
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Genet., 22 July 2022
Sec. Genomics of Plants and the Phytoecosystem
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.900253
This article is part of the Research TopicGenetics and Epigenetics: Plausible Role in Development of Climate Resilient CropsView all 17 articles
 B. S. Chandana1
B. S. Chandana1 Rohit Kumar Mahto1
Rohit Kumar Mahto1 Rajesh Kumar Singh1
Rajesh Kumar Singh1 Rebecca Ford2
Rebecca Ford2 Niloofar Vaghefi3
Niloofar Vaghefi3 Santosh Kumar Gupta4
Santosh Kumar Gupta4 Hemant Kumar Yadav5
Hemant Kumar Yadav5 Murli Manohar6
Murli Manohar6 Rajendra Kumar1*
Rajendra Kumar1*Epigenomics has become a significant research interest at a time when rapid environmental changes are occurring. Epigenetic mechanisms mainly result from systems like DNA methylation, histone modification, and RNA interference. Epigenetic mechanisms are gaining importance in classical genetics, developmental biology, molecular biology, cancer biology, epidemiology, and evolution. Epigenetic mechanisms play important role in the action and interaction of plant genes during development, and also have an impact on classical plant breeding programs, inclusive of novel variation, single plant heritability, hybrid vigor, plant-environment interactions, stress tolerance, and performance stability. The epigenetics and epigenomics may be significant for crop adaptability and pliability to ambient alterations, directing to the creation of stout climate-resilient elegant crop cultivars. In this review, we have summarized recent progress made in understanding the epigenetic mechanisms in plant responses to biotic and abiotic stresses and have also tried to provide the ways for the efficient utilization of epigenomic mechanisms in developing climate-resilient crop cultivars, especially in chickpea, and other legume crops.
Chickpea is the world’s second most significant grain legume, produced primarily in the tropics, subtropics, and temperate zones. It is a self-pollinated annual crop with a genomic size of 740 Mbp (Arumuganathan and Earle, 1991). Chickpeas are prized for their high levels of dietary proteins (20–30%), carbohydrates (40%), fibers (3–6%), and lipids (3–6%) (Pushpavalli et al., 2015). In addition, it is also a good source of fiber, minerals, vitamins, lysine, and sulfur-containing key amino acids. It is a resilient crop that is well adapted to stressful situations and is a god’s gift to tropical farmers. Chickpea yields on average around 780 kg and can reach up to 2.5 tons per hectare. Various biotic and abiotic stresses have a negative impact on chickpea yield and productivity. The principal factors that limit chickpea production in farmers’ fields include abiotic (drought, salinity, heat, and cold stresses), biotic (insect pests including pod borers, aphids—Aphis craccivora, leaf miner, bruchid, etc. and diseases like Fusarium wilt, Ascochyta blight, grey mold, and root rots) stresses. Every year, abiotic stresses cause roughly 6.4 million tons of crop output losses, with soil salinity being primary environmental stress (Jha et al., 2014). Soil salinity is a serious barrier to crop output, and it affects almost 80 million hectares of arable land worldwide (Flowers et al., 2010). Since chickpea is a winter crop, it is subjected to low-temperature stress (0–15°C) during the reproductive stage, which results in a significant loss of flowers and hence pods, reducing output potential by 30–40%. High temperature stress chickpea in late-sown crops, primarily during reproductive and pod filling stages and drought stress at several stages of development; terminal dryness, combined with heat stress during blooming and seed filling can reduce yield up to 70% due to drought and heat stress (Kudapa et al., 2014). Climate change is expected to increase the frequency of temperature extremes (cold and heat), as well as inconsistency in rainfall patterns, necessitating the development of stress-tolerant and climate-resilient chickpea cultivars with region-specific traits that perform well under drought, heat, and/or low-temperature stress. Chickpea production in harsh settings has been improved through a variety of methods, including genetic variability, genomic selection, molecular markers involving quantitative trait loci (QTLs), whole-genome sequencing, and transcriptomics study. Biotechnological technologies have improved our understanding of the genetic basis of chickpea stress tolerance as well as plant responses to abiotic challenges, allowing us to build stress-tolerant chickpeas. The immensity of the current task of maintaining or improving productivity in the face of growing salinity to fulfill yield demands has been clearly recognized and leads to nearly 70% increase in crop production as a top priority (Amin et al., 2016; Hong et al., 2016). So far, Mendelian-based genetic approaches and the selection of heritable target DNA sequences have provided significant genetic improvements in many crop species. In addition, a greater understanding and ability to select beneficial epigenetic and epigenomic changes are proposed to encompass a more efficient and holistic strategy for crop improvement. This is because epigenomic mechanisms are central to governing many plant stress responses, including through cell-autonomous epigenetic switching. This enables the registration and memory of unpredictable genetic signals. The term epigenetics was coined by Waddington (1942) that is used as an intermediate factor between the genotype and phenotype. During gene expression studies, there are various heritable changes occurred due to mitotic and meiotic divisions and are not coded in the DNA sequence itself (Tsaftaris and Polidoros, 2000). Heritable changes in gene expressions are independent of DNA sequence variation and steadily congenital from one generation to another (Berger et al., 2009; Chen et al., 2010a). Variations in the heritability of epigenetic marks (changes) occur during mitotic and meiotic cell divisions. Transient epigenetic changes are not heritable, stout ones, and mitotically transmitted with genome imprinting (Spillane et al., 2001). The meiotically generated epigenetic changes are heritable across the generations without the need for the original stimulus until they are lost or erased. Epigenetic repression is limited to one locus—the genes next to flowering locus C (FLC) are affected by the cold temperature, for example, which is common for many genes in Arabidopsis (Kilian et al., 2007). The loss may be because of genetic change, may be spontaneous (unknown reason), or submission to ambient. Those differ from those that induced the initial epigenetic alterations. The mitotically heritable changes that are not kept through meiosis (epigenetic variation in somatic cells) are lost irrespective of the certitude that mitosis usually perpetuates genetic constitution, such as heterosis is explicated as any edge observed in hybrids. The reverberations of heterosis appear to follow a preferably uncomplicated epigenetic presumption in plants. In hybrids, if the gene is entangled in growth, such as photosynthesis, the plant expressed enhanced vitality (Ni et al., 2008). Heritable epigenetic changes are also referred to as “epialleles,” where the epialleles of a locus are identical DNA sequences but display different epigenetic states and hence have an influence on a range of phenotypes (Richards, 2006). These may be classified into three categories based on relative dependence on the genotype: 1) Pure epialleles that are solely epigenetic and independent of the genetic variations; 2) Facilitated epialleles that partially depend on genetic variation. An example of epiallele transposon is that undergoes DNA methylation spreading into a gene after the insertion of an adjoining transposon. That will be passed across the generations and the changes include both genetic and epigenetic differences; and 3) Obligate epialleles, which are directly influenced by the genetic variants and co-segregate with the methylation variants (Woo et al., 2007). Epigenetic variations include various post-transcriptional histone modifications. These modifications include the activity of non-coding RNAs, histone variants, and DNA methylation, which showed drastic changes in plants’ response to biotic or abiotic stimuli by changing the transcriptional profile. The memory-directed modifications lead to improved capacity to withstand future stresses (Berr et al., 2011). Thus, epigenetic mechanisms influence the accessibility of DNA to enzymes, resulting in a wide range of gene expression and mRNA splicing. These processes add complexity to the traditional genotype-environment interaction for understanding phenotype expression and development since they are differentially sensitive to the environment (Burggren, 2020). Epigenetic pathways have recently been found as a mediator of this interaction, allowing fast phenotypic diversity in a variety of settings (Burggren, 2016). Under the ongoing climatic changes in agricultural adaptability and resilience to environmental changes, epigenetics has emerged as a major crop development method, ultimately leading to the generation of stable climate-smart crops. This has paved the path for crop breeding to take advantage of epigenetic diversity. Even though epigenetics mechanisms have not been demonstrated in many crop species, most mechanistic investigations are from model plant species. Thus, there is a need to understand how epigenetic mechanisms are linked to mortality predictions as a result of climate change, which affects a wide range of fields, from environmental conservation to climate change mitigation efforts, and is expected to be more frequent under a climate-change scenario (Amaral et al., 2020; González-Benito et al., 2020). Understanding the roles of epigenetics inducing stresses including histone modifications and DNA methylations helps to uncover the mechanisms that regulate plant-stress interactions and conditions (Chinnusamy and Zhu, 2009). In this review, we have attempted to summarize epigenetic contribution to agricultural adaptation in response to climate change, epigenomic mechanisms, and describe several characteristics in plants, problems in utilization and hypothesize with an objective of the future potential use of epigenetic variations in developing more resilient chickpea crop the staple food legume.
Understanding of the epigenetic regulatory machinery and mechanism in plants has most notably been achieved in Arabidopsis thaliana, (http://www.arabidopsis.org) a model species. Studies on crops, particularly maize, have led to a better understanding and more deep insight into the epigenetic phenomenon (Brink 1956). The implications for maize epigenetic research in the post-genomics era are manifold and it would be difficult to expect future discoveries that are yet to come. However, explanations for an epigenetic regulatory mechanism to stress response in staple crops are still not explored (Arefian et al., 2019; Khan et al., 2019). Along with diverse and overlapping epigenetic regulatory pathways in the maize genome, it could be the most important area of paramutation research for exploration (Chandler et al., 2010). Various reports indicate the novel contributions of the model plant, which have generally been provided in epigenetics and epigenomics. It was the first report in which the discrimination between euchromatin and heterochromatin was explained well based on cytological analyses (Heitz, 1929). Studies in tomato and maize gave heritable changes in expressions related to individual alleles with alternative states, a phenomenon known as paramutation, an inter-allelic interaction that leads to heritable changes in gene expression through mitotic and meiotic routes (Pikaard and Mittelsten, 2014). Paramutation was first explained in maize (Brink, 1956) and furnished the proof for non-Mendelian epigenetic transmissions in plants. The frequent eventuality of entities with changed flower conformity was first explained in the 18th century by Carl von Linne. It was recently observed and reported that a silenced epiallele handled these changes, which contain DNA identical to an expressed allele (Cubas et al., 1999). The innovative exertion of interchangeable ingredients in maize was reported by Barbara McClintock and others (1940), which disclosed the innumerable connections between genetic conduct and epigenetic rules. McClintock (1950) worked and published various reports on the transposable element in maize.
Although various ways regulate the gene expression process in the eukaryotes, DNA methylation is a usual epigenetic process by which cells are used to have switching control of genes in the “off” mode. In the last few years, researchers have unfolded the mechanism of DNA methylation, which led to the fact that methylation is a significant constituent in several cellular mechanisms, including embryonic growth, genomic imprinting, X-chromosome inactivation, and preservation of chromosome stoutness (Phillips, 2008). There are several reported mechanisms in which methylation plays a critical role. Researchers have also connected faults in the methylation process to a series of catastrophic outcomes, including innumerable human diseases (Kelkar et al., 2009). So, we can conclude that these studies help in developing bioinformatics tools that have wide applications across species kingdoms, including chickpeas.
In view of distinguishing differentiation between mammals and plants, it is important to consider the life cycle of plants. In mammals, fertilization is achieved by the fusion of two haploid cells produced by meiosis. However, in the case of plants, haploid (gametophyte) growth takes place that follows meiosis and precedes fertilization as presented in Figure 1. The male and female gametophytes are produced by mitotic divisions of the initial haploid meiotic products. In haploid gametophytes, loss of genetic or epigenetic information cannot be compensated for by information on homologous chromosomes. Unlike mammals, there is no evidence for a massive erasure of epigenetic marks during plant gametogenesis. Instead, repressive epigenetic marks in plant sperm and egg cells appear to be reinforced by specific trans-silencing RNAs produced in neighboring nuclei.
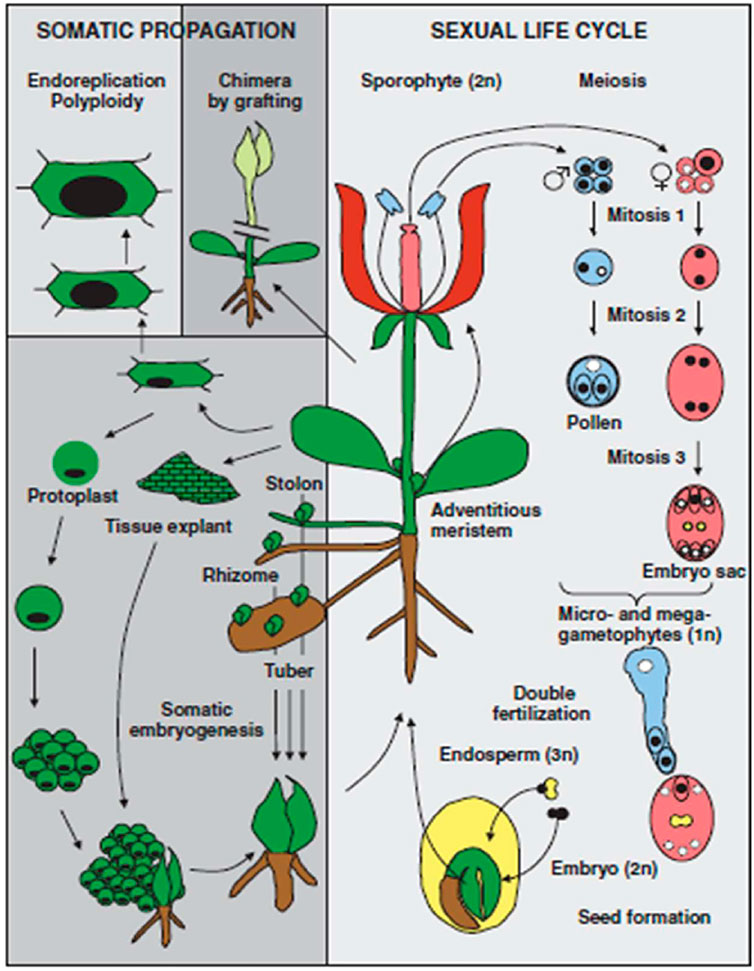
FIGURE 1. Unique aspects of the plant life cycle (Reproduced from Pikaard and Mittelsten, 2014).
The first distinguishing feature to notice is that haploid (gametophyte) development begins with meiosis and continues after conception. There is a loss of genetic or epigenetic information in genetically and metabolically active gametophytes (haploids) that cannot be replaced by homologous chromosomes. In required genes, harmful mutation events are chosen in contrast. Plant gametogenesis, in contrast to mammalian gametogenesis, lacks proof for genome imprinting. Rather, certain trans-silencing RNAs originating in neighboring nuclei appear to be equipped in plant sperm and egg cells to suppress epigenetic changes. That may reflect the process of epigenetic changes that occur during meiosis in plants (Pikaard and Mittelsten, 2014). A second distinguishing trait of plants is the lack of a clearly defined germ line during early embryogenesis. Germ cells are formed in the later stages of plant development. Floral organisms emerge at this stage as a result of transformations from vegetative organs to progeny cells. Meiosis and gametogenesis take place in these cells. Thus, epigenetic changes acquired by meristematic cells in response to plant interactions in the presence of a specific environment have the potential to be passed to germ line cells, reviewed by Pikaard and Mittelsten (2014).
The arbitrary introduction of transgenes or transposable elements can be achieved in plant genomes by the process of chemical and or physical mutagenesis (Pikaard and Mittelsten, 2014). In the case of A. thaliana, it is very easy to identify homozygous mutants, which were identified amongst thousands of progenies of a single mutagen-treated plant. Marker gene is the basis to screen the putative mutants in epigenetic regulators. The promoter region of the OsMYB91 gene was demethylated and rapid histone modifications at the OsMYB9 locus in rice account for salinity resistance (Zhu et al., 2015). Increased Asr1 and Asr2 gene expressions have been observed during drought-resistance in tomato plants. The expression was enhanced because of the demethylation of putative regulatory and transcribed regions (González and Álvarez, 2013).
The advances in the production of transgenic plants have, therefore, adequately supported epigenetic and epigenomic research. In collaboration with forwarding genetics, another approach, i.e., reverse genetics emphasizing changing gene functions is also feasible. The development of mutants or utilizing transgene-starting RNAi has made an easy way to knockout or knockdown the expression of candidate epigenetic regulator homologs that were previously identified in other organisms. Once, a particular epigenetic mutant is characterized, restrained screenings are usually fortuitous for recognizing interacting constituents or alternative pathways, as observed with Drosophila (Elgin and Reuter, 2013) as well as with mouse (Blewitt and Whitelaw, 2013). However, because of the availability of components of epigenetic regulation and exhaustive assemblage of introduced mutations in almost every gene, schematic mutagenesis, and comprehensive instinctive dissimilitude, A. thaliana has developed as a model plant used for epigenetic studies based on various researches as presented in Table 1 (Pikaard and Mittelsten, 2014).

TABLE 1. Model plant Arabidopsis thaliana and their epigenetic regulation (Pikaard and Mittelsten, 2014)
DNA methylation, histone/nonhistone alterations, and small RNA-mediated interference are the major mechanisms depicted in Figure 2, and explained as further in the following sections.
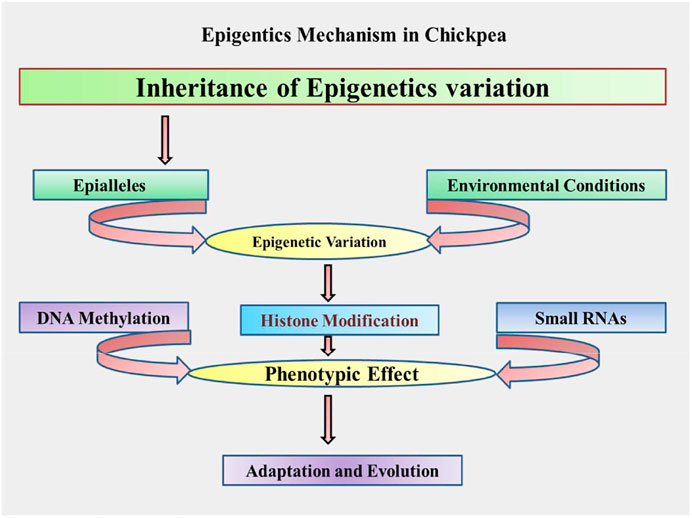
FIGURE 2. Proposed schematic mechanism/process of epigenetics in chickpea.
DNA methylation is the addition of methyl group at the fifth position of carbon in the DNA molecule of cytosine. DNA methyltransferase carries out the post-replicative modification of DNA known as methylation using S-adenosyl-l-methionine (SAMe) as a methyl donor. Due to this modification, DNA conformation, protein interactions, and chromatin structure were changed, conclusively changing their functional states. Various important biological processes were driven by DNA methylation. These are cell developmental stages, X-chromosome inactivation, transposon tagging, genomic imprinting, and gene silencing. In the case of plants, DNA methylation usually occurs at the positions of CG, CHG, and CHH (H = A, C, or T) and intricate unique DNA methyltransferase, viz, DNMT3B, DNMT1, DNMT2, and DNMT3A (Jurkowska and Jeltsch, 2016). In the case of animals, DNA methylation patterns are associated with the origin, growth, developmental pattern, and progression of cancer (Jones and Baylin, 2007). DNA methylation patterns vary throughout the developmental differentiation in cells and tissues (Barlow and Bartolomei, 2014). This process is carried out by a family of DNA methyltransferase enzymes in eukaryotes. This helps in transferring the methyl groups from the methyl donor SAMe to the cytosine. The resulting 5-methylcytosine (5 mC) is often repressive and leads to gene silencing.
