 Chloe Mighton1,2†Salma Shickh1,2†Vernie Aguda1,3Suvetha Krishnapillai1,2Ella Adi-Wauran1,2Yvonne Bombard1,2*
Chloe Mighton1,2†Salma Shickh1,2†Vernie Aguda1,3Suvetha Krishnapillai1,2Ella Adi-Wauran1,2Yvonne Bombard1,2*- 1Genomics Health Services Research Program, St. Michael’s Hospital, Unity Health Toronto, Toronto, ON, Canada
- 2Institute of Health Policy, Management and Evaluation, University of Toronto, Toronto, ON, Canada
- 3Centre for Medical Education, School of Medicine, Cardiff University, Cardiff, United Kingdom
Genomic medicine is expanding from a focus on diagnosis at the patient level to prevention at the population level given the ongoing under-ascertainment of high-risk and actionable genetic conditions using current strategies, particularly hereditary breast and ovarian cancer (HBOC), Lynch Syndrome (LS) and familial hypercholesterolemia (FH). The availability of large-scale next-generation sequencing strategies and preventive options for these conditions makes it increasingly feasible to screen pre-symptomatic individuals through public health-based approaches, rather than restricting testing to high-risk groups. This raises anew, and with urgency, questions about the limits of screening as well as the moral authority and capacity to screen for genetic conditions at a population level. We aimed to answer some of these critical questions by using the WHO Wilson and Jungner criteria to guide a synthesis of current evidence on population genomic screening for HBOC, LS, and FH.
Introduction
Genomic medicine is expanding from a focus on diagnosis at the patient level to prevention at the population level. As test costs fall, it could become feasible to screen pre-symptomatic individuals through public health-based approaches, rather than restricting testing to high-risk groups. Indeed, pilot initiatives in which hundreds of thousands of individuals will undergo genomic screening are being launched in health systems in the United States (U.S.) (Carey et al., 2016; Schwartz et al., 2018; Lacaze et al., 2019; Grzymski et al., 2020), the United Kingdom (U.K.) (Genomics England, 2021), and Australia (Rowley et al., 2019; Lacaze et al., 2022). Leading hereditary conditions for consideration in population screening include hereditary breast and ovarian cancer syndrome (HBOC), Lynch syndrome (LS), and familial hypercholesterolemia (FH). These conditions are prioritized for screening due to their under-ascertainment using current screening approaches and the availability of evidence-based interventions to reduce morbidity and mortality (Centers for Disease Control and Prevention OoPHG, 2022).
Traditional methods to identify cases with HBOC, LS, and FH include genetic testing for patients meeting clinical, ethnicity or family-history based criteria (Hampel et al., 2008; Schofield et al., 2014; Klančar et al., 2015; Tognetto et al., 2017; Groselj et al., 2018; Gupta et al., 2019; Daly et al., 2020; Kunnackal John et al., 2021; Zuurbier et al., 2021). However, these targeted approaches have been found to miss a substantial proportion of individuals who harbor pathogenic variants. For example, >50% of individuals with pathogenic BRCA1 and BRCA2 (BRCA1/2) variants are missed by family history-based criteria (Metcalfe et al., 2010a; Gabai-Kapara et al., 2014; Manchanda et al., 2015a). The availability of large-scale next-generation sequencing (NGS) strategies and preventive options for these conditions makes it increasingly feasible to screen pre-symptomatic individuals through public health-based approaches, rather than restricting testing to high-risk groups.
This raises anew, and with urgency, questions about the limits of screening as well as the capacity to screen for genetic conditions at a population level, or in other words, population genomic screening. We use the term “population genomic screening” to refer to germline DNA testing among an unselected, asymptomatic population with the aim of identifying individuals with pathogenic/likely pathogenic (henceforth, “pathogenic”) variants. Key issues to scaling up population genomic screening include the optimal testing approach, penetrance of these conditions in the general population, clinical effectiveness, cost-effectiveness, acceptability, health system capacity to implement such a program, ethical issues such as overdiagnosis, access challenges and equity.
Decisions about screening are expected to align with the World Health Organization principles of screening. These criteria, developed by Wilson and Jungner in 1968, inform decision-making around disease screening and generally include considerations of the nature of the disease, test characteristics, and the availability, effectiveness and acceptability of preventive interventions or treatments (Table 1) (Wilson and Jungner, 1968). Since its publication, Wilson and Jungner’s criteria have been widely accepted, modified and used by decision-makers across the world to guide screening decisions. Whereas the Wilson and Jungner criteria were developed for programs aiming to enable early detection and intervention for individuals with early stages of a disease, population genomic screening programs would identify those with a genetic predisposition to disease. The identification of a pathogenic variant in an asymptomatic individual through genetic screening is not equivalent to a clinical diagnosis of the associated disease (Murray, 2016; Murray et al., 2021). Given the complexity of policy decision-making for genetic tests and genetic screening programs, various frameworks and sets of decision criteria have been developed to guide these decisions (Sanderson et al., 2005; Burke and Zimmern, 2007; Andermann et al., 2008; Teutsch et al., 2009; Andermann et al., 2011; National Academies of Sciences Engineering and Medicine, 2017; Pitini et al., 2019). While these newer frameworks and decision criteria share core elements with Wilson and Jungner such as those related to the natural history of the condition, the effectiveness of the test, and effectiveness of preventive interventions, newer frameworks extend Wilson and Jungner’s criteria to include considerations related to implementation issues such as health service delivery, ethics, and equity. However, these more recent criteria for genomic evaluation have not been universally adopted, and different health systems vary in which criteria are used in policy decisions, if any. Given the lack of a universally accepted set of decision criteria for genomic screening, and the continued relevance of the fundamental principles of Wilson and Jungner, we will use the Wilson and Jungner criteria to guide a synthesis of the current evidence on population genomic screening for leading gene-condition pairs. In addition, we also discuss ethical and equity considerations. While these are absent from the original Wilson and Jungner criteria, they are increasingly important in decision frameworks for genomic screening programs (Andermann et al., 2008; Pitini et al., 2019) and are commonly considered across various frameworks and sets of decision criteria for genomic technologies (Burke and Zimmern, 2007; Andermann et al., 2008; Teutsch et al., 2009; Andermann et al., 2010; Botkin et al., 2010; Andermann et al., 2011). We highlight policy and practice issues as well as future research priorities to inform the design of population genomic screening programs to maximize population benefits and minimize harms.

TABLE 1. Wilson and Jungner’s principles for disease screening (Wilson and Jungner, 1968).
Is the condition sought an important health problem?
HBOC, LS and FH are characterized by their high penetrance, evidence-based interventions for prevention/treatment and subsequent benefits from the early detection, in line with fundamental principles of screening. The CDC Office of Public Health Genomics (OPHG) designates screening for HBOC, LS, and FH as Tier 1 genomic applications (Centers for Disease Control and Prevention OoPHG, 2022). Tier 1 genomic applications are those which could have a substantial, positive impact on public health based on: 1) A high prevalence of 1 in 200 for HBOC, 1 in 340 for LS and in 1 in 250 for FH in the general populations (exact frequency may vary in certain populations); 2) the under-ascertainment of current strategies; and, 3) established risk-reducing interventions that reduce morbidity and mortality (Abul-Husn et al., 2016; Akioyamen et al., 2017; Manickam et al., 2018; Grzymski et al., 2020; Manickam et al., 2021).
Is the natural history of the condition adequately understood?
The natural histories of HBOC, LS, and FH are relatively well understood. HBOC is caused by pathogenic variants in BRCA1/2 which confer substantially elevated risks for female breast cancer, ovarian cancer, and male breast cancer (in particular for BRCA2 carriers), in addition to increased risks for pancreatic cancer, prostate cancer, and melanoma (The Breast Cancer Linkage Consortium, 1999; Brose et al., 2002; Levine et al., 2003; Lindor et al., 2008; Lynch et al., 2009; Moran et al., 2012; Mavaddat et al., 2013; McKay et al., 2016). While pathogenic variants in other genes including PALB2, RAD51C, RAD51D, and BRIP1 also cause hereditary breast and ovarian cancer, we focus this review on BRCA1/2 because of the higher frequency of pathogenic variants in the population in these genes, and established clinical management guidelines (Manickam et al., 2018; National Comprehensive Cancer Network (NCCN), 2021a). LS is caused by pathogenic variants in mismatch repair (MMR) genes MLH1, MSH2, MSH6, and PMS2, as well as deletions in EPCAM which lead to silencing of MSH2. Affected individuals are at increased risk for colorectal cancer (CRC), endometrial cancer, ovarian cancer, and other cancers (Lindor et al., 2008; Senter et al., 2008; Baglietto et al., 2010; Bonadona et al., 2011; Giardiello et al., 2014). FH, caused by pathogenic variants in LDLR, PCSK9, and APOB, is characterized by elevated plasma low-density lipoprotein cholesterol (LDL-C) levels, which leads to risks for cardiovascular disease and premature mortality (Youngblom et al., 2016).
Two key issues that inform natural history are penetrance and age of onset. HBOC, LS and FH exhibit high but incomplete penetrance. Although the penetrance (the chance that an individual with the condition will manifest particular features) of the causative genes has been estimated in cohorts ascertained with strong personal and family history of disease, it has yet to be well-established in the general population (Murray et al., 2021). Some studies suggest penetrance in the general population may vary from estimates from family-based studies (Forrest et al., 2022). However, the risk to those identified through population screening will likely still be high enough to warrant clinical intervention, at least in BRCA1/2 carriers where there is substantial evidence demonstrating high penetrance even among unselected cases (Chatterjee et al., 2001; Chatterjee and Wacholder, 2001; Antoniou et al., 2005; Chatterjee et al., 2006; Kuchenbaecker et al., 2017; Chen et al., 2020). These studies highlight the importance of evaluating the appropriateness of population genomic screening and subsequent interventions, given the potential for overdiagnosis and overtreatment (to be discussed in a subsequent section, Ethical considerations). Adding another layer of complexity to risk prediction, other genetic factors, such as polygenic background, and non-genetic risk factors (e.g., diet, environmental exposures, and clinical risk factors) can also influence the penetrance of these conditions (Fahed et al., 2020).
Based on the age of onset and availability of age-appropriate preventive interventions, the optimal age to initiate screening will vary across target conditions. For example, surveillance and risk-reducing surgeries for HBOC and LS are recommended in adulthood (National Comprehensive Cancer Network (NCCN), 2021a; National Comprehensive Cancer Network (NCCN), 2021b), while pharmacologic treatment of FH can begin in childhood (Gidding et al., 2015). The health outcomes and costs of population screening programs will likely vary depending on the age at which screening and intervention is initiated. Specific considerations related to the target population for each condition are provided throughout the subsequent sections.
Is there a suitable test or examination?
