and ZG—Data collection: ZG and SL—Analysis and interpretation of results: ZG, SR, Z-XW, JL, and HA-K—Draft manuscript preparation: ZG, SR, SL, SW, MT, AA, JL, and HA-K.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Blank, C. E., Colver, D. C., Potter, A. M., McHugh, J., and Lorber, J. (1975). Physical and Mental Defect of Chromosomal Origin in Four Individuals of the Same Family. Trisomy for the Short Arm of 9. Clin. Genet. 7 (4), 261–273. doi:10.1111/j.1399-0004.1975.tb00328.x
Bouhjar, I. B., Hannachi, H., Zerelli, S. M., Labalme, A., Gmidène, A., Soyah, N., et al. (2011). Array-CGH Study of Partial Trisomy 9p without Mental Retardation. Am. J. Med. Genet. A 155a (7), 1735–1739. doi:10.1002/ajmg.a.34044
Czuchlewski, D. R., Farzanmehr, H., Robinett, S., Haines, S., and Reichard, K. K. (2011). t(9;22)(q34;q11.2) Is a Recurrent Constitutional Non-robertsonian Translocation and a Rare Cytogenetic Mimic of Chronic Myeloid Leukemia. Cancer Genet. 204 (10), 572–576. doi:10.1016/j.cancergen.2011.10.006
El-Fouly, M. H., Higgins, J. V., Kapur, S., Sankey, B. J., Matisoff, D. N., and Costa-Fox, M. (1991). DiGeorge Anomaly in an Infant with Deletion of Chromosome 22 and Dup(9p) Due to Adjacent Type II Disjunction. Am. J. Med. Genet. 38 (4), 569–573. doi:10.1002/ajmg.1320380415
Ganguly, B. B., Kadam, V., and Kadam, N. N. (2011). Clinical Expression of an Inherited Unbalanced Translocation in Chromosome 6. Case Rep. Genet. 2011, 396450. doi:10.1155/2011/396450
R. S. G. Gardner (Editor) (2004). Chromosome Abnormalities and Genetic Counseling 3rd Edition. 3 ed (New York: Oxford University Press).
Gribble, S. M., Prigmore, E., Burford, D. C., Porter, K. M., Ling Ng, B., Douglas, E. J., et al. (2005). The Complex Nature of Constitutional De Novo Apparently Balanced Translocations in Patients Presenting with Abnormal Phenotypes. J. Med. Genet. 42 (1), 8–16. doi:10.1136/jmg.2004.024141
Griffin, D. K., and Finch, K. A. (2005). The Genetic and Cytogenetic Basis of Male Infertility. Hum. Fertil. (Camb) 8 (1), 19–26. doi:10.1080/14647270400016407
Jacobs, P. A., Browne, C., Gregson, N., Joyce, C., and White, H. (1992). Estimates of the Frequency of Chromosome Abnormalities Detectable in Unselected Newborns Using Moderate Levels of Banding. J. Med. Genet. 29 (2), 103–108. doi:10.1136/jmg.29.2.103
Kochhar, P. K., and Ghosh, P. (2013). Reproductive Outcome of Couples with Recurrent Miscarriage and Balanced Chromosomal Abnormalities. J. Obstet. Gynaecol. Res. 39 (1), 113–120. doi:10.1111/j.1447-0756.2012.01905.x
Komatsu, H., Kihara, A., Komura, E., Mitsufuji, N., Tsujii, H., Kakita, S., et al. (2001). Combined Trisomy 9P and Shprintzen Syndrome Resulting from a Paternal T(9;22). Genet. Couns. 12 (2), 137–143.
McGoey, R. R., and Lacassie, Y. (2009). Paternal Balanced Reciprocal Translocation t(9;22)(q34.3;q11.2) Resulting in an Infant with Features of the 9q Subtelomere and the 22q11 Deletion Syndromes Due to 3:1 Meiotic Segregation and Tertiary Monosomy. Am. J. Med. Genet. A 149A (11), 2538–2542. doi:10.1002/ajmg.a.33078
Morin, S. J., Eccles, J., Iturriaga, A., and Zimmerman, R. S. (2017). Translocations, Inversions and Other Chromosome Rearrangements. Fertil. Steril. 107 (1), 19–26. doi:10.1016/j.fertnstert.2016.10.013
Ou, Z., Stankiewicz, P., Xia, Z., Breman, A. M., Dawson, B., Wiszniewska, J., et al. (2011). Observation and Prediction of Recurrent Human Translocations Mediated by NAHR between Nonhomologous Chromosomes. Genome Res. 21 (1), 33–46. doi:10.1101/gr.111609.110
Page, M. J., McKenzie, J. E., Bossuyt, P. M., Boutron, I., Hoffmann, T., Mulrow, C. D., et al. (2021). The PRISMA 2020 Statement: an Updated Guideline for Reporting Systematic Reviews. BMJ 372, n71. doi:10.1136/bmj.n71
Paththinige, C. S., Sirisena, N. D., Kariyawasam, U. G. I. U., and Dissanayake, V. H. W. (2019). The Frequency and Spectrum of Chromosomal Translocations in a Cohort of Sri Lankans. Biomed. Res. Int. 2019, 9797104. doi:10.1155/2019/9797104
Peterson, J. F., Pitel, B. A., Smoley, S. A., Smadbeck, J. B., Johnson, S. H., Vasmatzis, G., et al. (2019). Constitutional Chromosome Rearrangements that Mimic the 2017 World Health Organization "acute Myeloid Leukemia with Recurrent Genetic Abnormalities": A Study of Three Cases and Review of the Literature. Cancer Genet. 230, 37–46. doi:10.1016/j.cancergen.2018.11.005
Pivnick, E. K., Wilroy, R. S., Summitt, J. B., Tucker, B., Herrod, H. G., and Tharapel, A. T. (1990). Adjacent-2 Disjunction of a Maternal T(9;22) Leading to Duplication 9pter–--q22 and deficiency of 22pter–--q11.2. Am. J. Med. Genet. 37 (1), 92–96. doi:10.1002/ajmg.1320370121
Sanger, T. M., Olney, A. H., Zaleski, D., Pickering, D., Nelson, M., Sanger, W. G., et al. (2005). Cryptic duplication and deletion of 9q34.3--> qter in a family with a t(9;22)(q34.3;p11.2). Am. J. Med. Genet. A 138 (1), 51–55. doi:10.1002/ajmg.a.30922
Shuib, S., Abdul Latif, Z., Abidin, N. Z., Akmal, S. N., and Zakaria, Z. (2009). Inherited t(9;22) as the cause of DiGeorge syndrome: a case report. Malays J. Pathol. 31 (2), 133–136.