In plants including chickpeas, cytosine methylation is non-randomly distributed mostly to the repetitive regions that abundantly comprise transposable elements of the genome, centromeric frequencies, or multitudes of mute 45S or 5S rRNA gene recurrences. In addition, it further takes place by a handful of divergently controlled enhancers and within the protein-coding domains of tremendously conveyed genes in chickpeas (Zilberman et al., 2007). The eventual gene frame methylation is transformational preservation that plays a role during pre-mRNA splicing (Feng et al., 2010). DNA methylation plays immense contributions to various plant mechanisms, viz, gene silencing, imprinting of genes, plant immunity, escape from restriction enzymes, apomixis, etc. explained in the following sections.
The long-term silencing of the gene has been reported in DNA methylation patterns during reprogramming of seed differentiation and its functional relevancy in seed size and seed weight measurements in a large-seeded chickpea cultivar (JGK 3). The identified candidate genes involved in seed size/weight determination exhibited CG context hyper-methylation within the gene and manifold expression in JGK 3 provided insights into the role of DNA methylation in determining size, development, and weight of the seeds. The role of the RNA-dependent DNA methylation pathway has been shown by the gradual achievement of CHH-related DNA methylation in transposable elements (TEs) and by the elevated frequency of small RNAs in hyper-methylated TEs during the development of seed (Rajkumar et al., 2020).
An epigenetic phenomenon that causes genes to be expressed in a parent-of-origin-specific manner is known as genomic imprinting. This is an inheritance process that is independent of the classical Mendelian inheritance and involves DNA methylation along with histone methylation without altering the genetic sequences. Genome imprinting has been reported with Arabidopsis that pushes forward to the rapid escalation of the endosperm, a desirable characteristic (Berger, 2006). This mechanism can apply to commercial crops and hybrids for their regeneration so that we can overcome the current limitations of plant breeding for the perpetual maintenance of hybrid vigor for generations. In the central cell of the female gametophyte, the Demeter molecule-DNA glycosylase domain quantity increases, causing DNA demethylation of the transposons because of a reduction in MET1 methyltransferase. Upon transcription of the transposons, a 24-nucleotide small interfering RNA (siRNA) is produced, which moves to the egg cell and causes imprinting of genes. Similarly, in the vegetative cells of the male gametophyte, DNA demethylation of the transposons occurs because of an increase in the diameter molecule, and upon transcriptions of these transposons, a 21-nucleotide small interfering RNA is produced that moves to the sperm cell and causes imprinting of genes (Bratzel et al., 2012).
Another study has dissected five legumes namely chickpea, soybean, alfalfa, pigeon pea, and lotus indicating the putative role of DNA methylation in the development of inheritable gene silencing and recognized potential DNA MTases (Garg et al., 2014). Based on the domain organization, MTases have been categorized into four subfamilies in legumes, viz, MET, CMT, DRM, and DNA nucleotide methyltransferases (DNMT2). The DNMT2 is a transfer RNA (tRNA) MTase, whereas the first three MTases are a class of DNA MTases. Structural comparative studies of all the known MTases in mammals and plants have assigned biological functions to these MTases (Jurkowski and Jeltsch, 2011). There are various reports in legumes related to the exhaustive gene expression assays of MTases that provide pieces of evidence of their important role in various developmental processes. During the plant life cycle and response to various abiotic stresses, the critical roles of MTases are continued until their survival (Benedito et al., 2008).
Plant immunity is the built-in or catalyzed capability of plants to resist or avert a biological strike by pathogens. Molecules emancipated from pathogens are recognized by plant cell exterior sense organs; these receptors stimulate the specific showing cascades that facilitate to withstand the plant against pathogen infection. Roots activate specific tolerance mechanisms in response to elicitors such as molecular/pathogen-associated molecular patterns, (MAMPs/PAMPs), showing compounds (e.g., hormones), and plant defense activators (e.g., β-aminobutyric acid, BABA) (Zhang and Zhou, 2010). DNA methylation can have a critical role in plant immune responses to pathogens; for example, many defense genes in A. thaliana are modulated by DNA methylation starting defense reactions against Pseudomonas syringae pv tomato (Dowen et al., 2012).
Escape from Restriction Enzymes: Methylation-based modification interferes with the restriction process. It serves to protect bacterial chromosomal DNA against “self” restriction and is also responsible for the transient modification of those phages that escape restriction. It was reported in earlier studies that in bacteria, DNA methyltransferases provide a unique mechanism that handles the methylation of specific DNA sequences and is finally linked with epigenetic inheritance (Joseph and David, 2006). The restriction endonuclease recognizes a specifically marked DNA motif those are methylated by an analogs DNA methyltransferase when DNA methylation was primarily unzipped because of restriction-modification (R-M) forms. The R-M systems have been instigated as cellular protection, identifying incoming foreign DNA sequences (viral and another alien) for degradation. The methylation of foreign DNAs was based on specific recognition with related methylase of the same specification. The absolute methylation of the genome is enough to block double-strand DNA cut by the restriction enzyme when the restriction enzyme and its associated methylase both are expressed at levels in R-M systems. Owing to the cell demise that depreciated plasmid containing the EcoRV as post segregationally killing a plasmid comprising the type II R-M EcoRV pattern could not be substituted from the cells by a matchless plasmid (Nakayama and Kobayashi, 1998). The previous accomplishments have advised that R-M systems drive the features of selfish genes (Kobayashi et al., 1999; Kobayashi, 2001).
Plants reproduce by sexual or asexual means and asexual reproduction in plants is carried out by cloning apomixes for sexually reproducing plants including chickpea, fertilization-independent seed formation is not possible because fertilization is a prerequisite for the embryo sac to develop into seeds. For apomictic plants, fertilization is unnecessary because the genes responsible for the fertilization-independent seed formation produce the apomictic seeds (Hand and Koltunow, 2014). Apomixis was proposed to have developed to enable plant species to propagate under adverse environmental conditions. The previous study has found that reactive oxygen species (ROS) are produced in excess in plants under stress and they possess innate systems for scavenging/detoxifying after they have done their job. Polyamines (putrescine, spermidine, and spermine) are low molecular weight, polycationic aliphatic molecules that are well known for anti-senescence and anti-stress effects because of their antioxidant properties (Kumar and Singh, 2016).
DNA methylation, along with histone modifications that include histone acetylation, phosphorylation, methylation, and ubiquitination, contains the most identified epigenetic-associated mechanisms. Similar to DNA methylation, post-translational histone modifications do not possess the ability to influence the DNA nucleotide motif but might change its supply to the transcriptional system. Histone phosphorylation is another mechanism that is performed through histone modifications, often known for its creditability to DNA impairment in reaction to cell injury. The huge gene families in crop plants usually encipher histone-change enzymes by Berr et al. (2011), Deal and Henikoff (2011), and Lauria and Rossi (2011) and are explained in the following paragraphs.
Histone acetylation is carried out by histone acetyltransferases (HATs) enzymes, which mainly add acetyl groups to the histone tails and reduce positive charges and decrease the interaction of histones with DNA. HATs also facilitate transcription by enabling the DNA molecule more accessible to RNA polymerase II. Histone deacetylation is the reverse of histone acetylation and is carried out by Histone deacetylases (HDACs) enzymes. These HDACs remove acetyl groups from histone tails, increase the interaction of DNA, and repress transcription (Bird, 2007). Salinity and drought both are major environmental abiotic obstacles that cruelly affect overall global crop productivity and their nutritional quality (Sen et al., 2017). Plant-specific HD-Zip transcription agents are intricate in plant growth, development, and stresses. A novel HD-Zip (I) gene in chickpea, i.e. CaHDZ12, is expressed under water-deficit and salt-distress conditions. An improvement in the tolerance to osmotic stresses was observed in transgenic tobacco genotypes with over-expression of CaHDZ12. Silencing of CaHDZ12 resulted in escalated sensitivity to salt and drought-distresses in chickpeas. Epigenetic changes like histone acetylation at the CaHDZ12 promoting region have a critical role in distress-induced activation of this gene.
Histone methylation may cause the direct activation or repression of gene expressions and is carried out by histone methyltransferases (HMTs) having several classes including histone lysine methyltransferases (HKMTs), methylated lysine (k) residues, protein/arginine methyltransferase (PRMTs), and methylated arginine (R) residues. The trimethylation of histone H3 at lysine 4 (H3K4) is a specific region for transcription and demethylation to be carried on histone protein H3 to repress the transcription (Collins et al., 2019). Zentner & Henikoff (2013) reported that the addition of the methyl group from S-adenosyl-l-methionine (SAM) into lysine or arginine residues resulted in histone methylation. Histone methyltransferases (HMTs) are the enzyme that catalyzes this process (Bannister & Kouzarides, 2011). The DNA expression is changed through histone methylation by altering the engagement and the unbreakable controlling proteins adhered with the chromatin (Hyun et al., 2017). The histone lysine methylation occurs in mono-, di-, or tri-methylated forms, however, arginine methylation as mono- or di-methylated forms (Bannister & Kouzarides, 2011). The impact of histone methylation on DNA transcription depends on the numbers and to which residues methyl groups are getting added (Zentner & Henikoff, 2013). For example, methylations of H3K4, H3K36, and H3K79 are related to assiduous transcription, whereas the methylations of H3K9, H3K27, and H4K20 are related to tranquility (Black et al., 2012). The study conducted on DNA methylation and physio-biochemical analysis of chickpea in response to cold stress (CS) has reported that CS signals are converted as physiological changes as products of gene expression and are regulated by DNA methylation patterns (Rakei et al., 2015). The major roles of antioxidant enzymes (superoxide dismutase, catalase, ascorbate peroxidase, guaiacol peroxidase, and polyphenol oxidase) along with a noticeable ratio of changes in DNA methylation/demethylation patterns were often decisive factors in the preservation of cells against cold influenced oxidative distress.
In eukaryotic taxa, among the most conserved proteins include histone variants, linker histones, and non-histone proteins that are enciphered by highly superfluous gene families. The distinct categories of H2A and H3 histone variants, like animals also in plants based on their structure and function, have been identified (Henikoff and Smith, 2015). The physical properties of histone variants significantly affect their dynamic relations with DNA (Ingouff and Berger, 2010; Deal and Henikoff, 2011). The DNA damaged regions are discovered through phosphorylation of the H2AX variant and also assist during the recruitment of DNA repair proteins. A type of histone variant, H2A.Z exists mostly near the transcriptional start site of genes, most probably regulating transcription (Zilberman et al., 2008). The expression of this variant requires the initiative of an SWR1 chromatin-remodeling complex. Separation of DNA from H2A.Z comprising nucleosomes during heat stress is observed, which is followed by alterations in gene expression (Kumar and Wigge, 2010). During cell division, a particular histone H3 variant, i.e., CenH3 specifies the nucleosomes of centromeric regions and plays an important role in kinetochore assemblage, microtubule association, and chromosome segregation. H3.3, another histone variant having only a few different amino acids from its canonical H3 subunit, is predominantly found in regulatory regions. When specific linker histone proteins were downregulated as compensation, there is an upregulation of other histone variants. This results in a clear-cut phenotypic defective change with pleiotropy DNA hypomethylation (Jerzmanowski, 2007).
Several chickpea-specific histone-modifying enzymes as given further have been reported. The histone H2B is a core component of nucleosome that wraps up long compact DNA into chromatin, limiting DNA availability to the cellular mechanisms and using DNA as a template. Therefore, histones are an integral part of transcription regulation, chromosome stability, DNA repair, and DNA replication. A complex set of post-translational modifications of histones, known as histone code, and nucleosome remodeling help in regulating DNA accessibility. Histone H2B performs the molecular functions of DNA binding and protein heteromerization (https://www.uniprot.org/uniprot/Q9M3H6). It was reported that under heat stress conditions expression of six chromatin remodeling complex genes (SWR1) in diverse tissues of chickpea was based on nucleosome response through histone H2A.Z variants. A group of seven genes that are homologous to chromatin remodeling complexes (SWR1) of Arabidopsis was also identified in the chickpea genome. Three genes of chickpea homologs of photoperiod independent early flowering 1 (PIE), Actin associated protein (ARP6), two serrated leaves, and early flowering (SEF) for histone 2A variant-Z (H2A.Zs-a thermal sensor in plants) the three genes were analyzed for their appearances under heat distress and five diverse tissues. A significant role in chromatin remodeling complexes under heat stress conditions might be played by CarPIE1 gene. The entire three histone CarH2A.Z variants acted as potential candidate genes for the characterization of their specific function (Chidambaranathan et al., 2016). These chickpea-specific histone proteins are summarized in Table 2.
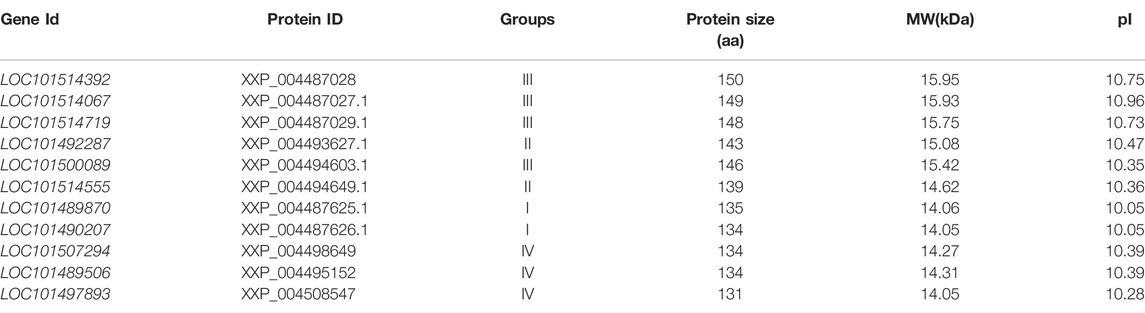
TABLE 2. Chickpea-specific histone proteins (Chidambaranathan et al., 2016).
Furthermore, various forms of histone modifications and their sites along with effects on transcriptional activity of genes are summarized in Table 3.
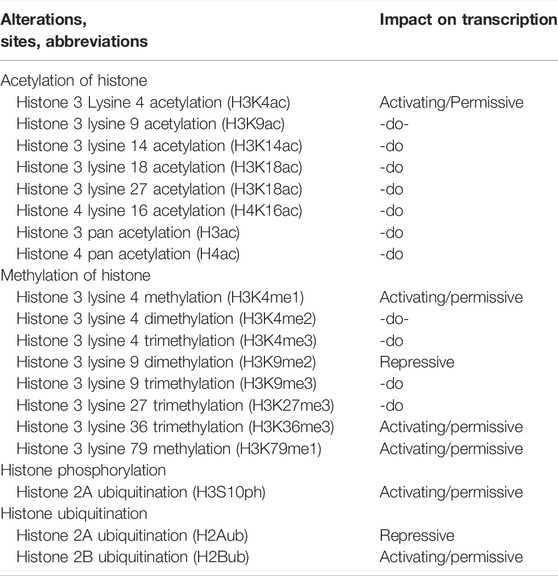
TABLE 3. Histone alterations, their sites and impacts on the activities of transcription.
The plant may continue to grow vegetatively through cell division during the cold period. When new seeds are produced, after the vernalization of the parent plant, the seeds are “reset.” The new plants they produce from the seeds will themselves have to go through their cold season before flowering. The key gene intricate in vernalization is referred to as FLOWERING LOCUS C or FLC. FLC encodes a protein known as a transcriptional repressor. It binds to other genes and stops them from getting switched on. These three genes FT, SOC1, and FD specifically regulate flowering in Arabidopsis thaliana, and show that the epigenetic status of FLC alters after a prolonged duration of cold. Experiments with mutated versions of epigenetic enzymes have shown that the changes in histone modifications at the FLC gene are critically important in controlling the flowering response. For example, there is a gene called SDG27 that adds methyl groups to the lysine amino acid at position four on histone H3, so it is an epigenetic writer that is associated with a vigorous gene expression. The SDG27 gene can be mutated experimentally so that it no longer encodes an active protein. Plants with this mutation have less of this active histone modification at the FLC gene promoter. They produce less FLC protein, and so are not so good at repressing the gene that triggers flowering. The SDG27 mutants flower earlier than the normal plants. Cold weather induces protein in plant cells called VIN3 that works as chromatin and can bind to the FLC promoter. When VIN3 binds to the FLC promoter, it alters the local structure of the chromatin instead of how tightly chromatin is wrapped up, making it often available to other proteins. Often, opening up chromatin leads to an increase in gene expression. However, in this case, VIN3 attracts yet another FD (FLOWERING DETERMINATE) enzyme that can add methyl groups at position 27 on lysine residue amino acid of histone H3 protein. This modification represses gene expression and is one of the most important methods that plant cell uses to switch off the FLC gene. Following cold weather, the cells in Arabidopsis thaliana produce a long RNA, which does not code for a protein called COLDAIR. The COLDAIR non-coding RNA is present, particularly in the FLC gene. When localized, it binds to the enzyme complex that creates the important repressive mark at position 27 on histone H3. COLDAIR, therefore, acts as a targeting mechanism for the enzyme complex. From these data, we can see that flowering plants use some of the same epigenetic machinery as many animal cells. These include histone protein alternations, and the utilization of long non-coding RNAs to target these changes. Earlier, it has been inferred that destabilization of the cells is a consequence of global DNA hypomethylation through DNMT1-depletion led, which ultimately leads to the production of aneuploids (Barra et al., 2012). In the case of aneuploids (45, XO; 46, XX; and 47,XXX) expression of DNA methyltransferase 1 and DNA methylation enzymatic gene showed a positive association with inactive X chromosomes (Rajpathak and Deobagkar, 2017).
To deduce and establish the critical role of histones in epigenetic regulation during the process of viral innate immunity, there is a great need for better grasping of these complicated interactions through the epigenetic lens, which may have therapeutic opportunities in the clinic. A grasping of the parts played by the key epigenetic controllers—chromatin remodeling and histone alterations—in atonements of chromatin candidness in the process of host defense against virus, how the RNA alteration m6A (N6-methyladenosine) influences basic features of hostvirus interplaying and conclusions with subsequent orchestrations for better understanding about epigenetic regulations in host and viruses’ contaminations are required (Xiao et al., 2021).