One element of test performance is validity, which encompasses both “analytic validity” (accuracy in detecting the target genetic variant) and “clinical validity” (accuracy in identifying patients with the target condition) (Bombard et al., 2013). Test selection for population genomic screening should consider what type of genetic variation primarily causes the target condition, and testing laboratories should be equipped to manage gene-specific technical challenges [e.g., PMS2 pseudogenes (Hegde et al., 2014; Li et al., 2015; Lee et al., 2021a)]. Several laboratory considerations for population genomic screening include whether to perform full gene sequencing or targeted variant testing, whether to test for only known pathogenic variants or also novel variants, and whether to perform deletion/duplication analysis in addition to sequence analysis; each of these decisions will impact test costs and post-test residual risk (Lu et al., 2019). NGS has very high analytic sensitivity and specificity for detecting single-nucleotide variants and small insertions/deletions (Baudhuin et al., 2015; Judkins et al., 2015; Toland et al., 2018), and could be coupled with gene-targeted deletion/duplication analysis to increase detection of disease-causing variants for HBOC, LS and FH (Petrucelli et al., 1998; Idos et al., 2004; Ison et al., 2014). Deletion/duplication analysis is necessary to identify disease-causing variants in EPCAM. The use of array-based genotyping in population genomic screening has been found to result in false positives and false negatives compared to NGS or Sanger sequencing (Blout Zawatsky et al., 2021; Bowling et al., 2021). For HBOC, in the Ashkenazi Jewish (AJ) population, there are three founder variants (BRCA1 c.68_69delAG, BRCA1 c.5266dupC and BRCA2 c.5946delT) which are prevalent in ∼2.5% (Roa et al., 1996) of the population. While these variants do account for the majority of pathogenic BRCA1/2 variants in the AJ population (Walsh et al., 2017), some BRCA1/2 carriers would be missed if targeted founder variant testing as opposed to NGS was used in population genomic screening among the AJ population (Rosenthal et al., 2015; Solano et al., 2018).
Another aspect of genetic test performance is variant interpretation (Richards et al., 2015). Key issues related to variant interpretation include variants of uncertain significance (VUS) (Burke et al., 192022), discordant variant interpretations between diagnostic laboratories (Garber et al., 2016; Harrison et al., 2017; Iacocca et al., 2018; Lebo et al., 2018; Amendola et al., 2020; Mighton et al., 2021a), variant reclassification over time (Macklin et al., 2018; Mersch et al., 2018; Slavin et al., 2018; Turner et al., 2018; Esterling et al., 2020; Chiang et al., 2021) and recontacting patients with updated results (e.g., changes from VUS to likely pathogenic or pathogenic which may warrant medical follow-up) (Otten et al., 2015; El Mecky et al., 2019). While these issues exist in standard clinical genetic testing, they will be magnified if genomic screening is conducted at a population scale, and will need to be considered in program design/implementation.
A further aspect of test performance is the positive predictive value (PPV), the probability that a patient with a positive result (a reported pathogenic or likely pathogenic variant) has the associated condition (Hagenkord et al., 2020). PPV depends on test characteristics (specificity, sensitivity) and condition prevalence (Akobeng, 2007; Oleske, 2010; Hagenkord et al., 2020). As HBOC, FH, and LS have a lower prevalence in the general population compared to populations ascertained based on family history, this would reduce the PPV of a positive result obtained from population genomic screening compared to a positive result from genomic testing among high-risk populations (Hagenkord et al., 2020). Estimates of PPV for Tier 1 conditions range from 80% to 91%, assuming 99.95% specificity and that one-third of the overall positive rate is likely pathogenic variants and two-thirds are pathogenic variants (Hagenkord et al., 2020). Increasing test specificity can increase the PPV, which laboratories could accomplish by adjusting the reporting cut-off between a positive and a negative result (Lu et al., 2019; Hagenkord et al., 2020). For example, reporting only high confidence likely pathogenic variants can increase specificity (Hagenkord et al., 2020).
Is there a recognizable latent or early symptomatic stage?
Among these three conditions, there is a pre-symptomatic state that is identifiable by molecular testing for pathogenic variants in the relevant genes (Youngblom et al., 2016; Petrucelli et al., 2022). Therefore, population genomic screening for HBOC, LS, and FH can be used to identify individuals with pathogenic variants in the causative genes who would not otherwise be identified through routine clinical care and could gain benefits from early intervention (Grzymski et al., 2020). Multiple studies have found that population genomic screening identifies carriers of pathogenic variants for HBOC, LS, and FH who were previously unaware of their variant (Buchanan et al., 2020; Grzymski et al., 2020; Abul-Husn et al., 2021; Lee et al., 2021b; Blout Zawatsky et al., 2021).
Hereditary breast and ovarian cancer
Population genomic screening methods have been found to identify a higher proportion of BRCA1/2 carriers than family-history and clinical criteria-based methods (Manchanda et al., 2015a; Manickam et al., 2018; Abul-Husn et al., 2019). In addition to their improved detection rate, BRCA1/2 screening programs suggest that penetrance of cancer in families of Ashkenazi Jewish ancestry identified through population screening programs is just as high as in families identified through traditional family history based or clinical criteria methods (Gabai-Kapara et al., 2014).
Lynch syndrome
Compared to traditional approaches for clinically ascertaining LS cases (e.g., tumor testing followed by germline testing among affected patients or family history-based approaches for unaffected cases (Hampel et al., 2008; Batte et al., 2014; Tognetto et al., 2017; Kahn et al., 2019), a potential benefit of population genomic screening is the identification of a greater number of pre-symptomatic patients which could allow for cancer prevention through enhanced surveillance, chemoprevention with aspirin, and surgical prevention with hysterectomy and bilateral salpingo-oophorectomy. Several studies have found that population genomic screening identified pre-symptomatic individuals with pathogenic variants in LS genes who were unaware of their variant and would be missed by standard approaches to case identification (Buchanan et al., 2020; Grzymski et al., 2020; Abul-Husn et al., 2021; Lee et al., 2021b; Blout Zawatsky et al., 2021).
Familial hypercholesterolemia
Evidence from clinical testing programs and population-based studies suggest that population genomic screening for FH will lead to benefits. These include increased case detection and short-term improvements, especially when conducted during the pediatric period, given the potential for early intervention through dietary cholesterol reduction, medication, and screening intensity (Smith et al., 2016). Systematic reviews and observational studies have found that universal lipid screening for FH among children and adolescents followed by targeted genetic testing, and cascade testing of relatives, are effective methods for identifying FH cases (Lozano et al., 2016a; Wald et al., 2016; Groselj et al., 2018; Lee et al., 2019; Matsunaga et al., 2021; Zuurbier et al., 2021). The availability and lower costs of lipid screening approaches raises questions about the necessity of using genomic screening as a first tier test to identify FH cases.
Are there accepted options for surveillance and prevention for high-risk populations?
There are various surveillance and prevention options endorsed by clinical practice guidelines to guide the management of individuals with HBOC, LS and FH.
Hereditary breast and ovarian cancer syndrome
Although there are guidelines for the management of patients with pathogenic variants in various HBOC genes (National Comprehensive Cancer Network (NCCN), 2021a; Tischkowitz et al., 2021; Manchanda et al., 2022), we are focusing on the Tier 1 genes, BRCA1 and BRCA2. In terms of prevention, bilateral prophylactic mastectomy and risk-reducing salpingo-oophorectomy are highly effective in preventing breast cancer and ovarian/fallopian tube cancers respectively in addition to reducing mortality, though a small residual risk for primary peritoneal cancer remains (National Comprehensive Cancer Network (NCCN), 2021a; Li et al., 2016; Honold and Camus, 2018; Finch et al., 2014).
Among females who decline or defer surgery, early detection options for female carriers of a disease-causing BRCA1/2 variant usually comprise of a combination of routine mammograms and breast MRIs for breast cancer risks, which are effective at detecting breast cancer among BRCA1/2-positive females. MRI is more sensitive than mammography in high-risk females (National Comprehensive Cancer Network (NCCN), 2021a; Warner et al., 2004; Kriege et al., 2004; Leach et al., 2005; Kuhl et al., 2005; Riedl et al., 2007; Sardanelli et al., 2007; Lowry et al., 2012; Lehman et al., 2016). Among high-risk females, MRI in combination with mammography has been found to be more sensitive than either modality alone (Warner et al., 2008; Mann et al., 2019) and to improve overall survival relative to mammography alone (Bae et al., 2020). In an observational cohort study of MRI in combination with mammography among unaffected female BRCA1/2 heterozygotes, the probability of dying of breast cancer within 20 years was 2% (Warner et al., 2020). For ovarian cancer risks, guidelines from the National Comprehensive Cancer Network (NCCN) suggest that transvaginal ultrasound and CA-125 may be offered at the clinician’s discretion to BRCA1/2 carriers who have not elected for risk-reducing salpingo-oophorectomy (National Comprehensive Cancer Network (NCCN), 2021a). However, these interventions are of uncertain benefit (National Comprehensive Cancer Network (NCCN), 2021a; Jacobs et al., 2016; Menon et al., 2009) and ovarian cancer screening with transvaginal ultrasound and CA-125 has not been demonstrated to reduce mortality (Menon et al., 2021).
Chemopreventive options are routinely offered in clinical practice given the evidence that they reduce breast cancer risk for all at-risk populations, including BRCA1/2 carriers (National Comprehensive Cancer Network (NCCN), 2021a; Gronwald et al., 2006; Narod et al., 2000).
For male carriers of BRCA1/2 pathogenic variants, recommendations consist of yearly screening with a digital rectal exam and prostate-specific antigen (PSA) blood test initiated by age 40–45 however limited data exists to support the effectiveness of additional screening (breast cancer) (National Comprehensive Cancer Network (NCCN), 2021a; Gao et al., 2019).
Studies with female AJ BRCA1/2 carriers identified through population screening indicate acceptability for and high uptake of risk-reducing strategies (Metcalfe et al., 2012; Lieberman et al., 2017). Long-term follow up supports improvements in psychological outcomes such as anxiety (Metcalfe et al., 2012; Manchanda et al., 2015a; Manchanda et al., 2020a; Morgan et al., 2021). In the general population, there is less evidence on the uptake of preventive strategies or outcomes. Several studies indicate that many BRCA1/2 carriers identified through population screening do undergo risk-reducing procedures such as surveillance or prophylactic surgery (Buchanan et al., 2020; Lee et al., 2021b; Elhanan et al., 2022). In some cases, HBOC-associated cancers were diagnosed because of the screening initiated based on the genomic screening results (Buchanan et al., 2020).