Tabet, A.-C., Verloes, A., and Pilorge, M. (2015). Complex nature of apparently balanced chromosomal rearrangements in patients with autism spectrum disorder. Mol. Autism 6, 19. doi:10.1186/s13229-015-0015-2
Tihy, F., Lemyre, E., Dallaire, L., and Lemieux, N. (2000). Supernumerary chromosome inherited from a maternal balanced translocation leading to pure trisomy 9p. Am. J. Med. Genet. 91 (5), 383–386. doi:10.1002/(sici)1096-8628(20000424)91:5<383::aid-ajmg12>3.0.co;2-g
Database: PubMed
Results: 45
Search: (“(9;22)” AND translocation AND (congenital OR constitutional OR inherited))
Database: Scopus
Results: 52
Search: TITLE-ABS-KEY(“9;22”) AND TITLE-ABS-KEY(translocation) AND TITLE-ABS-KEY((congenital OR constitutional OR inherited))
Methods for Cytogenetic Analysis, FISH, and CMA for Our Case Report
Conventional Cytogenetic Analysis for Our Case Report
Peripheral blood lymphocytes were cultured in PB-MAX™ medium supplemented with stimulant phytohemagglutinin (PHA) for 72 h. Metaphase chromosomes for Giemsa banding pattern by trypsin digestion with Wright stain (GTW banding) were prepared according to standard procedures. Metaphases were karyotyped with GenASI Cytogenetics Analysis System (Applied Spectral Imaging, Carlsbad, CA), and karyograms were described according to the International System for Human Cytogenetic Nomenclature 2021. Coordinates of cells with chromosome aberrations were documented, and the slides were subsequently prepared for metaphase FISH analysis.Interphase and Metaphase FISH (Fluorescence in situ Hybridization) Assays for Our Case Report
Interphase FISH was undertaken using BCR/ABL1/ASS1 Tri-Color DF FISH Probe (Vysis, Abbott Park, IL). Metaphase FISH was performed on the de-stained GTW slides. Standard FISH protocol was followed. FISH images were captured and analyzed with a GenASI FISHView Analysis System (Applied Spectral Imaging, Carlsbad, CA).Chromosome Microarray Analysis for Our Case Report
Single nucleotide polymorphisms (SNP) microarray analysis was performed using the Affymetrix Cytoscan HD platform which uses over 743,000 SNP probes and 1,953,000 NPCN probes with a median spacing of 0.88 kb. 250ng of total genomic DNA extracted from lymphocytes was digested with Nspl and then ligated to Nspl adaptors, respectively, and amplified using Titanium Taq with a GeneAmp PCR System 9700. PCR products were purified using AMPure beads and quantified using NanoDrop 8000. Purified DNA was fragmented and biotin labeled and hybridized to the Affymetric Cytosan HD GeneChip. Data was analyzed using Chromosome Analysis Suite. The analysis is based on the GRCh37/hg19 assembly.
Keywords: translocation, chromosome aberrations, Philadelphia chromosome, chromosome breakpoints, reproductive counseling
Citation: Gao Z, Rice SM, Wodoslawsky S, Long SC, Wang Z-X, Torkzaban M, Angarita Africano AM, Liu J and Al-Kouatly HB (2022) A Systematic Review of Reproductive Counseling in Cases of Parental Constitutional Reciprocal Translocation (9;22) Mimicking BCR-ABL1. Front. Genet. 13:921910. doi: 10.3389/fgene.2022.921910
Received: 16 April 2022; Accepted: 02 June 2022;
Published: 04 August 2022.
Edited by:
Gavin R. Oliver, Mayo Clinic, United StatesReviewed by:
Somprakash Dhangar, Indian Council of Medical Research (ICMR), IndiaCopyright © 2022 Gao, Rice, Wodoslawsky, Long, Wang, Torkzaban, Angarita Africano, Liu and Al-Kouatly. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huda B. Al-Kouatly, aHVkYS5hbC1rb3VhdGx5QGplZmZlcnNvbi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.