Similar to other eukaryotes, non-histone chromosomal proteins are also found in plants, which may assist epigenetic gene regulations, including HMG proteins. The HMGB family of proteins is the foremost assayed and varied subgroup of proteins in plants, members of whom differentiate in the level of expression, localization, style, and inter-playing with DNA along with other proteins. The partial sub-functionalization of individual family members results from mutation and abnormal expression showing their role in developmental stages and response to various stress stimuli (Pedersen and Grasser, 2010). The structure-specific recognition protein (SSRP1) indirectly contributed to the demethylation of DNA the recognized genes in the female gametophyte’s central cell (Ikeda et al., 2011). Yuan et al. (2011) have reported that the structure, assembly, and rejection of cohesion seem to be highly protected. There are only limited family members in plants that might have specific functionalities. It was also found that the defective meristem silencing 3, which was involved in de novo (DMS3/IDN1) is required for transcription of DNA-dependent RNA polymerase V (RNA Pol V) (Haag and Pikaard, 2011). This plays a very important role in the establishment of RdDM (RNA-controlled DNA methylation) proteins (Varshney et al., 2019a). The various mutant screens related to epigenetic regulators are the sites of REPLICATION FACTOR C1 and REPLICATION PROTEIN A2 (RPA2) (Elmayan et al., 2005; Kapoor et al., 2005). The role of this protein related to stem cell and meristem repair is also revealed when chromatin mutants having increased phenotypes and mutants having topoisomerase homolog MGOUN (MGO) functional loss were merged (Graf et al., 2010). This signified that many other non-histone proteins involved with DNA will also act as direct or indirect epigenetic regulators.
Short-term or long-term modifications in the nucleosomes' positioning and their connection with DNA were always required for replication, transcription, recombination, and maintenance. As a result, vigorous mechanisms on the chromatin amend DNA or protein modifications, incorporate alterations in nucleosome’s possession and, constitution, along with the attainability of the DNA to variegated proteins (Pikaard and Mittelsten, 2014).
Becker and Workman (2013) have reported that chromatin remodeling can be used for the relocation or dissociation of nucleosomes. It was first reported in yeast and was named after the respective processes that ATPases such as the SWI/SNF complexes have influenced the mutants. Many similar complexes were also reported in plants (Jerzmanowski, 2007). The functional information for very few putative chromatin remodelers has been got through genetic screens, the first recognized being DECREASE IN DNA METHYLATION1 (DDM1). The function of DDM1 included genome-wide decreased activity of methylation of DNA and H3K9me2, activating repetitive elements for transcription, and downregulation of many such genes. Therefore, the mutants for ddm1 exhibit severe defects in developmental and morphological growth that may reach an extreme in future generations. The amalgamation of epimutations and insertional mutations induced through reactivated transposons is the main reason behind the gradually diminished fitness of ddm1 mutants. In ddm1 mutants, epigenetic information is permanently deleted and can be restored through backcrosses with wild-type plants as epigenetic patterns at several loci, mainly because of the outcome of de novo methylation (Teixeira et al., 2009). DDM1 also shows in vitro ATP protease nucleosome moving initiative (Brzeski and Jerzmanowski, 2003). Mutants deficient in DDM1 and linker histone H1 originate when cytosine methylation deficiency occurs in ddm1 mutants (Zemach et al., 2013) signaling that the requirement of DDM1 is essential for the repair of methylation mechanics to discover DNA in nucleosomes consisting of core and linker histones. The defective RNA-mediated DNA Methylation 1 (DRD1) and CLASSY 1 (CLSY1), extremities of the SWI2/SNF2 family found in Arabidopsis, are unique to the plant kingdom including chickpea, and play a peculiar part in RNA-induced DNA methylation. Four additional SWI2/SNF2 derived proteins namely; BRAHMA (BRM), MINUSCULE (1, 2), and SPLAYED (SPD) are intricate in the RNA-led DNA methylation (Sang et al., 2012). Other than ATPases, quintessence parts of SWI/SNF remakes are also reported in plants, inclusive of one SNF5 homolog (BSH), two SWP73 homologs, and many SWI3 family members (AtSWI3 A-D) (Jerzmanowski, 2007). However, their direct contributions to plants are still unexplored. However, it has been unraveled that SWI3 interplays with RNA binding proteins lead to RNA-directed DNA methylation (Zhu et al., 2012).
The role of histone protein for drought and yield index (DYI) in chickpeas was studied, and it was disclosed that the development of functional molecular tags derived from the cis-regulatory sequence components of genes is crucial for their deployment and identification of several conserved non-coding SNPs (CNSNP). Among those, the two made-up natural haplotypes and alleles are derived from a histone H3 protein-coding gene and its transcriptional regulator NAC transcription factor (TF) anchoring the major QTLs and trans-acting eQTL controlling drought yield index (DYI) in chickpea (Sharma et al., 2019).
RNA interference (RNAi) is a technique in which tiny RNA molecules are combined with other molecules to target homologous DNA regions. They bring together the agents that alter chromatin, resulting in heterochromatin formation and gene suppression. Pre-transcriptional gene silencing can stop transcription from happening. As a result, DNA methylation at genomic locations corresponding to complex siRNA or miRNA is catalyzed by an enzyme complex. RNA interference in chickpea and other legume crops has been found to have a large number of drought-responsive miRNAs. In response to salt stress, 259 miRNAs were shown to be differentially expressed in the root tip of chickpea during drought and salinity stress, which were also seen in other legumes such as soybean root apex (Khandal et al., 2017). TIR1 (TRANSPORT INHIBITOR RESPONSE 1), an auxin receptor, and AUXIN RESPONSIVE FACTOR 10 (ARF10) and ARF16 are targets of miR393 and miR160 (Chen et al., 2011). Overexpression of miR160 causes unregulated cell division and a loss of gravity sensing at the root tip during primary root development (Mallory et al., 2005). MiR164 inhibited auxin signaling for lateral root initiation by targeting the transcription factor NAC1. ARF6 and ARF8, which are positive regulators of adventitious root growth, were targeted by miR167 (Gutierrez et al., 2009; Guo et al., 2005). Comparative miRNA expression profiling of Medicago truncatula (Medicago) in the root tip and elongation zone, as well as root-forming callus and non-root forming callus, revealed 107 miRNAs from 44 families expressed in these tissues and predicted conservation of some of the miRNA/target relationships seen in other species (Eyles et al., 2013). Overexpression of MiR396 in Medicago roots inhibits cell-cycle gene expression and limits root development (Bazin et al., 2013). miRNA expression analysis in normal soybean roots, as well as comparisons between phosphate-starved and phosphate-sufficient soybean roots, revealed some new miRNA/target interactions (Xu et al., 2013). In Arabidopsis, an increase of miR393, miR397b, and miR402 expression occurs under dehydration and salt stress, according to a study. miRNA has a vital function in controlling root growth under abiotic stresses (Ding et al., 2009; Lu et al., 2014). Drought stress increases the expression of miR398a/b and miR408 in the Medicago root (Trindade et al., 2010) and miR169g in rice roots (Zhao et al., 2007). In Arabidopsis, mi RNA165/166 regulates root development by targeting transcripts of leucine-zipper family proteins (Singh et al., 2014). In Medicago, overexpression of miR160 altered root development and nodule number (Bustos-Sanmamed et al., 2013). In another work, epigenetic modulation of drought stress in chickpea was investigated. They notably researched MicroRNAs (miRNAs), non-coding RNAs that have been identified as significant controllers of gene performances like BHLH23 operating at post-transcriptional stages, which are implicated in tolerance to water constraints, as well as extra abiotic distress. BHLH23 transcription factor, which encodes for low copper levels, was found to be downregulated, and another drought stress-responsive gene, APETALA2/Ethylene Response Factors (ERF/AP2), was found to have a lower expression profile in miR408 over-expressed chickpea plants when compared to vector control plants after stress treatment (Hajyzadeh et al., 2015).
Methylation of cytosine and modifications of histone plays an important role in the gene regulatory mechanisms of genes responsible for epigenetic changes in plants. These modulations serve as gene regulators during transcriptional activities. Post-transcriptional modifications in context to epigenetic regulation occur through targeted degradation of mRNA. Finally, translational repression occurs and post-transcriptional gene silencing (PTGS) of mRNAs acts as a defense molecule against several pathogens. These are viruses, bacteria, fungi, molds, and transgene (Ruiz and Voinnet, 2009; Vazquez et al., 2010). The small RNAs play an active role in transcriptional and post-transcriptional gene silencing in plants (Chapman and Carrington, 2007). The actions of these miRNAs or siRNAs in plants have a resemblance to eukaryotic biogenesis (Shabalina and Koonin, 2008). However, multiple pathways have been involved in the duplication and sub-functionalization of genes guiding miRNA or siRNA-mediated processes in plants as follows (Herr, 2005; Baulcombe, 2006).
i. Biogenesis process for the miRNAs that are complementary to the targeted sequences;
ii. A ropeway in which a miRNA activates the origin of secondary trans-acting siRNAs with no complementarities to the starting miRNAs;
iii. A route for siRNA-intervened abasement of infringed viral RNAs or transgene RNAs along with
iv. A route for siRNA-intervened methylation of DNA, transcriptional mute of transposons/viruses, and other genes.
Variegation of the core machinery for siRNA biogenesis along with function pinpoints the evolutionary process of the various small RNA suppressing mechanisms in plants (Vazquez et al., 2010). Plants, like fission yeast (S. pombe) and nematodes (C. elegans), also make use of RNA-dependent RNA polymerases in dsRNA production. Arabidopsis genome enciphers six unique RdRPs. The plant Dicer produces diversified sizes of small RNAs, viz, miRNAs of 21 nt (DCL1), or siRNAs of different sizes 23–24 nt (DCL3), 22 nt (DCL2), or 21 nt (DCL4). These diverse siRNAs differ in size but overlap in functions, due to their relatedness to variegated AGO protein that contains 10 extremities in Arabidopsis (Vaucheret, 2008).
Various proteins, such as AGO, Dicers, and RdRPs, were employed in a kind of permutations for accomplishing de novo methylation that occurs during the process of RdDM. It is initiated by the methyltransferases (DNA) of DRM grade. The exhaustive recruitment processes of DRM2 in DNA that takes place are still not conspicuous. The process is undertaken in green algae before plants prominently produce RNA Pols IV, and V, the complex configurations of RNA Pol II (Luo and Hall 2007; Tucker et al., 2010). RNA Pols II, IV, and V each have 12 basic parts in Arabidopsis, nearly half part of that is often for the above three explained polymerases and distorted by the interchangeable genes (Ream et al., 2009). The genes that originated through the reoccurrence of RNA Pol II subunit genes, bolstered with sub engaged for certain subunits enciphered the subunits that are specific to RNA Pols IV or V (Ream et al., 2009; Law et al., 2011).
Paramutation is an interaction between alleles of a gene in such a way that an allele is heritably affected by another allele. This phenomenon is explained nicely through the booster-1 (b-1) site in maize (Brink, 1956). A para mutable (B-l) allele (active allele) after getting affiliated with a para-mutagenic (B’) allele (inactive allele) becomes a paramount (B-l*) allele. Uniform DNA sequences for the two alleles at the b-1 locus are found but vary in the system of methylation (DNA). Para mutant allele itself displays as para mutagenic and is unchanged through one or more following generations. However, most alleles are neither para mutable nor para-mutagenic.
The above discussed various mechanisms and processes governing epigenetics can be adopted in chickpeas and presented through a schematic flow diagram depicted in Figure 2.
As presented in the Table 4, the epigenetic studies in biotic and abiotic stress response, DNA methylation is a crucial component in gene assertion control. The DNA methylation status in seven resistant and susceptible cultivars of chickpea for Fusarium oxysporum f. sp. was determined using the methylation-sensitive amplified polymorphism (MSAP) assay and 27, 468 DNA fragments were obtained, each of which represented a recognition site cleaved by one or both isoschizomers amplified using selected primers (Mohammadi et al., 2015). They showed DNA methylation patterns in leaves, stems, and roots from both controlled and inoculated plants, and found extensive cytosine methylation modifications in pathogen-treated/infected plants, but none in controls. Heterologous expression of WRKY40 promoter and its transcriptional regulation via epigenetic alteration controls the fusarium stress resistance. This expression aids in the prevention of bacterial infections spreading due to resistance (Chakraborty et al., 2018). The important function of the WRKY40 transcription factor in the susceptibility of chickpea to Fusarium oxysporum f. sp. ciceri race 1 (Foc1) and resistance to this strain (WR315) has been demonstrated. In a controlled and Fusarium-affected environment, the histone changes in two chickpea genotypes were evaluated using immunoblotting and real-time PCR techniques. In the process of resistance interaction with Foc1, location-specific Histone three lysine nine acetylation, a positive signal of transcription, becomes reinforced at the WRKY40 promoter. In Foc1-infected susceptible plants, the H3K9 Ac is reduced at the WRKY40 promoter. The salt tolerance mechanism in chickpea was studied using an epigenetic approach in FLIP 97-43C (salt-tolerant) and FLIP 97–196C (salt-susceptible), which aids in the discovery of proteins that regulate photosynthesis, distress responsiveness, and protein assimilation (Arefian et al., 2019).

TABLE 4. Epigenetic studies related to biotic and abiotic stresses in chickpea.
The cytosine residues in the DNA of pea root tips subjected to water deficit were investigated to see if there was a link between environmental stress and DNA methylation. Two complementary approaches were used to assess DNA methylation: (i) immunolabeling with a monoclonal antibody against 5-methylcytosine, and (ii) MSAP (Methylation-Sensitive Amplified Polymorphism) to see if methylation and demethylation in response to water deficit could be linked to specific DNA sequences (Labra et al., 2002).
Plant microRNAs were investigated in beans (Dela et al., 2019). They are generally transcribed in transcripts with a single microRNA precursor, which is processed by DICERLIKE 1 and associated proteins to produce a short RNA, which is then incorporated into an AGO-containing protein complex to direct silencing of an mRNA with a complementary target sequence. Certain microRNA loci have several precursor stem-loop structures, encoding multiple microRNAs in a single transcript that is one-of-a-kind example in which the evolutionarily conserved miR398a is encoded in the same transcript as the legume-specific miR2119. Other legumes showed the same dicistronic configuration as the common bean. The role of small RNAs in reaction to water stress was investigated in Phaseolus vulgaris, and it was discovered that mature miR398 and miR2119 are repressed in response to water deficit, but that they are functional since they target the mRNAs for CSD1 and ADH1, respectively. The down-regulation of miRNA with the consequences of upregulation of CSD1 and ADH1 genes in common beans and possibly in other legumes respond to water deprivation (Naya et al., 2014). In the case of cowpea, the homology search was used to predict miRNAs and their targets. Real-time quantitative PCR was used to confirm the identified cowpea miRNAs in the leaves and roots of drought-stricken cowpea plants. Target gene prediction reveals that a group of miRNA target genes is implicated in metabolic pathways associated with physiological changes caused by drought stress. We looked at the expression levels of some key genes involved in physiological responses to drought stress and discovered that differences in their expression levels corresponded to the various drought responses of drought-sensitive and drought-resistant cowpeas (Shui et al., 2013).
The legume miR1514a activates phasiRNA by modulating a NAC transcription factor transcript. MicroRNAs have been identified as post-transcriptional regulators implicated in stress responses in recent investigations. In Phaseolus vulgaris (common bean), miR1514a is a legume microRNA that is activated in response to drought stress and has varying levels of accumulation in roots during water deficit in two cultivars with different drought-resistance phenotypes. The role of miR1514 in the regulation of a NAC transcription factor gene via phasiRNA synthesis during response to drought has been reported in case of soybean (Sosa et al., 2017).
Many biotic and abiotic stress tolerance gene(s) in plants have been discovered through recent advances in next-generation sequencing (NGS) (Garg et al., 2016). Integration of omics-generated data from several platforms, such as transcriptomics, which is coupled with proteomics, and finally, metabolomics, is essential to close the genome-to-phenome gap in agricultural plants. These platforms and their data enable to identify the certain phenotypes based on genetic contribution (Choi, 2019). The use of the omics strategy to gather genomic information to influence various biological processes, as well as the discovery of differentially expressed genes in various environmental situations and positional cloning. This strategy can also be utilized in the targeted region with an mRNA or protein shift to uncover the role of connected genes associated with the trait of interest (Su et al., 2019). A comparison of salt stress generated FLIP 97-43C (salt-tolerant) and FLIP 97–196C (salt-susceptible) was undertaken to understand the salt tolerance mechanism in chickpea, which resulted in the identification of proteins regulating photosynthesis, distress responsiveness, and protein absorption (Arefian et al., 2019). The researchers discovered 134 proteins that were expressed differently in the extracellular matrix and during the dehydration response. During the comparative proteomics investigation of JG-62, these proteins were discovered in a variety of biological roles (Bhushan et al., 2007). Through a targeted metabolomics approach, Khan et al. (2019) identified key upregulated metabolites such as allot in, l-proline, l-arginine, and l-histidine, as well as downregulated metabolites such as alanine, choline, gamma-aminobutyric acid, and phenylalanine, that were differentially expressed under drought stress conditions. From sugars to organic acids, a total of 48 distinct metabolites were discovered. Under salt stress, 28 biogenic amino acids were expressed in chickpea cultivars with varying salt tolerance (Dias et al., 2015). The specified compounds were quantitatively analyzed using modern metabolomics techniques and GC and LC were integrated into a triple quadrupole mass spectrometer (GC-QqQ-MS and LC-QqQ-MS). As a result, the reports for drought tolerance mechanisms in chickpea genotypes, omics techniques, and crop production management were shown to be the best and most cost-effective.
The cultivated chickpea has a narrow genetic base (Varshney et al., 2013) and phenotypic plasticity (Berger, 2006). It is difficult to locate the stress-responsive and undeniably tolerant gene(s), especially when plants accept cross-talk to react to many concurrent distresses (Tuteja, 2007). The physiological and genomic screening revealed that there was a wide range of genetic differences among and within the tolerant and sensitive genotypes for salinity tolerance. For example, cold was included in tolerant-1 and inhibited in tolerant-2 during gene profiling using microarray aquaporin genes for drought, salinity, and homology-based induction for salinity, heat, and environmental stress (Mantri et al., 2007; Kotula et al., 2015; Kaur et al., 2008). Several haplotypes and significant numbers of alleles associated with agronomic parameters in chickpeas have been uncovered using genomic resources (Varshney et al., 2019a). Fine mapping of ‘QTL-hotspot’ for drought tolerance-related features for the region of 7.74 Mb–300 kb and chickpea bin mapping were done using genotyping-by-sequencing and skim-sequencing, respectively (Varshney et al., 2014; Jaganathan et al., 2015). A huge range of resources, including genetic, genomic, and transcriptome resources, have been created over the last decade as a result of developments in various NGS technologies, transforming the chickpea crop from an orphan to a genomic-rich resource (Varshney et al., 2009; Kudapa et al., 2014; Agarwal et al., 2016; Mashaki et al., 2018). In chickpea breeding projects, next-generation sequencing, high-throughput genotyping technologies, and cost-effective omics methods are critical. Translational genomics in crop breeding has been made possible by the availability of molecular markers, sequencing platforms, genotyping assays for low-to-high density, quality check panels, draught genome assemblies, and sequence-based genetic variants (Roorkiwal et al., 2014; Thudi et al., 2016; Varshney et al., 2019a; Rasheed et al., 2017; Varshney et al., 2021).