Lynch syndrome
For Lynch syndrome, there are strategies for early detection or prevention of CRC and gynaecological cancers. Early detection strategies in LS include recommendations for colonoscopy, endoscopy, and total body examinations (National Comprehensive Cancer Network (NCCN), 2021b; Stjepanovic et al., 2019). Surveillance colonoscopy is effective at reducing CRC burden and improving survival among LS patients (Dove-Edwin et al., 2002; Järvinen et al., 2009; Ladabaum et al., 2015; Stjepanovic et al., 2019), though the optimal intervals for surveillance and age to initiate screening are still areas of investigation (National Comprehensive Cancer Network (NCCN), 2021b; Stjepanovic et al., 2019; Järvinen et al., 2009; Dove-Edwin et al., 2002; Jenkins et al., 2015), especially among patients with PMS2 variants which may have lower penetrance (National Comprehensive Cancer Network (NCCN), 2021b; Lindor et al., 2006). There is observational evidence that prophylactic hysterectomy and/or bilateral salpingo-oophorectomy effectively reduce the incidence of gynaecological cancers among females with LS (Schmeler et al., 2006) and is routinely recommended for at-risk females (Crosbie et al., 2019); however, evidence on mortality is lacking. Chemoprevention with aspirin is also an option for LS risk management as there is evidence that aspirin reduces risk for CRC and other LS-associated cancers (Burn et al., 2011; Ait Ouakrim et al., 2015), however there is no evidence on the effect of aspirin on mortality (Rubenstein et al., 2015). Endometrial cancer screening has not been proven to benefit LS patients (National Comprehensive Cancer Network (NCCN), 2021b). However, it may be considered at the discretion of the clinician every 1–2 years in conjunction with endometrial biopsy, which is considered a sensitive and specific diagnostic test (National Comprehensive Cancer Network (NCCN), 2021b). Transvaginal ultrasound can be considered among postmenopausal females (National Comprehensive Cancer Network (NCCN), 2021b).
Evidence on the outcomes of population genomic screening for LS beyond detection rate is limited. Several studies of population genomic screening for LS have found that a proportion of individuals with pathogenic LS variants underwent risk-reducing procedures, including colonoscopy and prophylactic surgery (Buchanan et al., 2020; Lee et al., 2021b; Elhanan et al., 2022). Several individuals were diagnosed with LS-associated cancers because of follow-up initiated based on their genomic screening results (Buchanan et al., 2020). However, there is some literature that suggests the uptake of risk-reducing strategies is very low (< 10%) when patients are responsible for communicating their results to their clinicians (Elhanan et al., 2022).
Familial hypercholesterolemia
Management of heterozygous FH is aimed at primary prevention of atherosclerotic cardiovascular disease through lipid lowering pharmacological therapy, using statins, ezetimibe or PCSK9 inhibitors or other LDL lowering medications, with guidelines recommending initiation at ages 8–10 or earlier based on severity (Carroll et al., 2008; Gidding et al., 2015; Defesche et al., 2017; Kim et al., 2021). Trials have yet to directly compare cardiovascular disease outcomes associated with different pharmacologic treatments for heterozygous FH, and treatment recommendations therefore are based on surrogate outcomes including LDL cholesterol lowering and arterial imaging (Defesche et al., 2017). For example, a systematic review found that statins were effective at lowering LDL-C and total cholesterol (TC) concentration, but there was no evidence on the effect of screening on long term outcomes, such as lipid concentrations or cardiovascular outcomes in adulthood (Lozano et al., 2016a).
Evidence of clinical outcomes of population genomic screening for FH is emerging, but limited to short-term outcomes. Several studies have found that population genomic screening identified individuals with clinical manifestations of FH who were previously unaware of their condition (Buchanan et al., 2020; Lee et al., 2021b; Elhanan et al., 2022). In these studies, a proportion of individuals with pathogenic FH variants initiated risk-reducing strategies such as LDL-lowering medications (Buchanan et al., 2020; Lee et al., 2021b; Elhanan et al., 2022). In one study in which patients were tasked with informing their healthcare provider of their population genomic screening results, LDL-C levels improved in the short term for only 9% of patients with pathogenic FH-related variants, while the remainder exhibited no change in their clinical management (Elhanan et al., 2022).
Is there an agreed policy on whom to treat as patients?
There are evidence-based clinical practice guidelines for the management of individuals with pathogenic variants in genes for HBOC, LS, and FH, as described above. It is important to consider that the evidence used to develop these guidelines is largely from cases ascertained through standard diagnostic approaches, as opposed to through population screening-based ascertainment (Murray et al., 2021). Over time, as evidence on penetrance in unselected populations accumulates, management guidelines may need to be updated with specifications for how to manage individuals with disease risk identified through population genomic screening, given the potential reduced penetrance (Murray et al., 2021). This is less likely to be necessary for the genes included in this review than for moderate penetrance genes, given that the penetrance is likely still high in unselected populations and sufficient to warrant clinical intervention.
Is the test acceptable to the population?
Views among founder populations
Much of the evidence base for population-based genomic screening is from the three BRCA1/2 founder variants’ screening in the AJ population. Unselected population-based BRCA1/2-screening in the AJ population conducted in Israel (Gabai-Kapara et al., 2014; Lieberman et al., 2017a), Canada (Metcalfe et al., 2013), and the UK (Manchanda et al., 2015a) were found to be safe, acceptable, and feasible. In Israel, Poland, and the UK (Manchanda et al., 2019; Reisel et al., 2022), BRCA1/2-screening in the AJ population demonstrates high uptake (> 67%) and satisfaction rates (> 90%), with participants expressing positive attitudes towards the screening experience (Lieberman et al., 2017a). Within the AJ population, motivators for participation were reassurance, decreasing uncertainty, health empowerment, opportunity for risk reduction, and family planning (Lieberman et al., 2017b). Barriers for participation were fear of social and insurance discrimination, stigma, anxiety, and lack of physician awareness and support (Lehmann et al., 2002; Lieberman et al., 2017b). Established founder mutations for LS and FH may also offer a feasible opportunity for population-based genetic screening, however, very limited, if any, research has been done in those populations to determine the acceptability of such programs (Lahtinen et al., 2015; Ponti et al., 2015).
General public views
Current debate centers around whether the same screening principles and findings for populations with founder mutations can be expanded to all populations (Yurgelun et al., 2015; Foulkes et al., 2016). Outside of the AJ population, there is a paucity of research addressing public views and acceptability of a population-based genetic screening program for HBOC, LS, and FH. For HBOC, surveys of unselected females in the US (Rubinsak et al., 2019) and UK (Meisel et al., 2016) demonstrate high interest (> 82%) and acceptability for population-based BRCA1/2 screening. Quantitative and qualitative data from a pilot population genomic screening study predicting ovarian cancer risk demonstrate acceptability, feasibility, reduced cancer worry, and no adverse psychological impact (Gaba et al., 2020; Gaba et al., 2022). Universal genetic and cholesterol screening programs for FH in children demonstrated high uptake within the UK (Wald et al., 2016), and were acceptable to the Australian public (Bowman et al., 2019). Public and patient survey and qualitative interview results from the North America (Graham et al., 1998; Watkins et al., 2011), Europe (Berth et al., 2002), and Australia (Dunlop et al., 2021) indicate support for adult population genomic screening for LS.
Motivators for screening participation include eligibility for increased surveillance and treatment, and the benefits for family members (Ten Haaf et al., 2017). Barriers for screening participation include cost, genetic discrimination, test accuracy, and data confidentiality (Ten Haaf et al., 2017). Genetic discrimination, particularly in the context of insurance, employment, and social relationships (Wauters and Van Hoyweghen, 2016), remains a pervasive deterrent to screening amongst the public, despite the existence of policies to protect sensitive genetic information from misuse worldwide (Joly et al., 2017; Kim et al., 2021).
Providers’ view
Reported attitudes and views of population genomic screening at the provider level are scarce. Many international studies report that non-genetics specialist healthcare providers (Batra et al., 2002; Carroll et al., 2008; Menzin et al., 2010; Klitzman et al., 2013; Hauser et al., 2018) feel ill-equipped to discuss the benefits, limitations, and health implications of genetic testing for HBOC, LS (Hamilton et al., 2017; Laforest et al., 2019), and FH (Haga et al., 2019; Pang et al., 2020; Watts et al., 2021). Additional reported barriers to population genomic screening include implementation costs, misinterpretation of results, and the potential for increased patient anxiety (Shkedi-Rafid et al., 2013; De Simone et al., 2021). A potential benefit of population genomic screening is the removal of genetic testing eligibility criteria, which providers find overly complex (Klitzman et al., 2013; Laforest et al., 2019).
Is the cost of case-finding economically balanced in relation to possible expenditure on medical care as a whole?
Hereditary breast and ovarian cancer
Multiple modeling studies suggest population-based testing for BRCA1/2 would be more cost-effective than testing based on clinical criteria or family history from a health system perspective in high- and upper-middle income countries (Manchanda et al., 2018; Zhang et al., 2019; Manchanda et al., 2020b; Guzauskas et al., 2020), and cost-saving from a societal perspective (Manchanda et al., 2020b) in high- and upper-middle-income countries. In lower-middle income countries, cost-effectiveness depended on the cost of the test (Manchanda et al., 2020b; Meshkani et al., 2021). Models suggest it may be most cost-effective to initiate population screening among younger individuals (Zhang et al., 2019; Guzauskas et al., 2020). In the AJ population, economic evaluations indicate population genomic screening for BRCA1/2 variants would be cost-effective (Manchanda et al., 2015b; Manchanda et al., 2017).
Lynch Syndrome
For LS, economic evidence on population genomic screening among unaffected individuals is limited. A recent U.S.-based economic evaluation suggests that adult population genomic screening among unselected 30-years-old individuals for LS variants would likely be cost-effective at a $150,000 willingness-to-pay threshold (Guzauskas et al., 2022). In contrast, another study found that population genomic screening for LS in unaffected individuals at age 20, followed by cascade testing of first-degree relatives, would not be cost-effective compared to current practices (Dinh et al., 2011). An Australian economic evaluation found that population genomic screening for MLH1 and MSH2 for LS would be cost-effective if conducted as part of a multigene panel including BRCA1/2, but not if performed in isolation (Zhang et al., 2019).
Familial hypercholesterolemia
Multiple economic evaluations from the UK, Poland, Spain and Australia have found that population genomic screening for FH would be cost-effective from a healthcare system perspective (Marks et al., 2002; Lázaro et al., 2017; Pelczarska et al., 2018; Marquina et al., 2021), and one Australia-based evaluation suggests it would be cost saving from a societal perspective (Marquina et al., 2021). There is some evidence to suggest that greatest health gains could achieved by screening the youngest probands, however this would also be more costly (Pelczarska et al., 2018). Cascade testing of first- and second-degree relatives of identified patients with FH is also recommended and has been found to be highly cost-effective (Marks et al., 2002; Wonderling et al., 2004; Oliva et al., 2009; Nherera et al., 2011).
Are facilities for diagnosis and treatment available?