The advances in next generation sequencing (NGS) technologies have a lead impact on epigenomic research. The arrival of NGS technologies has introduced powerful sequencing methods–like, ChIP-Seq--to interrogate whole-genome histone modifications, improving on the conventional microarray-based method (ChIP-chip). More importantly, studies of DNA methylation and histone modification using NGS technologies have yielded new discoveries in plant biology too. The recent developments of third-generation sequencing technologies have shown promising results of directly sequencing methylated nucleotides and having the ability to differentiate between 5-methylcytosine and 5-hydroxymethylcytosine. The importance of 5-hydroxymethylcytosine remains largely unknown, but it has been found in various tissues. 5-hydroxymethylcytosine was particularly enriched at promoters and in intragenic regions (gene bodies) but was largely absent from non-gene regions in DNA from human brain frontal lobe tissue. The presence of 5-hydroxymethylcytosine in gene bodies was more positively correlated with gene expression levels. The importance of studying 5-methylcytosine and 5-hydroxymethylcytosine separately for their biological roles will become clearer when more efficient methods to distinguish them are available (Ku et al., 2011).
In contrast to histone modification profiling, a wide variety of approaches have been developed to profile DNA methylation utilizing next-generation sequencing platforms. Approaches to profile DNA methylation genome-wide can be broadly divided into those that rely on methylation-dependent enzymatic restriction, methyl-DNA enrichment, and direct bisulfite conversion (Fouse et al., 2010; Harris et al., 2010). Individual methods can also be combined to increase the resolution or efficiency of a single method. For example, a combination of MeDIP-seq and MRE-seq to profile both the methylated and unmethylated fractions of the genome (Maunakea et al., 2010).
Advances in next-generation sequencing (NGS) technology have considerably curtailed sequencing costs resulting in the evolution of genotyping methods from individual marker-to whole-genome sequencing-based genotyping. This has resulted in the development of large-scale genomic resources, including genome sequence assemblies, re-sequencing of a few thousand lines, high-resolution genetic maps, and a range of low-to high-density genotyping platforms. These genomic resources were used to find alleles and haplotypes linked to chickpea agronomic traits (Varshney et al., 2019b). Genetic diversity, population structure, domestication patterns, linkage disequilibrium, and the untapped genetic potential for chickpea improvement have all been studied using whole-genome re-sequencing (WGRS) (Varshney et al., 2019a). Varshney et al. (2021) conducted a study on molecular diversity in chickpeas to describe genomic diversity across cultivated and wild progenitors. They found chromosomal segments and genes that show signatures of selection during domestication, migration, and improvement. The chromosomal locations of deleterious mutations responsible for limited genetic diversity and decreased fitness were identified in elite germplasm along with the superior haplotypes for improvement-related traits. They found targets for purging deleterious alleles through genomics-assisted breeding and/or gene editing. We can use this sequence information to find the DNA methylation regions that are responsible for biotic and abiotic tolerance in the chickpea in future breeding approaches.
Epigenetic mechanisms have proven to have a role in enhancing plants’ resilience to environmental stresses, targeting varied traits, thus, giving a significant tool in breeding for climate-resilient crops. Epigenetic variation was applied for crop improvement to increase soybean yield (Raju et al., 2018). RNAi silencing of the plant-specific gene MutS HOMOLOG1 (MSH1) paved the way for the growth of epi-lines with variability for the arrangement of yield-associated traits in glasshouse and field trials. New epigenetic diversity indicted by MSH1 oppression was transmitted for at least three progenies. Similarly, the identification of epigenetic variations and regulatory mechanisms in chickpea plants, which impact important agronomic traits, can be exploited for epigenetic breeding for climate-resilient crops. The following schematic presentation as depicted in Figure 3 for the development of epigenetic data and tools will lead to breed of newer ep-breeds and varieties in the field and adapted to climatic changes.

FIGURE 3. Schematic integrated epigenetic data and tools will lead to epi-bred crops and new varieties in the field adapted to climate change (Modified from Kakoulidou et al., 2021).
Crop plants often have challenges of biotic and abiotic stresses, and they adopt sophisticated ways to acclimate and cope with these through the expression of specific genes. Changes in chromatin, histone, and DNA mostly serve the purpose of combating challenges and ensuring the survival of plants in stressful environments. Epigenetic changes, due to environmental stress, enable plants to remember a past stress event in order to deal with such challenges in the future. This heritable memory, called “plant stress memory”, enables plants to respond against stresses in a better and more efficient way, not only for the current plant in prevailing situations but also for future generations (Chao et al., 2021). Stress memory can also be described as a mechanism to enhance the resilience of crop plants (Walter et al., 2011), and the accumulation and changes in proteins (structural and regulatory) as transcription, translation, and transduction, which play an important role in the growth, development, and memory mechanisms of plants for stress resistance (Bruce et al., 2007; Janmohammadi et al., 2015, Marcos et al., 2018b). As the epigenetic modifications are environmentally accelerated, the phenotypic changes are mostly a reflection of a specific environmental interaction, and the changes adopted by the plant for a specific period may become permanent and heritable for future generations (Chinnusamy and Zhu, 2009; Verhoeven et al., 2010). Stress memory in plants is enhanced by up and downregulated sRNAs (miRNAs, siRNAs) they mainly downregulate negative regulators, upregulate positive regulators and regulate plant hormones, reactive oxygen species (ROS), and transcriptional factors in response to abiotic stress (Banerjee et al., 2017; Zhang et al., 2019).
Plant stress memory is achieved through the coordination of physiological, translational, transcriptional, and epigenetic activities in response to stress (Kinoshita and Seki, 2014; Hu et al., 2016). These regulatory processes can occur at any stage of plant development and are primarily controlled by epigenetic changes to phenotypically remodel for environmental stress (Rehman et al., 2015; Gallusci et al., 2017). Genetic diversity has been reduced as a result of intense breeding, and now epigenetic variation has arisen as a viable option for crop genetic improvement (Gallusci et al., 2017). There have been many developments for the quantification of epigenetic variations and their impact on the growth and development of plants, leading to improved yield and quality, and ultimately, this has opened another avenue for breeders to breed desirable agronomic characters successfully (Cortijo et al., 2014)]. In epigenetic modifications, DNA methylation plays an important role in gene regulation, expression, and stabilization (Lang et al., 2017). Various enzymes (DNA methyltransferase), targeted under different plant regulatory pathway systems, take part in the process to catalyze DNA methylation for a better and quicker response against biotic and abiotic stresses (Zhang et al., 2018). Epigenetic modification has the ability to memorize the event over a long time as a plant molecular memory and the ability to respond rapidly with heritable phenotypic characteristics as an inheritance system against environmental fluxes. Some extreme abiotic stress treatments can lead to plant genome reorganization (Klumpp et al., 2004; Molinier et al., 2006) but few reports are indicating that short-term stress causes a large number of genomic mutations (Lämke and Bäurle, 2017; Cruzan et al., 2018). More evidence supports the speculation that plant stress memory is mainly regulated by epigenetic pathways (Tang et al., 2014), which means changing the expression pattern of the entire genome to form a rebalanced genome expression system, without changing the genome sequence (Habu et al., 2001; Madlung and Comai, 2004).
Epigenetic variants can be produced through chemical treatment (5-azacytidine), epigenomic editing (TALENs), zinc finger nucleases, and the CRISPR/Cas system to counter biotic and abiotic stresses. There is an immense amount of care needed because targeted genes may be involved in complex and multiple pathways, which may cause complex and unexpected pleiotropic effects (Garg et al., 2015; Hung et al., 2018). All of these methods have tremendous scope for the creation of epigenetic variations (Kapazoglou et al., 2018) For successful breeding through epigenetic memory, it is necessary that variations should be inherited. DNA methylation changes and histone modifications are often reset during meiosis, meaning stable inheritance of the epigenetic mark is a problem in successful breeding goals achievement (Danchin et al., 2019).
Several reports revealed a correlation between the regulation of gene expression and changes in chromatin modifications in plants during stress exposure (Pandey et al., 2016). Epigenetic processes are crucial adaptive mechanisms that change the expression of genes in a heritable way without accompanying changes in DNA sequences (Chinnusamy and Zhu, 2009). Thus, heritable, but simultaneously reversible alterations in the transcriptional potential of cells are possible (Chen et al., 2010b). In a eukaryotic cell, the structure and function of chromatin depend upon several regulatory epigenetics. In plants such as Arabidopsis thaliana, Oryza sativa, and Zea mays, it was recently reported that hyper- or hypomethylation of DNA induced by abiotic stimuli can modulate the expression of stress-responsive genes (Wang et al., 2009; Van Dijk et al., 2010) mechanisms, including DNA methylation and histone modifications (Sahu et al., 2013). Epigenetics has a major role in symbiotic nitrogen fixation in chickpea, reported and experimentally proved by epigenetic regulations in the development of symbiotic root nodules of legume plants–EPISYM project. They have discovered that epigenetic regulations, involving plant DNA (de)methylation and small interfering RNA (siRNA) populations are essential to produce nitrogen-fixing nodules. They hypothesized that epigenetic regulations play an important role in gene expression reprogramming associated with nodule differentiation.
The accurate genetic assays of epigenetic variability and mapping of epigenetic quantitative trait loci (epiQTL) facilitated by the development of epi-RILs in Arabidopsis have provided a close association amongst epialleles and phenotypic characteristics. The epigenetics research in plants has taken a leap by employing epigenome-wide association study (EWAS) and epi-genotyping by sequencing (epi-GBS). Thus, chickpea improvement programs eventually can utilize the huge avenues provided by quantitative epigenetics to assay the contribution of epigenetic variability in trait control. Further, molecular breeding of important crop plants can be potentially facilitated by epigenome-editing tools, such as clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR/Cas9), for locus-specific DNA methylation (Vijay et al., 2020).
Several statistical methods exist to detect epigenetic variations and their impact on the phenotype or epiQTLs. The significance and accuracy of epiQTLs identification are affected by several factors like recombination, transgressive segregation, instability of epialleles, and parent-off-origin effect. These factors may create confounding effects during epiQTLs analysis and result in false positives or false negatives. To deal with these interrupting factors, Johannes and Colomé-Tatché (2011) advocated as most suitable population generated from the crosses between epigenetic isogenic lines. Tal et al. (2010) deduced covariances amongst kinships owing to epigenetic transmission and environmental effect and modeled the number of events for epigenetic reset amongst generations and environmental inductions, and estimated the heritable epigenetic variance along with the rate of transmission. Furthermore, the necessity of multiple replication-wise testing due to the occurrence of several false positives is the critical bottleneck of quantitative genetics. During genome-wide recognition of epigenetic variation to encounter the bottleneck of false positivity a statistical model was developed (Jaffe et al., 2017). The missing heritability contributed by epigenetic variability may be studied by employing these models but leaving aside epigenetic-induced phenotypic variability (Roux et al., 2011). Furthermore, another improved model was proposed to predict the proportion of genetic variation and estimate phenotypic variation explained by epigenetic variation and their effects on phenotypic values along with the interaction of genetic effects (additive and dominant) and epigenetics (Wang et al., 2012).
Epigenetic markers are randomly present with high frequencies in the genome and are stably inherited through generations. The identification of epiQTLs is facilitated by these characteristics that permit the utilization of epigenetic markers. Unlike QTLs where polymorphism for the DNA nucleotide sequence occurs, epiQTLs are epigenomic loci that differ in cytosine methylation patterns that control phenotypic variability. Cortijo et al (2014) recognized major epiQTLs explaining 60–90% heritability by employing ddm1-derived Arabidopsis epi-RILs for quantitative traits root length and flowering time. These epiQTLs were observed to be useful for artificial selection and were reproducible. Furthermore, on the basis of inheritance and recombination events using mutagenic accumulation lines epigenotype map (E-map) was constructed and 99.9% of epialleles were observed to be stable (Hofmeister et al., 2017). In another study employing methylation-sensitive amplified fragment length polymorphism (MS-AFLP) and retro transposon epimarkers epiQTLs for seven agronomic characters were recognized in Brassica (Long et al., 2011). During varied developmental, environmental, and transgenerational states highly stable epigenetic marks were observed. In Sorghum, employing MSAP genotyping approach and 122 methylation polymorphic loci E-map harboring methylation hotspots, was constructed. In soybean, localization of methyl QTL (QTLs associated with DNA methylation) was facilitated by employing differentially co-segregated methylated regions (DMRs) in RILs. Thus, the crop where genetic variability is the negligible stable inheritance of epialleles through the generations makes it a potential controller of phenotypic variability. However, till date very limited number of EWAS have been accomplished in plants, but employing somatic clones (diverse for mantled abnormality and oil yield), a locus MANTLED was localized where hypomethylation in LINE retro transposon pushes the alternate splicing and premature termination epigenetic modification associated with a mantled abnormality in oil palm (Ong-Abdullah et al., 2015).
Besides the genes inclusive of its genome, the genetic constitution of any organism also contains its epigenome including methyl classes to specific sequences within the DNA that work as epigenetic marks to minimize transcriptions, and thus the expressions of the linked genes. Several methods applied to change the plant’s epigenome contained mutagenesis, carcinogenesis, plant tissue culture, CRISPR/Cas9 genome editing, and RNAi, which are explained below.
The mutagenesis and carcinogenesis affecting DNA sequence and chromatin structure through Ethyl methane sulfonate (EMS) mutagenesis and associations with epigenetic changes have been explained (Yan et al., 2021) in rice. Whole-genome and re-sequenced data congregated from 52 rice EMS mutants facilitated mutation for altering DNA sequences and the probable linkages along with chromatin composition. Single nucleotide polymorphic sites (SNPs) along with genomic facets related to EMS anchored mutagenesis prejudices were unraveled. EMS, equated with natural SNPs available in the Rice 3K project, displayed a liking to G/C sites with flanking motifs higher in GC amounts. Efficacies of EMS mutagenesis and constituents of local dinucleotides along with trinucleotides were having associations. The prejudiced allocation of EMS indicted SNPs were affiliated in a positive direction with transposable element quantities, CpG numbers, and suppressive epigenetic markers but linked in the negative direction with active epigenetic markers and gene (s) displaying the euchromatin marker DNase I hypersensitive sites. Another example through which mutations created epigenesis was presented with Arabidopsis thaliana mutants originated straightway by changes in DNA methylation affecting transcription of the gene. The late-flowering mutant flowering Wageningen (FWA) created by ectopic demonstration of the FWA gene enciphers a homeodomain-containing transcription facet. In wild type, the escalating region of FWA is methylated DNA and FWA is not produced in vegetative tissues. When this methylated DNA is ousted from the ddm1 mutant, the FWA is noticed in vegetative tissues and causes late flowering. This late-flowering phenotypic form is also noticed in the mutant suggesting that silencing of FWA mainly depends on CG methylation (Soppe et al., 2000). In another recent study done at Indian Agricultural Research Institute, New Delhi, we treated Pusa 372- a high-yielding, and widely grown chickpea cultivar having moderate resistance/tolerance to major diseases with 0.3% EMS for 6 h at room temperature and found mutants with phenotypic variations for the increased number of pods (unpublished).
Tissue culture techniques are soul for any alteration at the genome level in crops. These techniques are also influenced its epigenome. The high-resolution maps of DNA methylation made in rice lines have reported that the regenerated plants have less methylation than control plants. The alterations were relatively over-represented around the promoter sequences of genes and affect gene expression. Critically, the plants’ offshoots also inherit the changes in methylation level (Hume et al., 2013). These aftermaths partly narrate the processes of somaclonal diversities that push forward epigenetic changes in the plants.
Gene silencing through RNA inference (RNAi) approach was widely used to better understand the gene function in plants. Repression of translation was achieved through post-transcriptional modifications using RNAi. Interestingly, many of the factors that mediate post-transcriptional silencing via RNAi also contribute to transcriptional gene suppression (Fire et al., 1998). In plants, RNA viruses were observed to guide DNA methylation of homologous genes along with introducing multiple transgene copies resulting in silencing (Napoli et al., 1990; Van der Krol et al., 1990).
The ribonucleoproteins (RNPs), containing Cas9 enzyme along with single-guide RNA (sgRNA) are successfully delivered using transformation methods or nanoparticle-based delivery approaches. The prime enzyme 4-coumarate ligase (4CL) involved in phenylpropanoid metabolism and responsible for the lignin biosynthesis process governs the congregation of lignin in distress stages. The 4CL along with the gene Reveille 7 (RVE7) linked with drought tolerance has been used for protoplast targeted mutagenesis in chickpeas. For the first time, chickpea protoplast was used as a transfection platform for CRISPR/Cas9-based genome editing in chickpeas (Badhan et al., 2021). The outcomes showed efficient editing got for the RVE7 gene in vivo compared with the 4CL gene.
Understanding genomic activities need site-specific modification at the loci via targeting systems (Papikian et al., 2019). Limited approaches for the desired manipulation of the epigenome present in plants were observed and adopted by the Cas9-system to design desired gene activation and DNA methylation in Arabidopsis.
Epigenetics and epigenomics display certain connotations as useful immense potentials along with numerous threats and challenges as stated by Springer and Schmitz (2017). Some of the potential connotations for utilization are summarized as given below.
As we mentioned above, the role of epigenetics in disease resistance (Sen et al., 2017), cold tolerance (Rakei et al., 2015), drought, salinity tolerance (Khandal et al., 2017), and manipulating the epigenome may provide a promising breeding strategy to enhance yield, disease resistance, or adaptation for changing environmental conditions in chickpea, as shown below.
i. DNA methylation patterns in cultivated chickpeas to understand the regulation of gene expression in different organs (Bhatia et al., 2018)
ii. Drought and salinity resistance by an epigenetic mechanism in chickpeas (Sen et al., 2017)
iv. DNA methylation and epigenetics mechanism on physio-biochemical analysis of chickpea in response to cold stress (Rakei et al., 2015)
v. The epigenetic mechanism to heat stress in chickpeas (Chidambaranathan et al., 2016)
vi. Role of epigenetics in drought yield index (Sharma et al., 2019)
Transgene methylation and transcriptional gene mutations are directly correlated to each other (Matzke et al., 1989; Park et al., 1996) mainly because of the association between methylation of the coding sequence and post-transcriptional gene oppression (Ingelbrecht et al., 1994). Although, the latest proof shows that a merging process gleaned from RNA interference is primary to both processes (Matzke and Matzke, 2004). The intricate and meticulous designing of the transgene constructs and intense dissection of transformants at the molecular level are the prerequisites of an efficient technique to avoid transgene silencing (Dewilde et al., 2000). Two dominant classes of transgene silencing, the first one results in position effects (Matzke et al., 2000) and the second one is silencing phenomena or homology-oriented gene silencing, HDGS (Meyer and Saedler, 1996). Some examples reported in plants are tobacco, transgenic tobacco (N. tabacum) that are constructed ectopically over-express AtMYB90v (Arabidopsis thaliana MYB 90) promoter gene in association with regulating anthocyanin production in Arabidopsis thaliana. Transgenic tobacco overexpressing AtMYB90 involved in anthocyanin biosynthesis showed siRNA-mediated silencing because of systemic acquired silencing (Velten et al., 2012).