Current models of genetics care are personnel- and time-intensive and not feasible at a population-scale. Key challenges include critical workforce shortages, which contribute to long wait times, a lack of genetics education among non-genetics specialist healthcare providers, and fragmentation of care (Suther and Kiros, 2009; Hann et al., 2017; Office of the Auditor General, 2017; Hoskovec et al., 2018; Stoll et al., 2018; Dragojlovic et al., 2020). These challenges persist in urban areas and are exacerbated in remote and under-served communities (Office of the Auditor General, 2017). Capacity to sustain population genomic screening must also include laboratory infrastructure, secure data storage, as well as bioinformatic and analytic pipelines to support population-scale genomic analyses (Kelly et al., 2021). There is a paucity of data on the availability and distribution of laboratory infrastructure and personnel including clinical laboratory geneticists and medical laboratory technicians (Dragojlovic et al., 2020). This is critical to understand as it will be variable across jurisdictions and will be important for decision-makers to determine how to deliver the program (i.e. the distribution of testing centres).
Is case finding a continuing process?
The possibility for variants to be reclassified over time means that case finding must be an ongoing process. Most reclassifications are from VUS to likely benign or benign, and reclassification of variants initially classified as pathogenic/likely pathogenic is very rare (Macklin et al., 2018; Mersch et al., 2018; Mighton et al., 2019). In the context of population genomic screening, reclassifications from VUS to pathogenic/likely pathogenic are particularly relevant, as an upgrade from VUS to pathogenic/likely pathogenic could impact medical management. This raises questions about the need for periodic reanalysis and recontact of patients for the return of updated results. The issues of reclassification and recontact already present practical and resource challenges in the context of targeted, clinical testing (Otten et al., 2015; El Mecky et al., 2019), and would be magnified if testing were implemented at the population scale. This is critical to note as non-European populations consistently have higher VUS rates due to lack of representation in databases, leading to higher rates of reclassification and the need for recontact in these populations (Popejoy and Fullerton, 2016; Slavin et al., 2019; Buchanan et al., 2020; Popejoy et al., 2020). There are currently variation in recontact guidelines and practices across jurisdictions, laboratories, and health systems (Bombard and Mighton, 2018; Sirchia et al., 2018), despite recontact being expected by patients (Linderman et al., 2016; Mighton et al., 2021b).
Ethical considerations
It is important to consider the potential harms and unintended consequences of population genomic screening. Early detection and preventive strategies for HBOC, LS, and FH such as high intensity surveillance, prophylactic surgeries, and pharmacotherapy are not without risks including exposure to radiation, false positives, surgical complications, and adverse drug reactions.
For HBOC, there is some observational evidence to suggest that exposure to diagnostic radiation, including mammography, at a young age is associated with increased risk for breast cancer among females with disease-causing BRCA1/2 variants (Pijpe et al., 2012). A systematic review of the harms of breast cancer screening among average-risk females found that harms included overdiagnosis (at rates of 11%–22% from randomized controlled trials [RCTs]) and false positive results which were associated with elevated anxiety, distress, and breast-cancer specific worry; however, the review only included females at average-risk and excluded those with pathogenic BRCA1/2 variants (Nelson et al., 2016). Psychological harms have been identified among BRCA1/2 carriers, related to false positives and living at risk for disease (Metcalfe et al., 2020). With respect to LS, a systematic review of colorectal cancer screening among average-risk individuals found serious adverse events from colonoscopy including perforations and major bleeds, but these events were uncommon in average-risk populations (Lin et al., 2021). However, high-risk patients such as those with LS were excluded from the review (Lin et al., 2021). For FH, the safety profile differs across pharmacologic therapies. For statins and PCSK9 inhibitors, RCTs have found that treatment-related adverse events did not significantly differ between therapy and placebo (Kastelein et al., 2015; Lozano et al., 2016b), though for statins there are sporadic reports of systemic, immunologic, and pain-related adverse events (Lozano et al., 2016b). Bile acid sequestrants have been commonly associated with adverse GI symptoms, and poor palatability (Lozano et al., 2016b).
Across all conditions, potential harms include genetic discrimination which can arise in a variety of settings. This includes insurance discrimination, which is especially relevant in countries such as the U.S. where much of the population must purchase private health insurances (Ridic et al., 2012; Maynard, 2013). Harms may also be caused when carriers face challenges in accessing risk-reducing strategies in jurisdictions without universal healthcare coverage or among historically underserved populations (e.g., rural populations) (Nguyen-Pham et al., 2014; Chandak et al., 2019; Villegas and Haga, 2019). This raises the question of whether it is ethical to offer population genomic screening in the absence of universal coverage of downstream risk-reducing management. Patient harm may also arise if patients who receive negative results from screening are falsely reassured and forego recommended scheduled screening for average risk populations (i.e., age- and family history-recommended screening) although recent evidence suggests this risk may be minimal (Burnell et al., 2022). Conversely, false positive results may lead to overdiagnosis and overtreatment, where patients and family members may undergo unnecessary investigations and potentially life-altering procedures such as prophylactic surgeries. Although these issues also affect patients undergoing family-history based testing, the higher rates of false positive results associated with population screening coupled with a larger number of patients undergoing genetic testing translates to a larger volume of patients who may receive inappropriate and unnecessary medical care.
At present, the balance of benefits and harms of population genomic screening are not well-characterized. This calls into question whether and to what degree the balance of benefits and harms of screening and subsequent interventions for HBOC, LS and FH should be discussed with patients to ensure informed decision-making. Likewise, it remains unclear how to meaningfully obtain informed consent at the population level given the diversity of literacy, health literacy, socioeconomic status, geography, and culture among screened populations. Genomic screening might not be desirable for all people based on their values and preferences, further highlighting the importance of informed decision-making.
Return of results at the population level presents a further issue. Genomic information is uncertain and complex; delivering this information may lead to adverse psychological outcomes (Mighton et al., 2021c). Among patients receiving positive results after genetic testing, there is evidence of increased risk of anxiety, distress and depression (Rew et al., 2010; Wade et al., 2010; Wade, 2019). Certain populations may face additional risks, such as children feeling a loss of autonomy and women who feel burdened with the responsibility of sharing results with relatives (Gaff et al., 2007; Wade et al., 2010; Wade, 2019). Moreover, parents become overprotective of genetically at-risk children and recognize a disruption of the parent-child relationship (Rew et al., 2010). Although these harms are typically rare and transient, genomic screening at a population level will result in a large number of individuals with psychological harms. In addition to high-quality genetic counseling support, there will also be a need for mental health professionals to support these patients and their families. Furthermore, how to manage VUS in population screening remains unresolved, though there is growing consensus that VUS should not be reported in screening contexts (Murray et al., 2021; Burke et al., 192022). An alternative approach is to examine strategies for return of VUS findings, reclassification, and follow-up, a focus of current investigation.
Equity
Equity is an important consideration. There are currently disparities in access to and outcomes of genetics services. Racialized and underserved populations often have lower referral rates, differential rates of service uptake, more frequent misdiagnoses or inconclusive test results, older age and more advanced disease stage of diagnosis, and higher mortality rates (Armstrong et al., 2005; Maddison et al., 2011; Cragun et al., 2015; Kerner et al., 2015; Purificacion et al., 2015; Manrai et al., 2016; Vohnout et al., 2016; Amrock et al., 2017; Landry and Rehm, 2018; Muller et al., 2018; Hendricks-Sturrup and Lu, 2019; Ndugga-Kabuye and Issaka, 2019; Ehrenberg et al., 2021). These disparities are present worldwide, highlighting the pervasiveness of health inequities and an urgency for strategies to address them prior to adoption of population screening, to avoid exacerbating these issues. In addition, many underserved populations have limited guidelines on risk factors or treatment recommendations, making it difficult for clinicians to provide appropriate and effective care (Hann et al., 2017). For example, there is a scarcity of guidelines for breast cancer screening in transgender individuals undergoing gender-affirming hormone therapy (Berro et al., 2020; Rolle et al., 2021).
Furthermore, availability of risk-reducing strategies is not consistent across jurisdictions. For example, the extent (if any) of reimbursement for these interventions will vary by healthcare systems, leading to out-of-pocket costs for high-risk individuals, likely exacerbating existing inequities for underserved populations and undermining the effectiveness of the screening program.
Gaps, future research, and key implications for practice and policy
Clinical effectiveness
There is considerable evidence that population genomic screening improves detection of individuals with pathogenic variants for HBOC, LS, and FH compared to family history or clinical criteria-based approaches, identifying individuals who would otherwise be missed. However, with the exception of BRCA1/2 screening in the AJ population, evidence on whether the improved detection rate translates into improved health outcomes (morbidity, mortality) is lacking (Table 2: Summary of key points and gaps). While there are guideline-endorsed, evidence-based strategies to reduce morbidity and mortality for these conditions, several studies suggest that only a proportion of individuals with pathogenic variants identified through population genomic screening approaches actually uptake the associated risk-reducing interventions (Elhanan et al., 2022). Furthermore, studies on clinical effectiveness and ongoing pilot studies (Foss et al., 2022) have primarily employed observational or retrospective designs which suffer from multiple sources of bias (e.g., missing data, loss to follow up) that could reduce the quality of the evidence. However, among the AJ population, there is substantial evidence to support population screening for BRCA1/2, including high acceptability, satisfaction, uptake of preventive strategies, in addition to improvements in long term outcomes and reduced costs (Metcalfe et al., 2010a; Metcalfe et al., 2010b; Metcalfe et al., 2012; Metcalfe et al., 2013; Gabai-Kapara et al., 2014; Manchanda et al., 2015a; Manchanda et al., 2015b; Manchanda et al., 2016; Lieberman et al., 2017a; Lieberman et al., 2017b; Manchanda et al., 2017; Manchanda et al., 2019; Manchanda et al., 2020a; Manchanda et al., 2020c; Reisel et al., 2022). Another gap in the literature is that some data has been generated from biobanks and return of secondary findings, which is not reflective of population genomic screening and its outcomes. There is a need for large-scale, prospective, purpose-built population genomic screening pilot studies designed to capture long-term outcomes (Table 3: Recommendations/future directions). While RCTs provide a higher level of evidence than observational studies (Brozek et al., 2009), it may not be warranted to screen only half the population given a lack of equipoise. However, RCTs could be conducted where appropriate, such as for refining the strategy of undertaking testing (e.g., comparing different models for obtaining consent or returning results).

TABLE 2. Summary of key points and gaps.
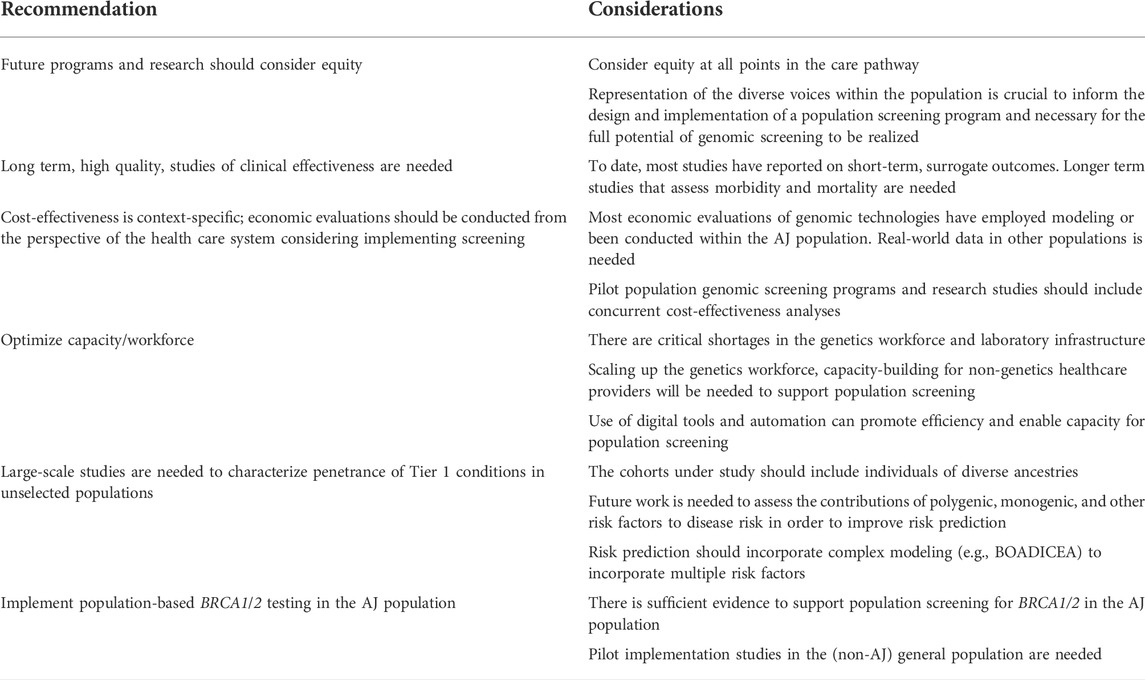
TABLE 3. Recommendations and future directions.
Acceptability
Successful implementation of population genomic screening depends on its acceptability to both the participants and providers, as it can reveal critical issues that can impact uptake, and program compliance (Screening programmes: A short guide, 2020). Much of the current evidence remains within the context of the AJ population for HBOC, which limits the transferability of these findings to the general population and for LS and FH contexts. Rigorous, evidence-based approaches to engage with the public and providers can include public deliberation (Siegel et al., 2013), discrete choice experiments (DCE) (Reed Johnson et al., 2013; Miller et al., 2015; Hauber et al., 2016; Marshall et al., 2016; Terris-Prestholt et al., 2019; Mighton et al., 2021b), or interviews and focus groups (Abelson et al., 2003). Diverse views on expectations and acceptance for the entire trajectory of population genomic screening (e.g., from invitation for screening to follow-up care) within the target jurisdiction, are required to justify the need and to inform the design and implementation of a public health program of this magnitude.
Economic evaluation
Economic evaluations of population genomic screening have had some limitations. Most have been conducted from the health system payer perspective, which is the perspective which typically informs health system decision-making. However, economic evaluations from a health system perspective do not capture out-of-pocket or indirect costs to patients and family members. More economic evaluations from societal perspectives that capture out-of-pocket and indirect costs borne by patients and family members are needed given the impact of results on relatives and their spill-over effects (Caro et al., 2012; Drummond et al., 2015; Husereau et al., 2022). Important contextual factors to consider include test costs and funding and implementation of healthcare (e.g., single-payer/universal healthcare systems vs. private health insurers). For example, in the US, where a large portion of funding is provided by various private insurers, implementation of a coordinated, public health screening program for the entire country will face challenges. Existing economic evaluations have used modeling to evaluate cost-effectiveness; yet models are limited by their assumptions and model inputs. Real-world evidence on the economic impacts of population genomic screening, is therefore needed. Furthermore, variations in cost-effectiveness thresholds exist between jurisdictions (e.g., $100,000/QALY gained). Decisions about population screening are highly context specific, and decision makers will also need to consider what the greatest public health priorities are in their jurisdiction.
Programme infrastructure and workforce
In order for population genomic screening to be feasible, there is a need to scale up the genomics workforce, build capacity among non-genetics healthcare providers, and incorporate alternative models of service delivery (Cragun et al., 2015; Peterson et al., 2020) such as mainstreaming (Hamilton et al., 2021; Scheinberg et al., 2021; McCuaig et al., 2021; Yoon et al., 2021; Piedimonte et al., 2020; O'Shea et al., 2021; Ramsey et al., 2022) and the use of digital tools (Manchanda et al., 2016; Bombard and Hayeems, 2020; Shickh et al., 2021; Lee et al., 2022). The use of digital decision support tools is particularly promising. There is increasing evidence that when combined with a brief genetic counseling session, they perform as well, if not better than traditional counseling at improving knowledge, satisfaction, risk perception, and communication between family members, while reducing time spent with HCP and costs (Manchanda et al., 2016; Bombard et al., 2020; Solomon et al., 2020; Bangash et al., 2022; Pande et al., 2022). Although tools have been developed for all three Tier 1 conditions, there are a larger number of tools, at more advanced stages of development and implementation for BRCA1/2 testing, compared to FH and Lynch syndrome (Manchanda et al., 2016; Bangash et al., 2022; Pande et al., 2022). Moreover, improvements in information technology infrastructure, bioinformatics pipelines, data security and corresponding workforce training would improve the management of population scale genetic data (Khoury et al., 2016; Kelly et al., 2021). It is critical that future research incorporates evaluations of alternative service delivery models, coordination and access of a putative population genomic screening program along with follow up care, both of which have been neglected in evaluation frameworks and the literature, but will inform the ultimate success of the programs (Andermann et al., 2008; Andermann et al., 2010; Pitini et al., 2019).
Equity
There are currently inequities in access to clinical genetics services, and any additional screening or innovations will only continue to serve populations with access to these services unless deliberate focus is placed on engagement and collaboration (Ford and Airhihenbuwa, 2010a; Ford and Airhihenbuwa, 2010b) across underserved populations. Representation of the diversity within the population is crucial to informing the design and implementation of a population screening program that is centered in the margins. Improvements in transparency, representation, and community collaboration must be prioritized at the outset (Lemke et al., 2010; Caulfield et al., 2014). Designing and implementing an accessible and inclusive population screening program offers opportunities to overcome well-characterized barriers of current genetic service models fueled by structural racism, medical distrust, and a history of eugenics (Fine et al., 2005; Ontario Ministry of Health and Long-Term Care, 2018; Fraiman and Wojcik, 2021). With more diverse participants engaging in genetic research, the diversity of genetic databases can improve, leading to more accurate variant interpretation and higher carrier identification for diverse communities (Landry et al., 2018). Until the benefits of screening are accessible to communities who have been historically underserved and marginalized, the full potential of population genomic screening cannot be realized.
Limitations
Our review has several limitations. This was not a systematic review, nor was a formal quality appraisal of studies conducted. Moreover, this review was limited to Tier 1 conditions-future research and evidence synthesis will be needed to address other actionable gene-condition pairs (e.g., other genes for hereditary breast and ovarian cancer including PALB2, RAD51C, RAD51D, and BRIP1 (Manchanda et al., 2018); TTR for hereditary transthyretin amyloidosis (Soper et al., 2021); endocrine tumour genes (Savatt et al., 2022); arrhythmia syndrome genes (Walsh et al., 2022)) and their suitability for population genomic screening.
Conclusion
Despite these limitations, our review suggests that there is evidence that population genomic screening for HBOC, LS, and FH would improve detection of individuals with pathogenic variants in the causative genes compared to traditional approaches to case ascertainment. For outcomes beyond detection rate, HBOC has the strongest support for population genomic screening, with evidence demonstrating clinical and cost-effectiveness in the general population; real world implementation studies in the general population are needed. In the AJ population, there is substantial evidence on acceptability, satisfaction, different models of implementation, psychological/quality of life outcomes, uptake of preventive strategies, and cost-effectiveness in support of population BRCA1/2 screening.
LS and FH both have preliminary evidence supporting population genomic screening, but major gaps remain in the literature. For FH, although there is evidence suggesting population genomic screening programs would have clinical and cost-effectiveness, the evidence on long-term outcomes is limited. Furthermore, the evidence on cost-effectiveness is limited to modelling studies. Real-world studies establishing cost-effectiveness and clinical effectiveness over longer follow-up periods are needed. Economic models suggest population genomic screening for LS may only be cost-effective at a very high cost-effectiveness threshold. Further evidence is critical to establish clinical effectiveness of screening for LS in asymptomatic individuals and cost-effectiveness in lower- and middle-income jurisdictions.
In addition to filling in the evidence gaps, ethical concerns such as potential overdiagnosis, as well as issues related to equity and access to testing and follow-up interventions will need to be considered at the program design stage. Adoption of population genomic screening will require major restructuring and investments to scale up the workforce, build capacity in non-genetics providers, adapt alternative delivery models (mainstreaming, digital tools), optimize IT infrastructure and prioritize an approach that is inclusive of historically underrepresented populations to ensure the full potential of population genomic screening can be realized.