Although epigenetics in multicellular organisms is the major mechanism for diversifications, with epigenetic motifs “reset” when organisms procreate, there were certain reflections of trans-generational epigenetic transmission, e.g., the phenomenon of para-mutation in maize (Brink, 1956). Epigenetic characters are multigenerational and eventually diminished over many generations. Then, there is a clear-cut maximum probability for explaining another aspect of evolution and adaptation. There are some speculations that the differential mutation rates associated with epigenetic features were taken as an advantage by the organisms that control the mutation rates of particular genes. Epigenetic changes have also been reflected to originate in reaction to environmental exposure; for example, epigenetic alterations are prevalent in inter-specific hybrids and polyploids. DNA methylation patterns after hybridization and/or polyploidization can be primarily changed by these re-patterning processes, as exemplified by studies in Brassica, Arabidopsis, Triticum, and Oryza. In these species, methylation-influenced AFLP assays provided widespread alterations in genomic methylation, including modifications in genes (Liu and Wendel, 2003).
A study unraveled DNA methylation systems in cultivated chickpea to explain the control of gene expression in variegated organs primarily by the methylating systems in leaf tissue of wild and cultivated chickpea. The results show a positive association of promoter hyper-methylation with increased transcript paucity through recognition of DMR of the genes governing flower development meant in cultivated chickpeas (Bhatia et al., 2018).
Epigenetic patterns in plants, once instituted, can be transmitted through the inheritance of epialleles across many generations (Kakutani, 2002). Such transmittable epigenetic alleles can be assumed as a novel source of polymorphism and may reproduce new phenotypes. Evaluating the significance of methylated epialleles in crop breeding requires the genetic variability in the selected population for the degrees that methylating modes influence superior phenotypes and the extent to which methylation is statically transmitted. DNA methylation was first reported in regeneration studies of crown gall tumor events in which phenotypic variability and methylation of T-DNA were linked (John and Amasino, 1989). The most interesting evidence suggests that a substantial proportion of somaclonal variation might be because of diverse, pre-existing epigenetic states’ result in the regeneration of individual somatic cells (Neuhuber et al., 2005).
Epigenetic initiation of DNA elements through transposable elements with Arabidopsis suggests epigenetic modifications may also be intricated in cytogenetic instability through changes of heterochromatin, and as a basis of phenotypic diversity through the modulation of gene functionalities (Kaeppler et al., 2000).
Epigenetics plays an important role in somaclonal variation, and chromatin modulation plays an important role in gene expression regulation and genome activities (Azizi et al., 2020). Some epigenetic modifications that induced intergenerational distress memory resistance in crop plants in addition to as presented in Table 5 are as below.
a) Chickpea-drought water and osmotic stress (Elkoca et al., 2007; Kilian et al., 2007)
b) Canola–Salt/drought, seed priming with NaCl, increased energy efficient utilization, and PGPR for halo-tolerant plant (Farhoudi et al., 2007)
c) Sugarcane–Drought/salinity, NaCl, and PEG-primed seeds (Marcos et al., 2018a)

TABLE 5. Intergenerational stress memory resistance development in crop plants through epigenetic modifications.
Heterosis is the superiority of the F1 hybrid phenotype over its parents. The phenomenon has been exploited extensively in agricultural breeding for decades and, despite its commercial impact; it has also improved crop performance tremendously. However, knowledge of the molecular basis underlying heterosis remains incomplete. Most studies have focused on finding genetic explanations, resulting in the classical dominance and overdominance models of heterosis (East, 1908; Shull, 1908; Bruce, 1910; Jones, 1917). Identification of better hybrids through the utilization of hybrid vigor in chickpea by assessing seven F1 hybrids inclusive of nine cultivars was executed (Ghfaffar et al., 2015). Paramount heterosis along with heterobeltiosis for plant height and subsidiary branches were observed in the cross K0014–10 × K0066-10, however, cross K0019–10 × K0031-10 performed the highest heterosis along with heterobeltiosis for principal branches and seed number plant−1, the cross K0014–10 × K0052-10 reflected the highest heterosis and heterobeltiosis with 33.18 and 30.84% for 100 seed weight and 97.37 and 76.47% for seed yield plant−1, discretely. Broad sense heritability for various characters observed varied from 63.14 to 77.18%. Remarkable heterosis, heritability, and genetic advance were recorded for pod number plant−1 that could be employed for identifying best segregate from crosses K0031–10 × K0052–10, K0019-10 × K0026-10, and K0019–10 × K0031-10. Best hybrids from the observation could be employed for the betterment of multiple traits by identifying single plants for varied characteristics.
Molecular profiling of superior hybrids reflected that their epigenomes are substantially remodeled to their parental lines, leading to epigenetic states that deviate from the expected mid-parent values. Extensive remodeling has been observed at the level of DNA methylation in Arabidopsis (Zhang et al., 2016), rice (He et al., 2010), pigeon pea (Zhang et al., 2006), broccoli (Li et al., 2018a), and rapeseed (Shen et al., 2017). It occurs either at regions where parents are differentially methylated (DMRs).
Irrespective of the immense potential of epigenetics for opening new avenues for utilization in crop improvement programs, there are certain challenges and threats as stated below.
A. Determination of Epigenetic State: The first challenge is to clearly define the basis of an epigenetic state. Whereas a DNA sequence is simply defined by the order of the four bases (A, C, G, and T), the exhaustive list of components that define given chromatin or epigenetic state is yet to be established. These components include methylation of cytosines and adenines, mono-, di- or tri-methylation, acetylation, phosphorylation, ubiquitination, etc of histones at various positions (e.g., H2AK119, H3K4, H3K9, H3K27, H3K36, etc) and long or short ncRNAs produced in cis or trans.
B. Determination of Chromatin Stability: The second important challenge is to define the stability of given chromatin. Three major levels of stability can be distinguished, as given below:
Transient Chromatin States: These chromatin states are specific to a different cell or established in immediate response to biotic or abiotic stress, and do not persist after the stimulus is removed.
Metastable Epigenetic States: These epigenetic states are started by specific stress or environmental inductions and can persist across multiple cell divisions after induces.
Inherited Epigenetic States: These epigenetic states are transmitted across multiple generations and are typically correlated with TEs or other repeat sequences. It is still unclear what role the environment plays in initiating or erasing these states.
Gene and environmental interactions as epigenetics are influenced by the environment and sometimes it leads to non-stable variations.
Epigenome sequencing methods are not well established.
Epigenetics has immense potential for opening new avenues for crop improvement programs stated as followings:
a) Variation: Considerable natural variability in the DNA methylation process exists within many plant species.
b) Stable Inheritance: Variability in DNA methylation can originate through processes and clonal propagation can propel epigenetic alleles.
c) Epigenome Engineering: New epigenome editing tools provide broader opportunities to create new epiallelic variants by altering the methylation of DNA or other modifications at the chromosome level. These tools can be used for crop improvement through epigenome engineering.
d) Emerging New Technologies: The development and application of methods for widespread epigenome profiling and engineering may generate new avenues for using the full potential of epigenetics in crop improvement.
e) Sources for Biotic and Abiotic Resistance: Epigenetics has become an important research focus at a time when rapid environmental changes are occurring. They enhance fitness extremely rapidly without depending on the slower process of natural selection through changing DNA-encoded genetic variants in plant populations.
f) Time and Cost-Effective: Epigenetics introduces as a time- and cost-effective tool in plants as a source of resistance against new future abiotic and biotic stresses.
g) Public and Producer’s Acceptance: Successful implementation of all crop enhancement approaches at the DNA level requires support from the public and government and epigenome editing does not change the genome sequence might ease the challenges of public acceptance for epigenetically modified products.
h) Equilibrium among important agronomic traits: Plants use epigenetic variation to reprogram their transcriptome in a precise and timely manner to maintain equilibrium amongst important agronomic traits.
Climate change is altering the predominance of varied environmental situations, and improved distress tolerance has become a primary breeding goal in chickpeas. In vivo situations, crops are usually concomitantly opposed by diverse biotic and abiotic distresses. Hence, grasping possible processes responsible for the occurrence of stresses has become a necessity for stable crop productivity. The epigenetic mechanisms play an important role in a classical plant breeding program, mainly by genetic heritability, hybrid vigor, plant-environment interactions, abiotic and abiotic stress tolerance, and yield stability performance of crop plants. A better and deep insight into epigenetic mechanisms might facilitate plant breeders in creating novel and more super crop varieties that can include natural phenotypic diversity. It is a very interesting fact that the environmental shielding effects of epigenetics are directly associated with those genes that play a very important role in the regulation of plant growth and yield in chickpeas and other crops. Furthermore, understanding the molecular bottom of trans-generational epigenetic transmission puts forward the development of epialleles identified for specific environmental status through combined and multidisciplinary efforts of researchers and targeted epigenetic modifications in genes of interest. Thus, epigenomics, either as exploitation of existing epigenomic variability or alteration of the epigenome, can complement conventional plant breeding to ensure global food security and sustainable agriculture.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
RK conceptualized and supervised the manuscript writing. BC, RM, and RS collected the related literature and contributed to the original writing. RK, RF, NV, SG, HY, and MM extended their help in the inference, review, and editing of the manuscript. All authors went through the final manuscript draft and approved it.
The study was supported by ICAR-Indian Agricultural Research Institute, New Delhi and DST-Science and Engineering Research Board through sanction order No.CRG2019006273. DBT-National Institute of Plant Genome Research, New Delhi, India; CSIR-National Botanical Research Institute, Lucknow, India; Griffith University, Australia; University of Southern Queensland, Queensland, Australia and Cornell University, Ithaca, New York, for providing necessary support for the study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors gratefully acknowledge the ICAR-Indian Agricultural Research Institute, New Delhi, India.
Agarwal, G., Garg, V., Kudapa, H., Doddamani, D., Pazhamala, L. T., Khan, A. W., et al. (2016). Genome-wide Dissection of AP2/ERF and HSP90 Gene Families in Five Legumes and Expression Profiles in Chickpea and Pigeonpea. Plant Biotechnol. J. 14, 1563–1577. doi:10.1111/pbi.12520
Amaral, J., Ribeyre, Z., Vigneaud, J., Sow, M. D., Fichot, R., Messier, C., et al. (2020). Advances and Promises of Epigenetics for Forest Trees. Forests 11, 976. doi:10.3390/f11090976
Amin, U. S. M., Biswas, S., Elias, S. M., Razzaque, S., Haque, T., Malo, R., et al. (2016). Enhanced Salt Tolerance Conferred by the Complete 2.3 Kb cDNA of the Rice Vacuolar Na+/H+ Antiporter Gene Compared to 1.9 Kb Coding Region with 5 ' UTR in Transgenic Lines of Rice. Front. Plant Sci. 7, 14. doi:10.3389/fpls.2016.00014
Arefian, M., Vessal, S., Malekzadeh-Shafaroudi, S., Siddique, K. H. M., and Bagheri, A. (2019). Comparative Proteomics and Gene Expression Analyses Revealed Responsive Proteins and Mechanisms for Salt Tolerance in Chickpea Genotypes. BMC Plant Biol. 19, 300. doi:10.1186/s12870-019-1793-z
Arumuganathan, K., and Earle, E. D. (1991). Nuclear DNA Content of Some Important Plant Species. Plant Mol. Biol. 9, 208–218. doi:10.1007/bf02672069
Azizi, P., Hanafi, M. M., Sahebi, M., Harikrishna, J. A., Taheri, S., Yassoralipour, A., et al. (2020). Epigenetic Changes and Their Relationship to Soma Clonal Variation: a Need to Monitor the Micro Propagation of Plantation Crops. Funct. Plant Biol. 47, 508–523. doi:10.1071/FP19077
Badhan, S., Ball, A. S., and Mantri, N. (2021). First Report of CRISPR/Cas9 Mediated DNA-free Editing of 4CL and RVE7 Genes in Chickpea Protoplasts. Int. J. Mol. Sci. 22, 396. doi:10.3390/ijms22010396
Banerjee, S., Sirohi, A., Ansari, A. A., and Gill, S. S. (2017). Role of Small RNAs in Abiotic Stress Responses in Plants. Plant gene. 11, 180–189. doi:10.1016/j.plgene.2017.04.005
Bannister, A., and Kouzarides, T. (2011). Regulation of Chromatin by Histone Modifications. Cell. Res. 21, 381–395. doi:10.1038/cr.2011.22
Barlow, D. P., and Bartolomei, M. S. (2014). Genomic Imprinting in Mammals. Cold Spring Harb. Perspect. Biol. 6, a018382. doi:10.1101/cshperspect.a018382
Barra, V., Schillaci, T., and Lentini, L. (2012). Bypass of Cell Cycle Arrest Induced by Transient DNMT1 Post-transcriptional Silencing Triggers Aneuploidy in Human Cells. Cell. Div. 7, 2. doi:10.1186/1747-1028-7-2
Baulcombe, D. C. (2006). Short Silencing RNA: The Dark Matter of Genetics? Cold Spring Harb. Symp. Quant. Biol. 71, 13–20. doi:10.1101/sqb.2006.71.052
Bazin, J., Khan, G. A., Combier, J. P., Bustos-Sanmamed, P., Debernardi, J. M., Rodriguez, R., et al. (2013). miR396 Affects Mycorrhization and Root Meristem Activity in the Legume Medicago Truncatula. Plant J. 74 (6), 920–934. doi:10.1111/tpj.12178
Becker, P. B., and Workman, J. L. (2013). Nucleosome Remodelling and Epigenetics. Cold Spring Harb. Perspect. Biol. 5, 017905. doi:10.1101/cshperspect.a017905
Benedito, V., Torez-Jerez, I., Murray, J., Andriankaja, A., Allen, S., Kakar, K., et al. (2008). A Gene Expression Atlas of the Model Legume Medicago Truncatula. Plant J. 55, 504–513. doi:10.1111/j.1365-313X.2008.03519.x
Berger, F. (2006). Endosperm: the Crossroad of Seed Development. Curr. Opin. Plant Biol. 6, 42–50. doi:10.1016/s1369526602000043
Berger, S. L., Kouzarides, T., Shiekhattar, R., and Shilatifard, A. (2009). An Operational Definition of Epigenetics. Genes. & Dev. 23, 81–783. doi:10.1101/gad.1787609
Berr, A., Shafiq, S., and Shen, W. H. (2011). Histone Modifications in Transcriptional Activation during Plant Development. Biochim. Biophys. Acta. 1809, 567–576. doi:10.1016/j.bbagrm.2011.07.001
Bhatia, H., Khemka, N., Jain, M., and Garg, R. (2018). Genome-wide Bisulphite-Sequencing Reveals Organ-specific Methylation Patterns in Chickpea. Sci. Rep. 8, 9704. doi:10.1038/s41598-018-27979-w
Bhushan, D., Pandey, A., Choudhary, M. K., Datta, A., Chakraborty, S., and Chakraborty, N. (2007). Comparative Proteomics Analysis of Differentially Expressed Proteins in Chickpea Extracellular Matrix during Dehydration Stress. Mol. Cell. Prot. 6, 1868–1884. doi:10.1074/mcp.M700015-MCP200
Black, J. C., Vanrechem, C., and Whetstine, J. R. (2012). Histone Lysine Methylation Dynamics: Establishment, Regulation, and Biological Impact. Mol. Cell. 48, 491–507. doi:10.1016/j.molcel.2012.11.006
Blewitt, M., and Whitelaw, E. (2013). The Use of Mouse Models to Study Epigenetics. Cold Spring Harb. Perspect. Biol. 5, 017939. doi:10.1101/cshperspect.a017939
Boucelha, L., and Djebbar, R. (2015). Influence of Different Pre-germination Treatments of Vigna Unguiculata (L.) Walp. Seeds on Germination Performance and Water Stress Tolerance. Biotechnol. Agron. Société Environ. 19, 160–172.