Author contributions
All authors contributed to conceptualizing, writing, editing and finalizing the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abelson, J., Forest, P. G., Eyles, J., Smith, P., Martin, E., and Gauvin, F. P. (2003). Deliberations about deliberative methods: Issues in the design and evaluation of public participation processes. Soc. Sci. Med. 57 (2), 239–251. doi:10.1016/s0277-9536(02)00343-x
Abul-Husn, N. S., Manickam, K., Jones, L. K., Wright, E. A., Hartzel, D. N., Gonzaga-Jauregui, C., et al. (2016). Genetic identification of familial hypercholesterolemia within a single U.S. health care system. Science 354 (6319), aaf7000. doi:10.1126/science.aaf7000
Abul-Husn, N. S., Soper, E. R., Braganza, G. T., Rodriguez, J. E., Zeid, N., Cullina, S., et al. (2021). Implementing genomic screening in diverse populations. Genome Med. 13 (1), 17. doi:10.1186/s13073-021-00832-y
Abul-Husn, N. S., Soper, E. R., Odgis, J. A., Cullina, S., Bobo, D., Moscati, A., et al. (2019). Exome sequencing reveals a high prevalence of BRCA1 and BRCA2 founder variants in a diverse population-based biobank. Genome Med. 1212 (1), 2. doi:10.1186/s13073-019-0691-1
Ait Ouakrim, D., Dashti, S. G., Chau, R., Buchanan, D. D., Clendenning, M., Rosty, C., et al. (2015). Aspirin, Ibuprofen, and the risk of colorectal cancer in lynch syndrome. J. Natl. Cancer Inst. 107 (9), djv170. doi:10.1093/jnci/djv170
Akioyamen, L. E., Genest, J., Shan, S. D., Reel, R. L., Albaum, J. M., Chu, A., et al. (2017). Estimating the prevalence of heterozygous familial hypercholesterolaemia: A systematic review and meta-analysis. BMJ Open 7 (9), e016461. doi:10.1136/bmjopen-2017-016461
Akobeng, A. K. (2007). Understanding diagnostic tests 1: Sensitivity, specificity and predictive values. Acta Paediatr. Acta Paediatr. 96 (3), 338–341. doi:10.1111/j.1651-2227.2006.00180.x
Amendola, L. M., Muenzen, K., Biesecker, L. G., Bowling, K. M., Cooper, G. M., Dorschner, M. O., et al. (2020). Variant classification concordance using the ACMG-AMP variant interpretation guidelines across nine genomic implementation research studies. Am. J. Hum. Genet. 11107 (5), 932–941. doi:10.1016/j.ajhg.2020.09.011
Amrock, S. M., Duell, P. B., Knickelbine, T., Martin, S. S., O'Brien, E. C., Watson, K. E., et al. (2017). Health disparities among adult patients with a phenotypic diagnosis of familial hypercholesterolemia in the CASCADE-FH™ patient registry. Atherosclerosis 267, 19–26. doi:10.1016/j.atherosclerosis.2017.10.006
Andermann, A., Blancquaert, I., Beauchamp, S., and Costea, I. (2011). Guiding policy decisions for genetic screening: Developing a systematic and transparent approach. Public Health Genomics 14 (1), 9–16. doi:10.1159/000272898
Andermann, A., Blancquaert, I., Beauchamp, S., and Déry, V. (2008). Revisiting Wilson and Jungner in the genomic age: A review of screening criteria over the past 40 years. Bull. World Health Organ. 86 (4), 317–319. doi:10.2471/blt.07.050112
Andermann, A., Blancquaert, I., and Déry, V. (2010). Genetic screening: A conceptual framework for programmes and policy-making. J. Health Serv. Res. Policy 15 (2), 90–97. doi:10.1258/jhsrp.2009.009084
Antoniou, A. C., Pharoah, P. D., Narod, S., Risch, H. A., Eyfjord, J. E., Hopper, J. L., et al. (2005). Breast and ovarian cancer risks to carriers of the BRCA1 5382insC and 185delAG and BRCA2 6174delT mutations: A combined analysis of 22 population based studies. J. Med. Genet. 42 (7), 602–603. doi:10.1136/jmg.2004.024133
Armstrong, K., Micco, E., Carney, A., Stopfer, J., and Putt, M. (2005). Racial differences in the use of BRCA1/2 testing among women with a family history of breast or ovarian cancer. JAMA. 293 (14), 1729–1736. doi:10.1001/jama.293.14.1729
Bae, M. S., Sung, J. S., Bernard-Davila, B., Sutton, E. J., Comstock, C. E., and Morris, E. A. (2020). Survival outcomes of screening with breast MRI in women at elevated risk of breast cancer. J breast imaging. Radiology 2 (1), 29–30. doi:10.1148/radiol.2019192339
Baglietto, L., Lindor, N. M., Dowty, J. G., White, D. M., Wagner, A., Gomez Garcia, E. B., et al. (2010). Risks of Lynch syndrome cancers for MSH6 mutation carriers. J. Natl. Cancer Inst. 102 (3), 193–201. doi:10.1093/jnci/djp473
Bangash, H., Makkawy, A., Gundelach, J. H., Miller, A. A., Jacobson, K. A., and Kullo, I. J. (2022). Web-based tool (FH family share) to increase uptake of cascade testing for familial hypercholesterolemia: Development and evaluation. JMIR Hum. Factors 9 (1), e32568. doi:10.2196/32568
Batra, S., Valdimarsdottir, H., McGovern, M., Itzkowitz, S., and Brown, K. (2002). Awareness of genetic testing for colorectal cancer predisposition among specialists in gastroenterology. Am. J. Gastroenterol. 97 (3), 729–733. doi:10.1111/j.1572-0241.2002.05556.x
Batte, B. A., Bruegl, A. S., Daniels, M. S., Ring, K. L., Dempsey, K. M., Djordjevic, B., et al. (2014). Consequences of universal MSI/IHC in screening ENDOMETRIAL cancer patients for Lynch syndrome. Gynecol. Oncol. 134 (2), 319–325. doi:10.1016/j.ygyno.2014.06.009
Baudhuin, L. M., Lagerstedt, S. A., Klee, E. W., Fadra, N., Oglesbee, D., Ferber, M. J., et al. (2014). Confirming variants in next-generation sequencing panel testing by sanger sequencing. J. Mol. Diagn 17 (4), 456–461. doi:10.1016/j.jmoldx.2015.03.004
Berro, T., Zayhowski, K., Field, T., Channaoui, N., and Sotelo, J. (2020). Genetic counselors' comfort and knowledge of cancer risk assessment for transgender patients. J. Genet. Couns. 29 (3), 342–351. doi:10.1002/jgc4.1172
Berth, H., Balck, F., and Dinkel, A. (2002). Attitudes toward genetic testing in patients at risk for HNPCC/FAP and the German population. Genet. Test. 6 (4), 273–280. doi:10.1089/10906570260471804
Blout Zawatsky, C. L., Shah, N., Machini, K., Perez, E., Christensen, K. D., Zouk, H., et al. (2021). Returning actionable genomic results in a research biobank: Analytic validity, clinical implementation, and resource utilization. Am. J. Hum. Genet. 108 (12), 2224–2237. doi:10.1016/j.ajhg.2021.10.005
Bombard, Y., Bach, P. B., and Offit, K. (2013). Translating genomics in cancer care. J. Natl. Compr. Canc. Netw. 11 (11), 1343–1353. doi:10.6004/jnccn.2013.0158
Bombard, Y., Clausen, M., Shickh, S., Mighton, C., Casalino, S., Kim, T. H. M., et al. (2020). Effectiveness of the genomics ADvISER decision aid for the selection of secondary findings from genomic sequencing: A randomized clinical trial. Genet. Med. 22 (4), 727–735. doi:10.1038/s41436-019-0702-z
Bombard, Y., and Hayeems, R. Z. (2020). How digital tools can advance quality and equity in genomic medicine. Nat. Rev. Genet. 21 (9), 505–506. doi:10.1038/s41576-020-0260-x
Bombard, Y., and Mighton, C. (2018). Recontacting clinical genetics patients with reclassified results: Equity and policy challenges. Eur. J. Hum. Genet. 27, 505–506. doi:10.1038/s41431-018-0313-1
Bonadona, V., Bonaïti, B., Olschwang, S., Grandjouan, S., Huiart, L., Longy, M., et al. (2011). Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA. 305 (22), 2304–2310. doi:10.1001/jama.2011.743
Botkin, J. R., Teutsch, S. M., Kaye, C. I., Hayes, M., Haddow, J. E., Bradley, L. A., et al. (2010). Outcomes of interest in evidence-based evaluations of genetic tests. Genet. Med. 12 (4), 228–235. doi:10.1097/GIM.0b013e3181cdde04
Bowling, K. M., Thompson, M. L., Gray, D. E., Lawlor, J. M. J., Williams, K., East, K. M., et al. (2021). Identifying rare, medically relevant variation via population-based genomic screening in Alabama: Opportunities and pitfalls. Genet. Med. 23 (2), 280–288. doi:10.1038/s41436-020-00976-z
Bowman, F. L., Molster, C. M., Lister, K. J., Bauskis, A. T., Garton-Smith, J., Vickery, A. W., et al. (2019). Identifying perceptions and preferences of the general public concerning universal screening of children for familial hypercholesterolaemia. Public Health Genomics 22 (1-2), 25–35. doi:10.1159/000501463
Brose, M. S., Rebbeck, T. R., Calzone, K. A., Stopfer, J. E., Nathanson, K. L., and Weber, B. L. (2002). Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. J. Natl. Cancer Inst. 94 (18), 1365–1372. doi:10.1093/jnci/94.18.1365
Brozek, J. L., Akl, E. A., Alonso-Coello, P., Lang, D., Jaeschke, R., Williams, J. W., et al. (2009). Grading quality of evidence and strength of recommendations in clinical practice guidelines. Part 1 of 3. An overview of the GRADE approach and grading quality of evidence about interventions. Allergy 64 (5), 669–677. doi:10.1111/j.1398-9995.2009.01973.x
Buchanan, A. H., Lester Kirchner, H., Schwartz, M. L. B., Kelly, M. A., Schmidlen, T., Jones, L. K., et al. (2020). Clinical outcomes of a genomic screening program for actionable genetic conditions. Genet. Med. 1122 (11), 1874–1882. doi:10.1038/s41436-020-0876-4
Burke, W., Parens, E., Chung, W. K., Berger, S. M., and Appelbaum, P. S. (1920). The challenge of genetic variants of uncertain clinical significance : A narrative review. Ann. Intern. Med. 175, 994–1000. doi:10.7326/M21-4109
Burke, W., and Zimmern, R. (2007). Moving beyond ACCE: An expanded framework for genetic test evaluation. Cambridge, United Kingdom: PHG Foundation. Available at: https://www.phgfoundation.org/report/moving-beyond-acce-an-expanded-framework-for-genetic-test-evaluation.
Burn, J., Gerdes, A. M., Macrae, F., Mecklin, J. P., Moeslein, G., Olschwang, S., et al. (2011). Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: An analysis from the CAPP2 randomised controlled trial. Lancet 378 (9809), 2081–2087. doi:10.1016/S0140-6736(11)61049-0
Burnell, M., Gaba, F., Sobocan, M., Desai, R., Sanderson, S., Loggenberg, K., et al. (2022). Randomised trial of population‐based BRCA testing in Ashkenazi jews: Long‐term secondary lifestyle behavioural outcomes. BJOG. doi:10.1111/1471-0528.17253
Carey, D. J., Fetterolf, S. N., Davis, F. D., Faucett, W. A., Kirchner, H. L., Mirshahi, U., et al. (2016). The Geisinger MyCode community health initiative: An electronic health record-linked biobank for precision medicine research. Genet. Med. 18 (9), 906–913. doi:10.1038/gim.2015.187
Caro, J. J., Briggs, A. H., Siebert, U., and Kuntz, K. M. (2012). Modeling good research practices--overview: A report of the ISPOR-SMDM modeling good research practices task force-1. Med. Decis. Mak. 32 (5), 667–677. doi:10.1177/0272989X12454577
Carroll, J. C., Cappelli, M., Miller, F., Wilson, B. J., GrunfEld, E., Peeters, C., et al. (2008). Genetic services for hereditary breast/ovarian and colorectal cancers - physicians' awareness, use and satisfaction. Community Genet. 11 (1), 43–51. doi:10.1159/000111639
Caulfield, T., Burningham, S., Joly, Y., Master, Z., Shabani, M., Borry, P., et al. (2014). A review of the key issues associated with the commercialization of biobanks. J. Law Biosci. 1 (1), 94–110. doi:10.1093/jlb/lst004
Centers for Disease Control and Prevention OoPHG (2022). Tier 1 genomics applications and their importance to public health.
Chandak, A., Nayar, P., and Lin, G. (2019). Rural-urban disparities in access to breast cancer screening: A Spatial Clustering analysis. J. Rural. Health 35 (2), 229–235. doi:10.1111/jrh.12308
Chatterjee, N., Kalaylioglu, Z., Shih, J. H., and Gail, M. H. (2006). Case-control and case-only designs with genotype and family history data: Estimating relative risk, residual familial aggregation, and cumulative risk. Biometrics 62 (1), 36–48. doi:10.1111/j.1541-0420.2005.00442.x
Chatterjee, N., Shih, J., Hartge, P., Brody, L., Tucker, M., and Wacholder, S. (2001). Association and aggregation analysis using kin-cohort designs with applications to genotype and family history data from the Washington Ashkenazi Study. Genet. Epidemiol. 21 (2), 123–138. doi:10.1002/gepi.1022
Chatterjee, N., and Wacholder, S. (2001). A marginal likelihood approach for estimating penetrance from kin-cohort designs. Biometrics 57 (1), 245–252. doi:10.1111/j.0006-341x.2001.00245.x
Chen, J., Bae, E., Zhang, L., Hughes, K., Parmigiani, G., Braun, D., et al. (2020). Penetrance of breast and ovarian cancer in women who carry a BRCA1/2 mutation and do not use risk-reducing salpingo-oophorectomy: An updated meta-analysis. JNCI Cancer Spectr. 4 (4), pkaa029. doi:10.1093/jncics/pkaa029
Chiang, J., Chia, T. H., Yuen, J., Shaw, T., Li, S. T., Binte Ishak, N. D., et al. (2021). Impact of variant reclassification in cancer predisposition genes on clinical care. JCO Precis. Oncol. (5), 577–584. doi:10.1200/po.20.00399
Cragun, D., Bonner, D., Kim, J., Akbari, M. R., Narod, S. A., Gomez-Fuego, A., et al. (2015). Factors associated with genetic counseling and BRCA testing in a population-based sample of young Black women with breast cancer. Breast Cancer Res. Treat. 151 (1), 169–176. doi:10.1007/s10549-015-3374-7
Crosbie, E. J., Ryan, N. A. J., Arends, M. J., Bosse, T., Burn, J., Cornes, J. M., et al. (2019). The Manchester International Consensus Group recommendations for the management of gynecological cancers in Lynch syndrome. Genet. Med. 21 (10), 2390–2400. doi:10.1038/s41436-019-0489-y
Daly, M. B., Pilarski, R., Yurgelun, M. B., Berry, M. P., Buys, S. S., Dickson, P., et al. (2020). NCCN guidelines Insights: Genetic/familial high-risk assessment: Breast, ovarian, and pancreatic, version 1.2020. J. Natl. Compr. Canc. Netw. 18 (4), 380–391. doi:10.6004/jnccn.2020.0017
De Simone, L. M., Arjunan, A., Vogel Postula, K. J., Maga, T., and Bucheit, L. A. (2021). Genetic counselors' perspectives on population-based screening for BRCA-related hereditary breast and ovarian cancer and Lynch syndrome. J. Genet. Couns. 30 (1), 158–169. doi:10.1002/jgc4.1305
Defesche, J. C., Gidding, S. S., Harada-Shiba, M., Hegele, R. A., Santos, R. D., and Wierzbicki, A. S. (2017). Familial hypercholesterolaemia. Nat. Rev. Dis. Prim. 3, 17093. doi:10.1038/nrdp.2017.93
Dinh, T. A., Rosner, B. I., Atwood, J. C., Boland, C. R., Syngal, S., Vasen, H. F. A., et al. (2011). Health benefits and cost-effectiveness of primary genetic screening for Lynch syndrome in the general population. Cancer Prev. Res. 4 (1), 9–22. doi:10.1158/1940-6207.CAPR-10-0262
Dove-Edwin, I., Boks, D., Goff, S., Kenter, G. G., Carpenter, R., Vasen, H. F. A., et al. (2002). The outcome of endometrial carcinoma surveillance by ultrasound scan in women at risk of hereditary nonpolyposis colorectal carcinoma and familial colorectal carcinoma. Cancer 94 (6), 1708–1712. doi:10.1002/cncr.10380
Dragojlovic, N., Borle, K., Kopac, N., Ellis, U., Birch, P., Adam, S., et al. (2020). The composition and capacity of the clinical genetics workforce in high-income countries: A scoping review. Genet. Med. 22 (9), 1437–1449. doi:10.1038/s41436-020-0825-2
Drummond, M., Sculpher, M., Claxton, K., Stoddart, G., and Torrance, G. (2015). Methods for the economic evaluation of health care programmes. 4th ed. Oxford University Press.
Dunlop, K., Rankin, N. M., Smit, A. K., Salgado, Z., Newson, A. J., Keogh, L., et al. (2021). Acceptability of risk-stratified population screening across cancer types: Qualitative interviews with the Australian public. Health Expect. 24 (4), 1326–1336. doi:10.1111/hex.13267
Ehrenberg, S., Walsh Vockley, C., Nelson, E., Baker, J., Arcieri, M., Lindenberger, J., et al. (2021). Under-referral of Plain community members for genetic services despite being qualified for genetic evaluation. J. Genet. Couns. 30, 1084–1090. doi:10.1002/jgc4.1395
El Mecky, J., Johansson, L., Plantinga, M., Fenwick, A., Lucassen, A., Dijkhuizen, T., et al. (2019). Reinterpretation, reclassification, and its downstream effects: Challenges for clinical laboratory geneticists. BMC Med. Genomics 1112 (1), 170. doi:10.1186/s12920-019-0612-6 29
Elhanan, G., Kiser, D., Neveux, I., Dabe, S., Bolze, A., Metcalf, W. J., et al. (2022). Incomplete penetrance of population-based genetic screening results in electronic health record. Front. Genet. 13, 866169. doi:10.3389/fgene.2022.866169
Esterling, L., Wijayatunge, R., Brown, K., Morris, B., Hughes, E., Pruss, D., et al. (2020). Impact of a cancer gene variant reclassification program over a 20-year period. JCO Precis. Oncol. 4doi, 944–954. doi:10.1200/PO.20.00020
Fahed, A. C., Wang, M., Homburger, J. R., Patel, A. P., Bick, A. G., Neben, C. L., et al. (2020). Polygenic background modifies penetrance of monogenic variants for tier 1 genomic conditions. Nat. Commun. 11 (1), 3635. doi:10.1038/s41467-020-17374-3
Finch, A. P., Lubinski, J., Moller, P., Singer, C. F., Karlan, B., Senter, L., et al. (2014). Impact of oophorectomy on cancer incidence and mortality in women with a BRCA1 or BRCA2 mutation. J. Clin. Oncol. 32 (15), 1547–1553. doi:10.1200/JCO.2013.53.2820
Fine, M. J., Ibrahim, S. A., and Thomas, S. B. (2005). The role of race and genetics in health disparities research. Am. J. Public Health 95 (12), 2125–2128. doi:10.2105/AJPH.2005.076588
Ford, C. L., and Airhihenbuwa, C. O. (2010). Critical race theory, race equity, and public health: Toward antiracism praxis. Am. J. Public Health 100 (1), S30–S35. doi:10.2105/AJPH.2009.171058
Ford, C. L., and Airhihenbuwa, C. O. (2010). The public health critical race methodology: Praxis for antiracism research. Soc. Sci. Med. 71 (8), 1390–1398. doi:10.1016/j.socscimed.2010.07.030
Forrest, I. S., Chaudhary, K., Vy, H. M. T., Petrazzini, B. O., Bafna, S., Jordan, D. M., et al. (2022). Population-based penetrance of deleterious clinical variants. JAMA 327 (4), 350–359. doi:10.1001/jama.2021.23686
Foss, K. S., O’Daniel, J. M., and Berg, J. S. (2022). The rise of population genomic screening: Characteristics of current programs and the need for evidence regarding optimal implementation. J. Pers. Med. 12 (5), 692. doi:10.3390/jpm12050692
Foulkes, W. D., Knoppers, B. M., and Turnbull, C. (2016). Population genetic testing for cancer susceptibility: Founder mutations to genomes. Nat. Rev. Clin. Oncol. 13 (1), 41–54. doi:10.1038/nrclinonc.2015.173
Fraiman, Y. S., and Wojcik, M. H. (2021). The influence of social determinants of health on the genetic diagnostic odyssey: Who remains undiagnosed, why, and to what effect? Pediatr. Res. 89 (2), 295–300. doi:10.1038/s41390-020-01151-5
Gaba, F., Blyuss, O., Liu, X., Goyal, S., Lahoti, N., Chandrasekaran, D., et al. (2020). Population study of ovarian cancer risk prediction for targeted screening and prevention. Cancers (Basel) 12 (5), E1241. doi:10.3390/cancers12051241
Gaba, F., Oxley, S., Liu, X., Yang, X., Chandrasekaran, D., Kalsi, J., et al. (2022). Unselected population genetic testing for personalised ovarian cancer risk prediction: A qualitative study using semi-structured interviews. Diagn. (Basel) 12 (5), 1028. doi:10.3390/diagnostics12051028
Gabai-Kapara, E., Lahad, A., Kaufman, B., Friedman, E., Segev, S., Renbaum, P., et al. (2014). Population-based screening for breast and ovarian cancer risk due to BRCA1 and BRCA2. Proc. Natl. Acad. Sci. U. S. A. 111 (39), 14205–14210. doi:10.1073/pnas.1415979111
Gaff, C. L., Clarke, A. J., Atkinson, P., Sivell, S., Elwyn, G., Iredale, R., et al. (2007). Process and outcome in communication of genetic information within families: A systematic review. Eur. J. Hum. Genet. 15 (10), 999–1011. doi:10.1038/sj.ejhg.5201883
Gao, Y., Goldberg, J. E., Young, T. K., Babb, J. S., Moy, L., and Heller, S. L. (2019). Breast cancer screening in high-risk men: A 12-year longitudinal observational study of male breast imaging utilization and outcomes. Radiology 11293 (2), 282–291. doi:10.1148/radiol.2019190971
Garber, K. B., Vincent, L. M., Alexander, J. J., Bean, L. J. H., Bale, S., and Hegde, M. (2016). Reassessment of genomic sequence variation to harmonize interpretation for personalized medicine. Am. J. Hum. Genet. 99 (5), 1140–1149. doi:10.1016/j.ajhg.2016.09.015
Genomics England (2021). Newborn Genomes programme. Genomics England. Available at: https://www.genomicsengland.co.uk/initiatives/newborns.
Giardiello, F. M., Allen, J. I., Axilbund, J. E., Boland, C. R., Burke, C. A., Burt, R. W., et al. (2014). Guidelines on genetic evaluation and management of lynch syndrome: A consensus statement by the US multi-society task force on colorectal cancer. Am. J. Gastroenterol. 109 (8), 1159–1179. doi:10.1038/ajg.2014.186
Gidding, S. S., Champagne, M. A., de Ferranti, S. D., Defesche, J., Ito, M. K., Knowles, J. W., et al. (2015). The agenda for familial hypercholesterolemia: A scientific statement from the American heart association. Circulation 132 (22), 2167–2192. doi:10.1161/CIR.0000000000000297
Graham, I. D., Logan, D. M., Hughes-Benzie, R., Evans, W. K., Perras, H., McAuley, L. M., et al. (1998). How interested is the public in genetic testing for colon cancer susceptibility? Report of a cross-sectional population survey. Cancer Prev. Control 2 (4), 167–172.