Bratzel, F., Yang, C., Angelova, A., López-Torrejón, G., Koch, M., del Pozo, J. C., et al. (2012). Regulation of the New Arabidopsis Imprinted Gene AtBMI1C Requires the Interplay of Different Epigenetic Mechanisms. Mol. Plant 5, 260–269. doi:10.1093/mp/ssr078
Brink, R. A. (1956). A Genetic Change Associated with the R Locus in Maize Which Is Directed and Potentially Reversible. Genetics 41, 872–889. doi:10.1093/genetics/41.6.872
Bruce, A. B. (1910). The Mendelian Theory of Heredity and the Augmentation of Vigor. Science 32, 627–628. doi:10.1126/science.32.827.627-a10.1126/science.32.827.627.b
Bruce, J. A., Matthes, M., C., Napier, J. A., and Pickett, J. A. (2007). Stressful “Memories” of Plants: Evidence and Possible Mechanisms. Plant Sci. 173, 603–608. doi:10.1016/j.plantsci.2007.09.002
Brzeski, J., and Jerzmanowski, A. (2003). Deficiency in DNA Methylation 1 (DDM1) Defines a Novel Family of Chromatin-Remodeling Factors. J. Biol. Chem. 278, 823–828. doi:10.1074/jbc.M2092602010.1074/jbc.m209260200
Burggren, W. (2016). Epigenetic Inheritance and its Role in Evolutionary Biology: Re-evaluation and New Perspectives. Biology 5 (2), 24. doi:10.3390/biology5020024
Burggren, W. (2020). Phenotypic Switching Resulting from Developmental Plasticity: Fixed or Reversible? Front. Physiol. 10. doi:10.3389/fphys.2019.01634
Bustos-Sanmamed, P., Mao, G., Deng, Y., Elouet, M., Khan, G. A., Bazin, J. R. M., et al. (2013). Overexpression of miR160 Affects Root Growth and Nitrogen-Fixing Nodule Number in Medicago Truncatula. Funct. Plant Biol. 40, 1208–1220. doi:10.1071/FP13123
Chakraborty, J., Ghosh, P., Sen, S., and Das, S. (2018). Epigenetic and Transcriptional Control of Chickpea WRKY40 Promoter Activity under Fusarium Stress and its Heterologous Expression in Arabidopsis Leads to Enhanced Resistance against Bacterial Pathogen. Plant Sci. 276, 250–267. doi:10.1016/j.plantsci.2018.07.014
Chandler, V. L., Eggleston, W. B., and Dorweiler, J. E. (2010). Paramutation in Maize. Plant Mol. Biol. 43, 121–145. doi:10.1023/a:1006499808317
Chapman, E. J., and Carrington, J. C. (2007). Specialization and Evolution of Endogenous Small RNA Pathways. Nat. Rev. Genet. 884–896. doi:10.1038/nrg2179
Chao, S., Kazim, A., Kan, Y., Sajid, F., and Richard, D. (2021). Plants Exploration of Epigenetics for Improvement of Drought and Other Stress Resistance in Crops. Rev. Plants 10 (6), 1226. doi:10.3390/plants10061226
Chen, H. M., Chen, L. T., Patel, K., Li, Y. H., Baulcombe, D. C., and Wu, S. H. (2010b). 22-Nucleotide RNAs Trigger Secondary siRNA Biogenesis in Plants. Proc. Natl. Acad. Sci. U. S. A. 107, 15269–15274. doi:10.1073/pnas.10017310.1073/pnas.1001738107
Chen, M., Lv, S., and Meng, Y. (2010a). Epigenetic Performers in Plants. Dev. Growth Differ. 52, 555–566. doi:10.1111/j.1440-169x.2010.01192.x
Chen, R., Li, M., Zhang, H., Duan, L., Sun, X., Jiang, Q., et al. (2019). Continuous Salt Stress-Induced Long Non-coding RNAs and DNA Methylation Patterns in Soybean Roots. BMC Genom 20, 730. doi:10.1186/s12864-019-6101-7
Chen, Z. H., Bao, M. L., Sun, Y. Z., Yang, Y. J., Xu, X. H., Wang, J. H., et al. (2011). Regulation of Auxin Response by miR393-Targeted Transport Inhibitor Response Protein 1 Is Involved in Normal Development in Arabidopsis. Plant Mol. Biol. 77, 619–629. doi:10.1007/s11103-011-9838-1
Chidambaranathan, P., Jagannadham, P. T. K., Satheesh, V., Jain, P. K., and Srinivasan, R. (2016). Expression Analysis of Six Chromatin and Remodeling Complex Genes (SWR1) in Chickpea in Different Tissues and during Heat Stress. Indian J. Genet. 76, 47–56. doi:10.5958/0975-6906.2016.00007.9
Chinnusamy, V., and Zhu, J. K. (2009). Epigenetic Regulation of Stress Responses in Plants. Curr.Opin. Plant Biol. 12, 133–139. doi:10.1016/j.pbi.2008.12.006
Choi, H. K. (2019). Translational Genomics and Multi-Omics Integrated Approaches as a Useful Strategy for Crop Breeding. Genes. Genom 41 (2), 133–146. doi:10.1007/s13258-018-0751-8
Collins, B. E., Greer, C. B., and Coleman, B. C. (2019). Histone H3 Lysine K4 Methylation and its Role in Learning and Memory. Epigenetics Chromatin 12, 7. doi:10.1186/s13072-018-0251-8
Cortijo, S., Wardenaar, R., Colome-Tatche, M., Gilly, A., Etcheverry, M., Labadie, K., et al. (2014). Mapping the Epigenetic Basis of Complex Traits. Science 343, 1145–1148. doi:10.1126/science.1248127
Cruzan, M. B., Streisfeld, M. A., and Schwoch, J. A. (2018). Phenotypic Effects of Somatic Mutations Accumulating during Vegetative Growth. bioRxiv. doi:10.1007/s10682-022-10188-3
Cubas, P., Vincent, C., and Coen, E. (1999). An Epigenetic Mutation Responsible for Natural Variation in Floral Symmetry. Nature 401, 157–161. doi:10.1038/43657
Danchin, E., Pocheville, A., Rey, O., Pujol, B., and Blanchet, S. (2019). Epigenetically Facilitated Mutational Assimilation: Epigenetics as a Hub within the Inclusive Evolutionary Synthesis. Biol. Rev. 94, 259–282. doi:10.1111/brv.12453
Deal, R. B., and Henikoff, S. (2011). Histone Variants and Modifications in Plant Gene Regulation. Curr.Opin. Plant Biol. 14, 116–122. doi:10.1016/j.pbi.2010.11.005
Dela, R. C., Covarrubias, A. A., and Reyes, J. L. (2019). A Dicistronic Precursor Encoding miR398 and the Legume-specific miR2119 Coregulates CSD1 and ADH1 mRNAs in Response to Water Deficit. Plant, Cell. & Environ. 42, 133–144. doi:10.1111/pce.13209
Dewilde, C., Vanhoudt, H., and Debuck, S. (2000). Plants as Bioreactors for Protein Production: Avoiding the Problem of Transgene Silencing. Plant. Mol. Biol. 43, 347–359. doi:10.1023/a:1006464304199
Dias, D. A., Hill, C. B., Jayasinghe, N. S., Atieno, J., Sutton, T., and Roessner, U. (2015). Quantitative Profiling of Polar Primary Metabolites of Two Chickpea Cultivars with Contrasting Responses to Salinity. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 1000, 1–13. doi:10.1016/j.jchromb.2015.07.002
Ding, D., Zhang, L., Wang, H., Liu, Z., Zhang, Z., and Zheng, Y. (2009). Differential Expression of miRNAs in Response to Salt Stress in Maize Roots. Ann. Bot. 3 (1), 29–38. doi:10.1093/aob/mcn205
Dowen, R. H., Mattia, P., Robert, J. S., Ryan, L., Jill, M. D., Joseph, R. N., et al. (2012). Widespread Dynamic DNA Methylation in Response to Biotic Stress. PNAS 109, 12858–12859. doi:10.1073/pnas.1209329109
East, E. M. (1908). “Inbreeding in Corn,” in Report of the Connecticut Agricultural Experiment Station for the Year 1907 (New Haven: Connecticut Agricultural Experiment Station), 419–428.
Elgin, S. C. R., and Reuter, G. (2013). Position Effect Variegation, Heterochromatin Formation, and Gene Silencing in Drosophila. Cold Spring Herb. Perspect. Biol. 5, 017780. doi:10.1101/cshperspect.a017780
Elkoca, E., Haliloglu, K., Esitken, A., and Ercisli, S. (2007). Hydro- and Osmopriming Improve Chickpea Germination. Acta. Agric. Scand. Sect. b. Soil Plant Sci. 57, 193–200. doi:10.1080/09064710600914087
Elmayan, T., Proux, F., and Vaucheret, H. (2005). Arabidopsis RPA2: A Genetic Link Among Transcriptional Gene Silencing, DNA Repair, and DNA Replication. Curr. Biol. 15, 1919–1925. doi:10.1016/j.cub.2005.09.044
Eskandari, H., and Kazemi, K. (2011). Effect of Seed Priming on Germination Properties and Seedling Establishment of Cowpea (Vigna Sinensis). Not. Sci. Biol. 3, 113–116. doi:10.15835/nsb346338
Eyles, R. P., Williams, P. H., Ohms, S. J., Weiller, G. F., Ogilvie, H. A., Djordjevic, M. A., et al. (2013). microRNA Profiling of Root Tissues and Root Forming Explant Cultures in Medicago Truncatula. Planta 238, 91–105. doi:10.1007/s00425-013-1871-7
Farhoudi, R., Sharifzadeh, F., Poustini, K., Makkizadeh, M. T., and Kochakpor, M. (2007). The Effects of NaCl Priming on Salt Tolerance in Canola (Brassica Napus) Seedlings Grown under Saline Conditions. Seed Sci. Technol. 35, 754–759. doi:10.15258/sst.2007.35.3.23
Feng, S., Cokus, S. J., Zhang, X., Chen, P. Y., Bostick, M., Goll, M. G., et al. (2010). Conservation and Divergence of Methylation Patterning Inplants and Animals. Proc. Natl. Acad. Sci. 107, 8689–8694. doi:10.1073/pnas.1002720107
Fire, A., Xu, S., Montgomery, M. K., Kostas, S. A., Driver, S. E., and Mello, C. C. (1998). Potent and Specific Genetic Interference by Double-Stranded RNA in Caenorhabditis elegans. Nature 391806811. doi:10.1038/35888
Flowers, T. J., Galal, H. K., and Bromham, L. (2010). Evolution of Halophytes: Multiple Origins of Salt Tolerance in Land Plants. Funct. Plant Biol. 37 (7), 604–612. doi:10.1071/FP09269
Fouse, S., D., Nagarajan, R., P., and Costello, J. F. (2010). Genome-scale DNA Methylation Analysis. Epigenomics 2, 105–117. doi:10.2217/epi.09.35
Gallusci, P., Dai, Z., Génard, M., Gauffretau, A., Leblanc-Fournier, N., Richard-Molard, C., et al. (2017). Epigenetics for Plant Improvement. Curr. Knowl. Model. Ave. Trends Plant Sci 22, 610–623. doi:10.1016/j.tplants.2017.04.009
Garg, R., Narayana, C. V., Shankar, R., and Jain, M. (2015). Divergent DNA Methylation Patterns Associated with Gene Expression in Rice Cultivars with Contrasting Drought and Salinity Stress Response. Sci. Rep. 5, 14922. doi:10.1038/srep14922
Garg, R., Shankar, R., Thakkar, B., Kudapa, H., Krishnamurthy, L., Mantri, N., et al. (2016). Transcriptome Analyses Reveal Genotype- and Developmental Stage-specific Molecular Responses to Drought and Salinity Stresses in Chickpea. Sci. Rep. 6, 19228. doi:10.1038/srep19228
Garg, R., Tiwari, R., Tiwari, S., and Goyal, S. (2014). Genomic Survey, Gene Expression Analysis and Structural Modeling Suggest Diverse Roles of DNA Methyltransferases in Legumes. PLoS One 9, e88947. doi:10.1371/journal.pone.0088947
Ghfaffar, A., Hussain, N., Asiam, M., Hussain, K., and Israd, M. (2015). Identification of Superior Hybrid through Exploitation of Hybrid through Exploitation of Hybrid Vigour in Chickpea (Cicer Arietinum L.). Int. J. Adv. Res. Biol.Sci. 2 (2), 180–184.
González, P. H., and Álvarez, M. P. (2013). Epigenetics and its Implications for Psychology. Psicothema 25 (1), 3–12. doi:10.7334/psicothema.2012.327
González-Benito, M., E., Ibáñez, M. Á., Pirredda, M., Mira, S., and Martín, C. (2020). Application of the MSAP Technique to Evaluate Epigenetic Changes in Plant Conservation. Int. J. Mol. Sci. 21, 7459. doi:10.3390/ijms21207459
Graf, P., Dolzblasz, A., Wuerschum, T., Lenhard, M., Pfreundt, U., and Laux, T. (2010). MGOUN1 Encodes an Arabidopsis Type IB DNA Topoisomerase Required in Stem Cell Regulation and to Maintain Developmentally Regulated Gene Silencing. Plant Cell. 22, 716–728. doi:10.1105/tpc.109.068296
Guo, H. S., Xie, Q., Feia, J. F., and Chuad, N. H. (2005). MicroRNA Directs mRNA Cleavage of the Transcription Factor NAC1 to Downregulate Auxin Signals for Arabidopsis Lateral Root Development. Plant Cell. 17 (5), 1376–1386. doi:10.1105/tpc.105.030841
Gutierrez, L., Bussell, J. D., Pacurar, D. I., Schwambach, J., Pacurar, M., and Bellini, C. (2009). Phenotypic Plasticity of Adventitious Rooting in Arabidopsis Is Controlled by Complex Regulation of AUXIN RESPONSE FACTOR Transcripts and microRNA Abundance. Plant Cell. 21, 3119–3313. doi:10.1105/tpc.108.064758
Haag, J. R., and Pikaard, C. S. (2011). Multi Subunit RNA Polymerases IV and V: Purveyors of Non-coding RNA for Plant Gene Silencing. Nat. Rev. Mol. Cell. Biol. 12, 483–492. doi:10.1038/nrm3152
Habu, Y., Kakutani, T., and Paszkowski, J. (2001). Epigenetic Developmental Mechanisms in Plants: Molecules and Targets of Plant Epigenetic Regulation. Curr. Opin. Genet. Dev. 11, 215–220. doi:10.1016/s0959-437x(00)00182-9
Hajyzadeh, M., Turktas, M., Khawar, K., and Unver, T. (2015). miR408 over Expression Causes Increased Drought Tolerance in Chickpea. Gene 555, 186–193. doi:10.1016/j.gene.2014.11.002
Hand, M. L., and Koltunow, A. M. (2014). The Genetic Control of Apomixis: Asexual Seed Formation. Genetics 197, 441–450. doi:10.1534/genetics.114.163105
Harris, R. A., Ting, W., and Cristian, C. (2010). Sequence-based Profiling of DNA Methylation: Comparisons of Methods and Catalogue of Allelic Epigenetic Modifications. NBiotechnol 28, 1097–1105. doi:10.1200/jco.2009.27.7111
He, G., Zhu, X., Elling, A. A., Chen, L., Wang, X., Guo, L., et al. (2010). Global Epigenetic and Transcriptional Trends Among Two Rice Subspecies and Their Reciprocal Hybrids. Plant Cell. 22, 17–33. doi:10.1105/tpc.109.072041
Heitz, E. (1929). Heterochromatin, Chromocentren, Chromomeren.(Vorlaufige Mitteilung.). erichte Dtsch. Bot. Schen. Gesell. Schaft. 47 (4), 274–284. doi:10.1111/j.1438-8677.1929.tb01609.x
Henikoff, S., and Smith, M. M. (2015). Histone Variants and Epigenetics. Cold Spring Harb. Perspect. Biol. 125 (3), 346–364. doi:10.1101/cshperspect.a019364
Herr, A. J. (2005). Pathways through the Small RNA World of Plants. FEBS Lett. 579, 5879–5888. doi:10.1016/j.febslet.2005.08.040
Hofmeister, B. T., Lee, K., Rohr, N. A., Hall, D.,W., and Schmitz, R., J. (2017). Stable Inheritance of DNA Methylation Allows Creation of Epigenotype Maps and the Study of Epiallele Inheritance Patterns in the Absence of Genetic Variation. Genome Biol. 18, 155. doi:10.1186/s13059-017-128810.1186/s13059-017-1288-x
Hong, Y., Zhang, H., Huang, L., Li, D., and Song, F. (2016). Overexpression of a Stress-Responsive NAC Transcription Factor Gene ONAC022 Improves Drought and Salt Tolerance in Rice. Front. Plant Sci. 7, 4. doi:10.3389/fpls.2016.00004
Hu, T., Jin, Y., Li, H., Amombo, E., and Fu, J. (2016). Stress Memory Induced Transcriptional and Metabolic Changes of Perennial Ryegrass (Lolium Perenne) in Response to Salt Stress. Physiol. Plant. 156, 54–69. doi:10.1111/ppl.12342
Hume, S., Bo, D., Stacey, A. S., Suhua, F., Maria, B., Matteo, P., et al. (2013). Plants Regenerated from Tissue Culture Contain Stable Epigenome Changes in Rice. ELife 2, e00354. doi:10.7554/eLife.00354
Hung, Y. H., Liu, F., Zhang, X. Q., Xiao, W., and Hsieh, T., F. (2018). Sexual and Non-sexual Reproduction: Inheritance and Stability of Epigenetic Variations and Consequences for Breeding Application. Adv. Bot. Res. 88, 117–163. doi:10.1016/bs.abr.2018.09.002
Hyun, K., Jeon, J., and Park, K. (2017). Writing, Erasing and Reading Histone Lysine Methylations. Exp. Mol. Med. 49, e324. doi:10.1038/emm.2017.11
Ikeda, Y., Kinoshita, Y., Susaki, D., Ikeda, Y., Iwano, M., Takayama, S., et al. (2011). HMG Domain Containing SSRP1 Is Required for DNA Demethylation and Genomic Imprinting in Arabidopsis. Dev. Cell. 21, 589–596. doi:10.1016/j.devcel.2011.08.013
Ingelbrecht, I., Vanhoudt, H., Vanmontagu, M., and Depicker, A. (1994). Post-Transcriptional Silencing of Reporter Transgenes in Tobacco Correlates with DNA Methylation. Proc. Natl. Acad. Sci. USA. 91, 10502–10506. doi:10.1073/pnas.91.22.10502
Ingouff, M., and Berger, F. (2010). Histone3 Variants in Plants. Chromosoma 119, 27–33. doi:10.1007/s00412-009-0237-1
Jaffe, A. E., Feinberg, A. P., Irizarry, R. A., and Leek, J. T. (2017). Significance Analysis and Statistical Dissection of Variably Methylated Regions. Biostatistics 13, 166–178. doi:10.1093/biostatistics/kxr013
Jaganathan, D., Thudi, M., Kale, S., Azam, S., Roorkiwal, M., Gaur, P. M., et al. (2015). Genotyping-by-sequencing Based Intra-specific Genetic Map Refines a ‘‘QTL-Hotspot” Region for Drought Tolerance in Chickpea. Mol. Genet. Genomics 290, 559–571. doi:10.1007/s00438-014-0932-3
Janmohammadi, M., Zolla, L., and Rinalducci, S. (2015). Low Temperature Tolerance in Plants: Changes at the Protein Level. Phyto Chem. 117, 76–89. doi:10.1016/j.phytochem.2015.06.003
Jerzmanowski, A. (2007). SWI/SNF Chromatin Remodeling and Linker Histones in Plants. Biochim. Biophys. Acta 1769, 330–345. doi:10.1016/j.bbaexp.2006.12.003
Jha, U. C., Chaturvedi, S. K., Bohra, A., Basu, P. S., Khan, M. S., and Barh, D. (2014). Abiotic Stresses, Constraints and Improvement Strategies in Chickpea. Plant Breed. 133 (2), 163–178. doi:10.1111/pbr.12150
Jisha, K., C., and Puthur, J.,T. (2014). Halopriming of Seeds Imparts Tolerance to NaCl and PEG Induced Stress in Vigna Radiata (L.) Wilczek Varieties. Physiol. Mol. Biol. Plants. 303, 312. doi:10.1007/s12298-014-0234-6
Johannes, F., and Colomé-Tatché, M. (2011). Quantitative Epigenetics through Epigenomic Perturbation of Isogenic Lines. Genetics 188, 215–227. doi:10.1534/genetics.111.127118
John, M. C., and Amasino, R. M. (1989). Extensive Changes in DNA Met Hylation Patterns Accompany Activation of a Silent T-DNA Ipt Gene in Agrobacterium Tumefaciens-transformed Plant Cells. Mol. Cell. Biol. 9, 4298–4303. doi:10.1128/mcb.910.4298-4303.198910.1128/mcb.9.10.4298
Jones, D. F. (1917). Dominance of Linked Factors as a Means of Accounting for Heterosis. Proc. Natl. Acad. Sci. U. S. A. 3, 310–312. doi:10.1073/pnas.3.4.310
Jones, P. A., and Baylin, S. B. (2007). The Epigenomics of Cancer. Cell. 128, 683–692. doi:10.1016/j.cell.2007.01.029
Joseph, C., and David, L. (2006). Epigenetic Gene Regulation in the Bacterial World. Am. Soc. Microbiol. Rev. 70, 830–856. doi:10.1128/MMBR.00016-06
Jurkowska, R. Z., and Jeltsch, A. (2016). Enzymology of Mammalian DNA Methyltransferases. Adv. Exp. Med. Biol. 945, 87–122. doi:10.1007/978-3-319-43624-1-510.1007/978-3-319-43624-1_5
Jurkowski, T. P., and Jeltsch, A. (2011). On the Evolutionary Origin of Eukaryotic DNA Methyltransferases and Dnmt 2. PLoS One 6, e28104. doi:10.1371/journal.pone.0028104
Kaeppler, S. M., Kaeppler, H. F., and Rhee, Y. (2000). Epigenetic Aspects of Soma Clonal Variation in Plants. Plant Mol. Biol. 43, 179–188. doi:10.1023/A:1006423110134
Kakoulidou, I., Avramidou, E. V., Baránek, M., Brunel-Muguet, S., Farrona, S., Johannes, F., et al. (2021). Epigenetics for Crop Improvement in Times of Global Change. Biol. (Basel) 210 (8), 766. doi:10.3390/biology10080766
Kakutani, T. (2002). Epi-alleles in Plants: Inheritance of Epigenetic Information over Generations. Plant Cell. Physiol. 43, 1106. doi:10.1093/pcp/pcf131
Kapazoglou, A., Ganopoulos, I., Tani, E., and Tsaftaris, A. (2018). “Chapter Nine—Epigenetics, Epigenomics and Crop Improvement,” in Advances in Botanical Research. Editor M. Kuntz (New York, NY, USA: Academic Press), 86, 287–324. doi:10.1016/bs.abr.2017.11.007
Kapoor, A., Agarwal, M., Andreucci, A., Zheng, X., Gong, Z., Hasegawa, P. M., et al. (2005). Mutations in a Conserved Replication Protein Suppress Transcriptional Gene Silencing in a DNA-methylation-independent Manner in Arabidopsis. Curr. Biol. 15, 1912–1918. doi:10.1016/j.cub.2005.09.013
Kaur, H., Shukla, R. K., Yadav, G., Chattopadhyay, D., and Majee, M. (2008). Two Divergent Genes Encoding L-Myo-Inositol 1-phosphate Synthase1 (CaMIPS1) and 2 (CaMIPS2) Are Differentially Expressed in Chickpea. Plant Cell. Environ. 31 (11), 1701–1716. doi:10.1111/j.1365-3040.2008.0187710.1111/j.1365-3040.2008.01877.x
Kaur, N. (2000). Effect of Osmo- and Hydropriming of Chickpea Seeds on Seedling Growth and Carbohydrate Metabolism under Water Deficit Stress. Plant Growth Regul. 37, 17–22. doi:10.1023/A:1020310008830
Kaur, S., Gupta, A., K., and Kaur, N. (2002). Effect of Osmo- and Hydropriming of Chickpea Seeds on Seedling Growth and Carbohydrate Metabolism under Water Deficit Stress. Plant Growth Regul. 37, 17–22. doi:10.1023/a:1020310008830
Kelkar, A., Thakur, V., Ramaswamy, R., and Deobagkar, D. (2009). Characterisation of Inactivation Domains and Evolutionary Strata in Human X Chromosome through Markov Segmentation. PloS One 4, e7885. doi:10.1371/journal.pone.0007885
Khan, N., Bano, A., Rahman, M. A., Guo, J., Kang, Z., and Babar, M. A. (2019). Comparative Physiological and Metabolic Analysis Reveals a Complex Mechanism Involved in Drought Tolerance in Chickpea (Cicer Arietinum L.) Induced by PGPR and PGRs. Sci. Rep. 9, 2097. doi:10.1038/s41598-019-38702-8
Khandal, H., Parween, S., Roy, R., Meena, M. K., and Chattopadhyay, D. (2017). MicroRNA Profiling Provides Insights into Post-transcriptional Regulation of Gene Expression in Chickpea Root Apex under Salinity and Water Deficiency. Sci. Rep. 7, 4632. doi:10.1038/s41598-017-04906-z
Kilian, J., Whitehead, D., Horak, J., Wanke, D., Weinl, S., Batistic, O., et al. (2007). The at Gen Express Global Stress Expression Data Set: Protocols, Evaluation and Model Data Analysis of UV-B Light, Drought and Cold Stress Responses. Plant J. 50, 347–363. doi:10.1111/j.1365-313X.2007.03052.x
Kinoshita, T., and Seki, M. (2014). Epigenetic Memory for Stress Response and Adaptation in Plants. Plant Cell. Physiol. 55, 1859–1863. doi:10.1093/pcp/pcu125
Klumpp, A., Ansel, W., Fomin, A., Schnirring, S., and Pickl, C. (2004). Influence of Climatic Conditions on the Mutations in Pollen Mother Cells of Tradescantia Clone 4430 and Implications for the Trad-MCN Bioassay Protocol. Hereditas 141, 142–148. doi:10.1111/j.1601-5223.2004.01806.x
Kobayashi, I. A., Nobusato, N., Kobayashi, T., and Uchiyama, I. (1999). Shaping the Genome Restriction-Modification Systems as Mobile Genetic Elements. Curr. Opin. Genet. Dev. 9, 649–656. doi:10.1016/s0959-437x(99)00026-x
Kobayashi, I. (2001). Behavior of Restriction-Modification Systems as Selfish Mobile Elements and Their Impact on Genome Evolution. Nucleic Acids Res. 29, 3742–3756. doi:10.1093/nar/29.18.3742
Kotula, L., Khan, H. A., Quealy, J., Turner, N. C., Vadez, V., Siddique, K. H., et al. (2015). Salt Sensitivity in Chickpea (Cicer Arietinum L.): Ions in Reproductive Tissues and Yield Components in Contrasting Genotypes. Plant Cell. Environ. 38 (8), 1565–1577. doi:10.1111/pce.12506
Ku, C. S., Naidoo, N., Wu, M., and Soong, R. (2011). Studying the Epigenome Using Next Generation Sequencing. J.Med. Genet. 48 (11), 721–730. Epub 2011 Aug 8. PMID: 21825079. doi:10.1136/jmedgenet-2011-100242
Kudapa, H., Azam, S., Sharpe, A. G., Taran, B., Li, R., Deonovic, B., et al. (2014). Comprehensive Transcriptome Assembly of Chickpea (Cicer Arietinum L.) Using Sanger and Next Generation Sequencing Platforms: Development and Applications. PLoS One 9 (1), e86039. doi:10.1371/journal.pone.0086039
Kumar, S., and Singh, A. (2016). Epigeneticregulation of Abiotic Stress Tolerance in Plants. Adv. Plants Agric. Res. 5, e00179. doi:10.15406/apar.2016.05.00179
Kumar, S. V., and Wigge, P. A. (2010). H2A.Z-containing Nucleosomes Mediate the Thermo Sensory Response in Arabidopsis. Cell. 140, 136–147. doi:10.1016/j.cell.2009.11.006
Labra, M., Ghiani, A., Citterio, S., Sgorbati, S., Sala, F., Vannini, C., et al. (2002). Analysis of Cytosine Methylation Pattern in Response to Water Deficit in Pea Root Tips. Plant Biol. 4 (06), 694–699. doi:10.1055/s-2002-37398
Lämke, J., and Bäurle, I. (2017). Epigenetic and Chromatin-Based Mechanisms in Environmental Stress Adaptation and Stress Memory in Plants. Genome Biol. 18, 124. doi:10.1186/s13059-017-1263-6
Lang, Z., Wang, Y., Tang, K., Tang, D., Datsenka, T., Cheng, J., et al. (2017). Critical Roles of DNA Demethylation in the Activation of Ripening-Induced Genes and Inhibition of Ripening-Repressed Genes in Tomato Fruit. Proc. Natl. Acad. Sci. USA. 114, E4511. doi:10.1073/pnas.1705233114
Lauria, M., and Rossi, V. (2011). Epigenetic Control of Gene Regulation in Plants. Biochim. Biophys. Acta 1809, 369–378. doi:10.1016/j.bbagrm.2011.03.002
Law, J. A., Vashisht, A. A., Wohlschlegel, J. A., and Jacobsen, S. E. (2011). SHH1, a Homeodomain Protein Required for DNA Methylation, as Well as RDR2, RDM4, and Chromatin Remodeling Factors, Associate with RNA Polymerase IV. PLoS Genet. 7, e1002195. doi:10.1371/journal.pgen.10021910.1371/journal.pgen.1002195
Li, H., Yuan, J., Wu, M., Han, Z., Li, L., Jiang, H., et al. (2018a). Transcriptome and DNA Methylomereveal Insights into Yield Heterosis in the Curds of Broccoli (Brassica oleracea L Var. Italic). BMC Plant. Biol. 18, 168. doi:10.1186/s12870-018-1384-4
Li, S., W., Zeng, X., Y., Leng, Y., Feng, L., and Kang, X., H. (2018b). Indole-3-butyric Acid Mediates Antioxidative Defense Systems to Promote Adventitious Rooting in Mung Bean Seedlings under Cadmium and Drought Stresses. Ecotoxicol. Environ. Saf. 161, 332–341. doi:10.1016/j.ecoenv.2018.06.003
Liu, B., and Wendel, J. (2003). Epigenetic Phenomena and the Evolution of Plant Allopolyploids. Mol. Phylogenet. Evol. 29, 365–379. doi:10.1016/s1055-7903(03)00213-6
Long, Y., Xia, W., Li, R., Wang, J., Shao, M., Feng, J., et al. (2011). Epigenetic QTL Mapping in Brassica Napus. Genetics 189, 1093–1102. doi:10.1534/genetics.111.131615
Lu, Y. B., Yang, L. T., Qi, Y. P., Li, Y., Li, Z., Chen, Y. B., et al. (2014). Identification of Boron-Deficiency-Responsive microRNAs in Citrus Sinensis Roots by Illumina Sequencing. BMC Plant Biol. 14, 123. doi:10.1186/1471-2229-14-123
Luo, J., and Hall, B. D. (2007). A Multi Step Process Gave Rise to RNA Polymerase IV of Land Plants. J. Mol. Evol. 64, 101–112. doi:10.1007/s00239-006-0093-z
Madlung, A., and Comai, L. (2004). The Effect of Stress on Genome Regulation and Structure. Ann. Bot. 94, 481–495. doi:10.1093/aob/mch172
Mallory, A. C., Bartel, D. P., and Bartel, B. (2005). MicroRNA-directed Regulation of Arabidopsis AUXIN RESPONSE FACTOR17 Is Essential for Proper Development and Modulates Expression of Early Auxin Response Genes. Plant Cell. 17 (5), 1360–1375. doi:10.1105/tpc.105.031716
Mantri, N. L., Ford, R., Coram, T. E., and Pang, E. C. (2007). Transcriptional Profiling of Chickpea Genes Differentially Regulated in Response to High-Salinity, Cold and Drought. BMC Genomics 8, 303. doi:10.1186/1471-2164-8-303
Marcos, F. C. C., Silveira, N. M., Marchiori, P. E. R., Machado, E. C., Souza, G. M., Landell, M. G. A., et al. (2018a). Drought Tolerance of Sugarcane Propagules Is Improved when Origin Material Faces Water Deficit. PLoS One 13 (12), e0206716. doi:10.1371/journal.pone.0206716
Marcos, F. C. C., Silveira, N. M., Mokochinski, J. B., Sawaya, A. C. H. F., Marchiori, P. E. R., Machado, E. C., et al. (2018b). Drought Tolerance of Sugarcane Is Improved by Previous Exposure to Water Deficit. J. Plant Physiol. 223, 9–18. doi:10.1016/j.jplph.2018.02.001
Mashaki, K. M., Garg, V., Ghomi, A. A. N., Kudapa, H., Chitikineni, A., Nezhad, K. Z., et al. (2018). RNA-seq Analysis Revealed Genes Associated with Drought Stress Response in Kabuli Chickpea (Cicer Arietinum L.). PLoS ONE 13, e0199774. doi:10.1371/journal.pone.0199774
Matzke, M. A., and Matzke, A. J. M. (2004). Planting the Seeds of a New Paradigm. PloS Biol. 2, 582–586. doi:10.1371/journal.pbio.0020133
Matzke, M. A., Mette, M. F., and Matzke, A. J. (2000). Transgene Silencing by the Host Genome Defense: Implications for the Evolution of Epigenetic Control Mechanisms in Plants and Vertebrates. Plant Mol. Biol. 43, 401–415. doi:10.1023/a:1006484806925
Matzke, M. A., Primig, M., Trnovsky, J., and Matzke, A. J. M. (1989). Reversible Methylation and Inactivation of Marker Genes in Sequentially Transformed Tobacco Plants. EMBO J. 8, 643–649. PMCID: PMC40085. doi:10.1002/j.1460-2075.1989.tb03421.x
Maunakea, A., K., Nagarajan, R., P., and Bilenky, M. (2010). Conserved Role of Intragenic DNA Methylation in Regulating Alternative Promoters. Nature 466, 253–257. doi:10.1038/nature09165
McClintock, B. (1950). The Origin and Behavior of Mutable Loci in Maize. Proc. Natl. Acad. Sci. 36, 344–355. doi:10.1073/pnas.36.6.344
Meyer, P., and Saedler, H. (1996). Homology-dependent Gene Silencing in Plants. Annu. Rev. Plant Physiol. 47, 23–48. doi:10.1146/annurev.arplant.47.1.23z
Mohammadi, P., Bahramnejada, B., Badakhshana, H., and Kanouni, H. (2015). DNA Methylation Changes in fusarium Wilt Resistant and Sensitive Chickpea Genotypes (Cicer Arietinum L.). Physiol. Mol. Plant Pathol. 91, 72–80. doi:10.1016/j.pmpp.2015.06.001
Molinier, J., Ries, G., Zipfel, C., and Hohn, B. (2006). Transgeneration Memory of Stress in Plants. Nature 442, 1046–1049. doi:10.1038/nature05022
Mouradi, M., Bouizgaren, A., Farissi, M., Latrach, L., Qaddoury, A., and Ghoulam, C. (2016). Seed Osmopriming Improves Plant Growth, Nodulation, Chlorophyll Fluorescence and Nutrient Uptake in Alfalfa (Medicago Sativa L.)—Rhizobia Symbiosis under Drought Stress. Sci. Hortic. 213, 232–242. doi:10.1016/j.scienta.2016.11.002
Nakayama, Y., and Kobayashi, I. (1998). Restriction-modification Gene Complexes as Selfish Gene Entities: Roles of a Regulatory System in Their Establishment, Maintenance, and Apoptotic Mutual Exclusion. Proc. Natl. Acad. Sci. U. S. A. 95, 6442–6447. doi:10.1073/pnas.95.11.6442
Napoli, C., Lemieux, C., and Jorgensen, R. (1990). Introduction of a Chimeric Chalcone Synthase Gene into Petunia Results in Reversible Co-suppression of Homologous Gene in Trans. Plant Cell. 2, 279–289. doi:10.1105/tpc.2.4.279
Naya, L., Paul, S., Valdes Lopez, O., Mendoza Soto, A. B., Soto, A. B., Nova Franco, B., et al. (2014). Regulation of Copper Homeostasis and Biotic Interactions by microRNA 398b in Common Bean. PLoS ONE 9, e84416. doi:10.1371/journal.pone.0084416
Neuhuber, B., Himes, B. T., Shumsky, J. S., Gallo, G., and Fischer, I. (2005). Axon Growth and Recovery of Function Supported by Human Bone Marrow Stromal Cells in the Injured Spinal Cord Exhibit Donor Variations. Brain Res. 1035, 73–85. doi:10.1016/j.brainres.2004.11.055
Ni, Z., Kim, E. D., Ha, M., Lackey, E., Liu, J., Zhang, Y., et al. (2009). Altered Circadian Rhythms Regulate Growth Vigour in Hybrids And Allopolyploidss. Nature 457, 327–331. doi:10.1038/nature07523
Ong-Abdullah, M., Ordway, J. M., Jiang, N., Ooi, S. E., Kok, S. Y., Sarpan, N., et al. (2015). Loss of Karma Transposon Methylation Underlies the Mantled Soma Clonal Variant of Oil Palm. Nature 525, 533–537. doi:10.1038/nature15365
Pandey, G., Sharma, N., Sahu, P. P., and Prasad, M. (2016). Chromatin-Based Epigenetic Regulation of Plant Abiotic Stress Response. Curr. Genomics 17 (6), 490–498. doi:10.2174/1389202917666160520103914
Papikian, A., Liu, W., Gallego-Bartolomé, J., and Jacobsen, S. E. (2019). Site-specific Manipulation of Arabidopsis Loci Using CRISPR-Cas9 Sun Tag Systems. Nat. Commun. 10, 729. doi:10.1038/s41467-019-08736-7
Park, C. L., Cohen, L. H., and Murch, R. L. (1996). Assessment and Prediction of Stress-Related Growth. J. Pers. 64, 71–105. doi:10.1111/j.1467-6494.1996.tb0081510.1111/j.1467-6494.1996.tb00815.x
Pedersen, D. S., and Grasser, K. D. (2010). The Role of Chromosomal HMGB Proteins in Plants. Biochim. Biophys. Acta 1799, 171–174. doi:10.1016/j.bbagrm.2009.11.004
Pikaard, C. S., and Mittelsten, S. O. (2014). Epigenetic Regulation in Plants. Cold Spring Harb. Perspect. Biol. 6, a019315. doi:10.1101/cshperspect.a019315
Pushpavalli, R., Krishnamurthy, L., Thudi, M., Gaur, P. M., Rao, M. V., Siddique, K. M. H., et al. (2015). Two Key Genomic Regions Harbour QTLs for Salinity Tolerance in ICCV 2 X JG 11 Derived Chickpea (Cicer Arietinum L.) Recombinant Inbred Lines. BMC Plant Biol. 15, 124. doi:10.1186/s12870-015-0491-8
Rajkumar, M. S., Gupta, K., and Khemka, N. K. (2020). DNA Methylation Reprogramming during Seed Development and its Functional Relevance in Seed Size/weight Determination in Chickpea. Commun. Biol. 3, 340. doi:10.1038/s42003-020-1059-1
Rajpathak, S. N., and Deobagkar, D. D. (2017). Micro RNAs and DNA Methylation Are Regulatory Players in Human Cells with Altered X Chromosome to Autosome Balance. Sci. Rep. 7, 43235. doi:10.1038/srep43235
Raju, S. K. K., Shao, M. R., Sanchez, R., Xu, Y. Z., Sandhu, A., Graef, G., et al. (2018). An Epigenetic Breeding System in Soybean for Increased Yield and Stability. Plant Biotechnol. J. 16, 1836–1847. doi:10.1111/pbi.12919
Rakei, A., Maali, A. R., Zeinali, H., and Ranjbar, M. (2015). DNA Methylation and Physio-Biochemical Analysis of Chickpea in Response to Cold Stress. Protoplasma 253, 61–76. doi:10.1007/s00709-015-0788-3
Rasheed, A., Hao, Y., Xia, X., Khan, A., Xu, Y., Varshney, R. K., et al. (2017). Crop Breeding Chips and Genotyping Platforms: Progress, Challenges, and Perspectives. Mol. Plant 10, 1047–1064. doi:10.1016/j.molp.2017.06.008
Ream, T. S., Haag, J. R., Wierzbicki, A. T., Nicora, C. D., Norbeck, A. D., Zhu, J. K., et al. (2009). Sub Unit Compositions of the RNA-Silencing Enzymes Pol IV and Pol V Reveal Their Origins as Specialized Forms of RNA Polymerase II. Mol. Cell. 33, 192–203. doi:10.1016/j.molcel.2008.12.015
Rehman, H., Iqbal, H., Basra, S. M. A., Afzal, I., Farooq, M., Wakeel, A., et al. (2015). Seed Priming Improves Early Seedling Vigor, Growth and Productivity of Spring Maize. J. Integr. Agric. 14, 1745–1754. doi:10.1016/s2095-3119(14)61000-5
Richards, E. J. (2006). Inherited Epigenetic Variation Revisiting Soft Inheritance. Nat. Rev. Genet. 7, 395–401. doi:10.1038/nrg1834
Roorkiwal, M., vonWettberg, E. J., Upadhyaya, H. D., Warschefsky, E., Rathore, A., and Varshney, R. K. (2014). Exploring Germplasm Diversity to Understand the Domestication Process in Cicer Spp. Using Snp and Dart Markers. PLoS One 9 (7), e102016. doi:10.1371/journal.pone.0102016
Ruiz, F. V., and Voinnet, O. (2009). Roles of Plant Small RNAs in Biotic Stress Responses. Annu. Rev. Plant. Biol. 60, 485–510. doi:10.1146/annurev.arplant.043008.092111
Roux, F., Colome Tatche, M., Edelist, C., Warenaar, R., Guerche, P., Hospital, F., et al. (2011). Genome-Wide Epigenetic Perturbation Jumpstarts Patterns of Heritable Variation Found in Nature. Genetics 188, 1015–1017.
Sahu, P. P., Pandey, G., Sharma, N., Puranik, S., Muthamilarasan, M., and Prasad, M. (2013). Epigenetic Mechanism of Plant Stress Responses and Adaptation. Plant Cell. Rep. 32 (8), 1151–1159. doi:10.1007/s00299-013-1462-x
Sang, Y., Silvaortega, C. O., Wu, S., Yamaguchi, N., Wu, M. F., Pfluger, J., et al. (2012). Mutations in Two Non-canonical Arabdopsis SWI2/SNF2 Chromatin Remodelling ATPases Cause Embryogenesis and Stem Cell Maintenance Defects. Plant J. 72, 1000–1014. doi:10.1111/tpj.12009
Sani, E., Herzyk, P., Perrella, G., Colot, V., and Amtmann, A. (2013). Hyperosmotic Priming of Arabidopsis Seedlings Establishes a Long-Term Somatic Memory Accompanied by Specific Changes of the Epigenome. Genome Biol. 14, R59. doi:10.1186/gb-2013-14-6-r59
Sen, S., Chakraborty, J., Ghosh, P., Basu, D., and Das, S. (2017). Chickpea WRKY70 Regulates the Expression of a Homeodomain-Leucine Zipper (HD-Zip) I Transcription Factor CaHDZ12, Which Confers Abiotic Stress Tolerance in Transgenic Tobacco and Chickpea. Plant Cell. Physiol. 58, 1934–1952. doi:10.1093/pcp/pcx126
Shabalina, S. A., and Koonin, E. V. (2008). Origins and Evolution of Eukaryotic RNA Interference. Trends Ecol. Evol. 23, 578–587. doi:10.1016/j.tree.2008.06.005
Sharma, A., Basu, U., Malik, N., Daware, A., Thakro, V., Narnoliya, L., et al. (2019). Genome-wide Cis-Regulatory Signatures for Modulation of Agronomic Traits as Exemplified by Drought Yield Index (DYI) in Chickpea. Funct. Integr. Genomics 19, 973–992. doi:10.1007/s10142-019-00691-2
Shen, Y., Sun, S., Hua, S., Shen, E., Ye, C., Cai, D., et al. (2017). Analysis of Transcriptional and Epigenetic Changes in Hybrid Vigor of Allopolyploid Brassica Napus Uncovers Key Roles for Small RNAs. Plant. J. 91, 874–893. doi:10.1111/tpj.13605
Shui, X. R., Chen, Z. W., and Li, J. X. (2013). MicroRNA Prediction and its Function in Regulating Drought-Related Genes in Cowpea. Plant Sci. 210, 25–35. doi:10.1016/j.plantsci.2013.05.002
Shull, G. H. (1908). The Composition of a Field of Maize. Am. Breed. Assoc. Rep. 4, 296–301. doi:10.1093/jhered/os-4.1.296
Singh, A., Singh, S., Panigrahi, K., C., Reski, R., and Sarkar, A., K. (2014). Balanced Activity of microRNA166/165 and its Target Transcripts from the Class III Homeodomain-Leucine Zipper Family Regulates Root Growth in Arabidopsis thaliana. Plant Cell. Rep. 33 (6), 945–953. doi:10.1007/s00299-014-157310.1007/s00299-014-1573-z
Slaughter, A., Daniel, X., Flors, V., Luna, E., Hohn, B., and MauchMani, B. (2012). Descendants of Primed Arabidopsis Plants Exhibit Resistance to Biotic Stress. Plant Physiol. 158, 835. doi:10.1104/pp.111.191593
Soppe, W. J., Jacobsen, S. E., Alonso-Blanco, C., Jackson, J. P., Kakutani, T., Koornneef, M., et al. (2000). The Late Flowering Phenotype of Fwa Mutants Is Caused by Gain-Of-Function Epigenetic Alleles of a Homeodomain Gene. Mol. Cell. 6, 791–802. doi:10.1016/s1097-2765(05)00090-0
Sosa, V. G., Palomar, M., Covarrubias, A. A., and Reyes, J. L. (2017). The Legume miR1514a Modulates a NAC Transcription Factor Transcript to Trigger phasiRNA Formation in Response to Drought. J. Exp. Bot. 68 (8), 2013–2026. PMID: 28338719; PMCID: PMC5429018. doi:10.1093/jxb/erw380
Spillane, C., Steimer, A., and Grossniklaus, U. (2001). Apomixis in Agriculture: the Quest for Clonal Seeds. Sex. Plant Reprod. 14, 179–187. doi:10.1007/s00497-001-0117-1
Springer, N., and Schmitz, R. (2017). Exploiting Induced and Natural Epigenetic Variation for Crop Improvement. Nat. Rev. Genet. 18, 563–575. doi:10.1038/nrg.2017.45
Su, Z., Bernardo, A., Tian, B., Chen, H., Wang, S., Ma, H., et al. (2019). A Deletion Mutation in TaHRC Confers Fhb1 Resistance to Fusarium Head Blight in Wheat. Nat. Genet. 51, 1099–1105. doi:10.1038/s41588-019-0425-8
Tang, X. M., Tao, X., Wang, Y., Ma, D. W., Li, D., Yang, H., et al. (2014). Genomics. Analysis of DNA Methylation of Perennial Ryegrass under Drought Using the Methylation-Sensitive Amplification Polymorphism (MSAP) Technique. Mol. Genet. 289, 1075–1084. doi:10.1007/s00438-014-0869-6
Teixeira, F. K., Heredia, F., Sarazin, A., Roudier, F., Boccara, M., Ciaudo, C., et al. (2009). A Role for RNAi in the Selective Correction of DNA Methylation Defects. Science 323, 1600–1604. doi:10.1126/science.1165313
Thudi, M., Chitikineni, A., Liu, X., He, W., Roorkiwal, M., Yang, W., et al. (2016). Recent Breeding Programs Enhanced Genetic Diversity in Both Desi and Kabuli Varieties of Chickpea (Cicer Arietinum L.). Sci. Rep. 6, 38636. doi:10.1038/srep38636
Trindade, I., Capitao, C., Dalmay, T., Fevereiro, M., P., and Santos, D., M. (2010). miR398 and miR408 Are Up-Regulated in Response to Water Deficit in Medicago Truncatula. Planta 231 (3), 705–716. doi:10.1007/s00425-009-1078-0.38636
Tsaftaris, A. S., and Polidoros, A. N. (2000). DNA Methylation and Plant Breeding. Plant Breed. Rev. 18, 87–176. doi:10.1002/9780470650158.ch3
Tucker, S. L., Reece, J., Ream, T. S., and Pikaard, C. S. (2010). Evolutionary History of Plant Multi Subunit RNA Polymerases IV and V: Subunit Origins via Genome-wide and Segmental Gene Duplications, Retro Transposition, and Lineage-specific Sub Functionalization. Cold Spring Harb. Symp. Quant. Biol. 75, 285–297. doi:10.1101/sqb.2010.75.037
Tuteja, N. (2007). Mechanisms of High Salinity Tolerance in Plants. Methods Enzymol. 428, 419–438. doi:10.1016/S0076-6879(07)28024-3
Umezawa, T., Shimizu, K., Kato, M., and Ueda, T. (2000). Enhancement of Salt Tolerance in Soybean with NaCl Pretreatment. Physilogia Plant 110, 59–63. doi:10.1034/j.1399-3054.2000.110108.x
Van der Krol, A. R., Mur, L. A., Beld, M., Mol, J. N., and Stuitje, A. R. (1990). Flavonoid Genes in Petunia: Addition of a Limited Number of Gene Copies May Lead to a Suppression of Gene Expression. Plant Cell. 2, 291–299. doi:10.1105/tpc.2.4.291
Van Dijk, K., Ding, Y., Malkaram, S., Riethoven, J. J., Liu, R., Yang, J., et al. (2010). Dynamic Changes in Genome-wide Histone H3lysine 4 Methylation Patterns in Response to Dehydration Stress in Arabidopsis thaliana. BMC Plant Biol. 10, 238. doi:10.1186/1471-2229-10-238
Varshney, R.,K., Thudi, M., Nayak, S., N., Gaur, P., M., Kashiwagi, J., Krishnamurthy, L., et al. (2014). Genetic Dissection of Drought Tolerance in Chickpea (Cicer Arietinum L.). Theor. Appl. Genet. 127, 445–462. doi:10.1007/s00122-013-2230-6
Varshney, R. K., Hiremath, P., J., Lekha, P., Kashiwagi, J., Balaji, J., Deokar, A. A., et al. (2009). A Comprehensive Resource of Drought- and Salinity- Responsive ESTs for Gene Discovery and Marker Development in Chickpea (Cicer Arietinum L.). BMC Genomics 10, 523. doi:10.1186/1471-2164-10-523
Varshney, R. K., Mohan, S. M., Gaur, P. M., Gangarao, N. V., Pandey, M. K., Bohra, A., et al. (2013). Achievements and Prospects of Genomics-Assisted Breeding in Three Legume Crops of the Semi-arid Tropics. Biotechnol. Adv. 31 (8), 1120–1134. doi:10.1016/j.biotechadv.2013.01.001
Varshney, R. K., Pandey, M., Bohra, K. A., Singh, V. K., Thudi, M., et al. (2019b). Toward the Sequence-Based Breeding in Legumes in the Post-genome Sequencing Era. Theor. Appl. Genet. 132, 797–816. doi:10.1007/s00122-018-3252-x
Varshney, R. K., Roorkiwal, M., Sun, S., Bajaj, P., Chitikineni, A., Thudi, M., et al. (2021). A Chickpea Genetic Variation Map Based on the Sequencing of 3,366 Genomes. Nature 599, 622–627. Available at: www.nature.com/articles/s41586-021-04066-1. doi:10.1038/s41586-021-04066-1
Varshney, R. K., Thud, M., Roorkiwal, M., He, W., Upadhyaya, H. D., Yang, W., et al. (2019a). Resequencing of 429 Chickpea Accessions from 45 Countries Provides Insights into Genome Diversity, Domestication and Agronomic Traits. Nat. Genet. 51, 857–864. doi:10.1038/s41588-019-0401-3
Vaucheret, H. (2008). Plant Argonautes. Trends Plant Sci. 13, 350–358. doi:10.1016/j.tplants.2008.04.007
Vazquez, F., Legrand, S., and Windels, D. (2010). The Biosynthetic Pathways and Biological Scopes of Plant Small RNAs. Trends Plant Sci. 15, 337–345. doi:10.1016/j.tplants.2010.04.001
Velten, J., Cakir, C., Youn, E., Chen, J., and Cazzonelli, I. C. (2012). Transgene Silencing and Transgene-Derived siRNA Production in Tobacco Plants Homozygous for an Introduced AtMYB90 Construct. PLoS One 7, e30141. doi:10.1371/journal.pone.0030141
Verhoeven, K. J. F., Jansen, J. J., Van Dijk, P. J., and Biere, A. (2010). Stress-induced DNA Methylation Changes and Their Heritability in Asexual Dandelions. New Phytol. 185, 1108–1118. doi:10.1111/j.1469-8137.2009.03121.x
Vijay, G., Zinta, G., kumar, S., and Jaswal, V. (2020). Quantitative Epigenetics: A New Avenue for Crop Improvement. Epigenomes 4 (4), 25. doi:10.3390/epigenomes404002
Walter, J., Nagy, L., Hein, R., Rascher, U., Beierkuhnlein, C., Willner, E., et al. (2011). Do plants Remember Drought? Hints towards a Drought-Memory in Grasses. Environ. Exp. Bot. 71, 34–40. doi:10.1016/j.envexpbot.2010.10.020
Wang, X., Elling, A. A., Li, X., Li, N., Peng, Z., He, G., et al. (2009). Genome-Wide and Organ-specific Landscapes of Epigenetic Modifications and Their Relationships to mRNA and Small RNA Transcriptomes in Maize. Plant Cell. 21, 1053–1069. doi:10.1105/tpc.109.065714
Wang, Z., Wang, Z., Wang, J., Sui, Y., Zhang, J., Liao, D., et al. (2012). A Quantitative Genetic and Epigenetic Model of Complex Traits. BMC Bioinforma. 13, 274. doi:10.1186/1471-2105-13-274
Woo, H. R., Pontes, O., Pikaard, C. S., and Richards, E. J. (2007). VIM1, a Methylcytosine-Binding Protein Required for Centromeric Hetero Chromatinization. Genes. Dev. 21, 267–277. doi:10.1101/gad.1512007
Xiao, W., Huawei, X., Shengde, L., Lili, C., and Fuping, Y. (2021). Epigenetic Regulation in Antiviral Innate Immunity. Eur. J. Immunol. 51, 1641–1651. doi:10.1002/eji.202048975
Xu, F., Liu, Q., Chen, L., Kuang, J., Walk, T., Wang, J., et al. (2013). Genome-wide Identification of Soybean microRNAs and Their Targets Reveals Their Organ-Specificity and Responses to Phosphate Starvation. BMC Genomics 14, 66. doi:10.1186/1471-2164-14-66
Yan, W., Deng, X. W., Yang, C., and Tang, X. (2021). The Genome-wide EMS Mutagenesis Bias Correlates with Sequence Context and Chromatin Structure in Rice. Front. Plant Sci. 12, 370–462. doi:10.3389/fpls.2021.579675
Yuan, L., Yang, X., and Makaroff, C. A. (2011). Plant Cohesins, Common Themes and Unique Roles. Curr. Prot. Pept. Sci. 12 (2), 93–104. doi:10.2174/138920311795684904
Zemach, A., Kim, M. Y., Hsieh, P. H., Coleman, D. D., Eshed, W. L., Thao, K., et al. (2013). The Arabidopsis Nucleosome Remodeler DDM1 Allows DNA Methyltransferases to Access H1-Containing Heterochromatin. Cell. 153, 193–205. doi:10.1016/j.cell.2013.02.033
Zentner, G. E., and Henikoff, S. (2013). Regulation of Nucleosome Dynamics by Histone Modifications. Nat. Struct. Mol. Biol. 20, 259–266. doi:10.1038/nsmb.2470
Zhang, H., Lang, Z., and Zhu, J. K. (2018). Dynamics and Function of DNA Methylation in Plants. Nat. Rev. Mol. Cell. Biol. 19, 489–506. doi:10.1038/s41580-018-0016-z
Zhang, J., and Zhou, J. M. (2010). Plant Immunity Triggered by Microbial Molecular Signatures. Mol. Plant 3, 783–793. doi:10.1093/mp/ssq035
Zhang, M., An, P., Li, H., Wang, X., Zhou, J., Dong, P., et al. (2019). The miRNA-Mediated Post-Transcriptional Regulation of Maize in Response to High Temperature. Int. J. Mol. Sci. 20, 1754. doi:10.3390/ijms20071754
Zhang, Q., Wang, D., Lang, Z., He, L., Yang, L., Zeng, L., et al. (2016). Methylation Interactions in Arabidopsis Hybrids Require RNA-Directed DNA Methylation and Are Influenced by Genetic Variation. Proc. Natl. Acad. Sci. USA. 113, E4248–E4256. doi:10.1073/pnas.1607851113
Zhang, X., Yazaki, J., Sundaresan, A., Cokus, S., Chan, S. W., Chen, H., et al. (2006). Genome-wide High Resolution Mapping and Functional Analysis of DNA Methylation in Arabidopsis. Cell. 126, 1189–1201. doi:10.1016/j.cell.2006.08.003
Zhao, B., Liang, R., Ge, L., Li, W., Xiao, H., Lin, H., et al. (2007). Identification of Drought-Induced microRNAs in Rice. Biochem. Biophys. Res. Commun. 354 (2), 585–590. doi:10.1016/j.bbrc.2007.01.022
Zhu, N., Cheng, S., Liu, X., Du, H., Dai, M., Zhou, D. X., et al. (2015). The R2R3-type MYB Gene OsMYB91 Has a Function in Coordinating Plant Growth and Salt Stress Tolerance in Rice. Plant Sci. 236, 146–156. doi:10.1016/j.plantsci.2015.03.023
Zhu, Y., Rowley, M. J., Bohmdorfer, G., and Wierzbicki, A. T. (2012). A SWI/SNF Chromatin-Remodeling Complex Acts in Noncoding RNA-Mediated Transcriptional Silencing. Mol. Cell. 49, 298–309. doi:10.1016/j.molcel.2012.11.011
Zilberman, D., Coleman, D. D., Ballinger, T., and Henikoff, S. (2008). HistoneH2A.Z and DNA Methylation Are Mutually Antagonistic Chromatin Marks. Nature 456, 125–129. doi:10.1038/nature07324
Keywords: chickpea (Cicer arietinum L.), epigenomics, DNA methylation, RNAi- RNA interference, mutagenesis, climate resilience, sustainability
Citation: Chandana BS, Mahto RK, Singh RK, Ford R, Vaghefi N, Gupta SK, Yadav HK, Manohar M and Kumar R (2022) Epigenomics as Potential Tools for Enhancing Magnitude of Breeding Approaches for Developing Climate Resilient Chickpea. Front. Genet. 13:900253. doi: 10.3389/fgene.2022.900253
Received: 20 March 2022; Accepted: 10 June 2022;
Published: 22 July 2022.
Edited by:
Abhishek Bohra, Indian Institute of Pulses Research (ICAR), IndiaReviewed by:
Ali Raza, Fujian Agriculture and Forestry University, ChinaCopyright © 2022 Chandana, Mahto, Singh, Ford, Vaghefi, Gupta, Yadav, Manohar and Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajendra Kumar, cmFqZW5kcmFrNjRAeWFob28uY28uaW4=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.