Gronwald, J., Tung, N., Foulkes, W. D., Offit, K., Gershoni, R., Daly, M., et al. (2006). Tamoxifen and contralateral breast cancer in BRCA1 and BRCA2 carriers: An update. Int. J. Cancer 118 (9), 2281–2284. doi:10.1002/ijc.21536
Groselj, U., Kovac, J., Sustar, U., Mlinaric, M., Fras, Z., Podkrajsek, K. T., et al. (2018). Universal screening for familial hypercholesterolemia in children: The Slovenian model and literature review. Atherosclerosis 277, 383–391. doi:10.1016/j.atherosclerosis.2018.06.858
Grzymski, J. J., Elhanan, G., Morales Rosado, J. A., Smith, E., Schlauch, K. A., Read, R., et al. (2020). Population genetic screening efficiently identifies carriers of autosomal dominant diseases. Nat. Med. 26 (8), 1235–1239. doi:10.1038/s41591-020-0982-5
Gupta, S., Provenzale, D., Llor, X., Halverson, A. L., Grady, W., Chung, D. C., et al. (2019). NCCN guidelines Insights: Genetic/familial high-risk assessment: Colorectal, version 2.2019. J. Natl. Compr. Canc. Netw. 17 (9), 1032–1041. doi:10.6004/jnccn.2019.0044
Guzauskas, G. F., Garbett, S., Zhou, Z., Spencer, S. J., Smith, H. S., Hao, J., et al. (2020). Cost-effectiveness of population-wide genomic screening for hereditary breast and ovarian cancer in the United States. JAMA Netw. Open 3 (10), e2022874. doi:10.1001/jamanetworkopen.2020.22874
Guzauskas, G. F., Jiang, S., Garbett, S., Zhou, Z., Spencer, S. J., Snyder, S. R., et al. (2022). Cost-effectiveness of population-wide genomic screening for Lynch syndrome in the United States. Genet. Med. 24, 1017–1026. doi:10.1016/j.gim.2022.01.017
Haga, S. B., Kim, E., Myers, R. A., and Ginsburg, G. S. (2019). Primary care physicians' knowledge, attitudes, and experience with personal genetic testing. J. Pers. Med. 9 (2), E29. doi:10.3390/jpm9020029
Hagenkord, J., Funke, B., Qian, E., Hegde, M., Jacobs, K. B., Ferber, M., et al. (2020). Design and reporting considerations for genetic screening tests. J. Mol. Diagn. 22 (5), 599–609. doi:10.1016/j.jmoldx.2020.01.014
Hamilton, J. G., Abdiwahab, E., Edwards, H. M., Fang, M. L., Jdayani, A., and Breslau, E. S. (2017). Primary care providers' cancer genetic testing-related knowledge, attitudes, and communication behaviors: A systematic review and research agenda. J. Gen. Intern. Med. 32 (3), 315–324. doi:10.1007/s11606-016-3943-4
Hamilton, J. G., Symecko, H., Spielman, K., Breen, K., Mueller, R., Catchings, A., et al. (2021). Uptake and acceptability of a mainstreaming model of hereditary cancer multigene panel testing among patients with ovarian, pancreatic, and prostate cancer. Genet. Med. 1123 (11), 2105–2113. doi:10.1038/s41436-021-01262-2
Hampel, H., Frankel, W. L., Martin, E., Arnold, M., Khanduja, K., Kuebler, P., et al. (2008). Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J. Clin. Oncol. 26 (35), 5783–5788. doi:10.1200/JCO.2008.17.5950
Hann, K. E. J., Freeman, M., Fraser, L., Waller, J., Sanderson, S. C., Rahman, B., et al. (2017). Awareness, knowledge, perceptions, and attitudes towards genetic testing for cancer risk among ethnic minority groups: A systematic review. BMC Public Health 17 (1), 503. doi:10.1186/s12889-017-4375-8
Harrison, S. M., Dolinsky, J. S., Knight Johnson, A. E., Pesaran, T., Azzariti, D. R., Bale, S., et al. (2017). Clinical laboratories collaborate to resolve differences in variant interpretations submitted to ClinVar. Genet. Med. 19 (10), 1096–1104. doi:10.1038/gim.2017.14
Hauber, A. B., González, J. M., Groothuis-Oudshoorn, C. G., Prior, T., Marshall, D. A., Cunningham, C., et al. (2016). Statistical methods for the analysis of discrete choice experiments: A report of the ISPOR conjoint analysis good research practices task force. Value Health 19 (4), 300–315. doi:10.1016/j.jval.2016.04.004
Hauser, D., Obeng, A. O., Fei, K., Ramos, M. A., and Horowitz, C. R. (2018). Views of primary care providers on testing patients for genetic risks for common chronic diseases. Health Aff. 37 (5), 793–800. doi:10.1377/hlthaff.2017.1548
Hegde, M., Ferber, M., Mao, R., Samowitz, W., and Ganguly, A. (2014). ACMG technical standards and guidelines for genetic testing for inherited colorectal cancer (Lynch syndrome, familial adenomatous polyposis, and MYH-associated polyposis). Genet. Med. 16 (1), 101–116. doi:10.1038/gim.2013.166
Hendricks-Sturrup, R. M., and Lu, C. Y. (2019). Understanding implementation challenges to genetic testing for familial hypercholesterolemia in the United States. J. Pers. Med. 9 (1). doi:10.3390/jpm9010009
Honold, F., and Camus, M. (2018). Prophylactic mastectomy versus surveillance for the prevention of breast cancer in women's BRCA carriers. Medwave 18 (4), e7161. doi:10.5867/medwave.2018.04.7160
Hoskovec, J. M., Bennett, R. L., Carey, M. E., DaVanzo, J. E., DoughertyM., , Hahn, S. E., et al. (2018). Projecting the supply and demand for certified genetic counselors: A workforce study. J. Genet. Couns. 27 (1), 16–20. doi:10.1007/s10897-017-0158-8
Husereau, D., Drummond, M., Augustovski, F., de Bekker-Grob, E., Briggs, A. H., Carswell, C., et al. (2022). Consolidated health economic evaluation reporting standards (CHEERS) 2022 explanation and elaboration: A report of the ISPOR CHEERS II good practices task force. Value Health. 25 (1), 10–31. doi:10.1016/j.jval.2021.10.008
Iacocca, M. A., Chora, J. R., Carrié, A., Freiberger, T., Leigh, S. E., Defesche, J. C., et al. (2018). ClinVar database of global familial hypercholesterolemia-associated DNA variants. Hum. Mutat. 1139 (11), 1631–1640. doi:10.1002/humu.23634
Ison, H. E., Clarke, S. L., and Knowles, J. W. (2014). “Familial hypercholesterolemia,” in GeneReviews® [Internet]. Editors M. P. Adam, D. B. Everman, and G. M. Mirzaa (Seattle, WA: University of Washington, Seattle), 1993–2022. Available at: https://www.ncbi.nlm.nih.gov/books/NBK174884/
Idos, G., and Valle, L. (2004). “Lynch Syndrome,” in GeneReviews® [Internet]. Editors M. P. Adam, D. B. Everman, and G. M. Mirzaa (Seattle, WA: University of Washington, Seattle), 1993–2022. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1211/
Jacobs, I. J., Menon, U., Ryan, A., Gentry-Maharaj, A., Burnell, M., Kalsi, J. K., et al. (2016). Ovarian cancer screening and mortality in the UK collaborative trial of ovarian cancer screening (UKCTOCS): A randomised controlled trial. Lancet 387 (10022), 945–956. doi:10.1016/S0140-6736(15)01224-6
Järvinen, H. J., Renkonen-Sinisalo, L., Aktán-Collán, K., Peltomäki, P., Aaltonen, L. A., and Mecklin, J. P. (2009). Ten years after mutation testing for lynch syndrome: Cancer incidence and outcome in mutation-positive and mutation-negative family members. J. Clin. Oncol. 27 (28), 4793–4797. doi:10.1200/JCO.2009.23.7784
Jenkins, M. A., Dowty, J. G., Ait Ouakrim, D., Mathews, J. D., Hopper, J. L., Drouet, Y., et al. (2015). Short-term risk of colorectal cancer in individuals with lynch syndrome: A meta-analysis. J. Clin. Oncol. 33 (4), 326–331. doi:10.1200/JCO.2014.55.8536
Joly, Y., Feze, I. N., Song, L., and Knoppers, B. M. (2017). Comparative approaches to genetic discrimination: Chasing Shadows? Trends Genet. 33 (5), 299–302. doi:10.1016/j.tig.2017.02.002
Judkins, T., Leclair, B., Bowles, K., Gutin, N., Trost, J., McCulloch, J., et al. (2015). Development and analytical validation of a 25-gene next generation sequencing panel that includes the BRCA1 and BRCA2 genes to assess hereditary cancer risk BMC Cancer 15 (5), 299–302. doi:10.1186/s12885-015-1224-y
Kahn, R. M., Gordhandas, S., Maddy, B. P., Baltich Nelson, B., Askin, G., Christos, P. J., et al. (2019). Universal endometrial cancer tumor typing: How much has immunohistochemistry, microsatellite instability, and MLH1 methylation improved the diagnosis of Lynch syndrome across the population? Cancer 125 (18), 3172–3183. doi:10.1002/cncr.32203
Kastelein, J. J., Ginsberg, H. N., Langslet, G., Hovingh, G. K., Ceska, R., Dufour, R., et al. (2015). ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur. Heart J. 1436 (43), 2996–3003. doi:10.1093/eurheartj/ehv370
Kelly, M. A., Leader, J. B., Wain, K. E., Bodian, D., Oetjens, M. T., Ledbetter, D. H., et al. (2021). Leveraging population-based exome screening to impact clinical care: The evolution of variant assessment in the Geisinger MyCode research project. Am. J. Med. Genet. C Semin. Med. Genet. 187 (1), 83–94. doi:10.1002/ajmg.c.31887
Kerner, J., Liu, J., Wang, K., Fung, S., Landry, C., Lockwood, G., et al. (2015). Canadian cancer screening disparities: A recent historical perspective. Curr. Oncol. 22 (2), 156–163. doi:10.3747/co.22.2539
Khoury, M. J., Iademarco, M. F., and Riley, W. T. (2016). Precision public health for the era of precision medicine. Am. J. Prev. Med. 50 (3), 398–401. doi:10.1016/j.amepre.2015.08.031
Kim, H., Ho, C. W. L., Ho, C. H., Athira, P. S., Kato, K., De Castro, L., et al. (2021). Genetic discrimination: Introducing the asian perspective to the debate. NPJ Genom. Med. 6 (1), 54. doi:10.1038/s41525-021-00218-4
Klančar, G., Grošelj, U., and Kovač, J. (2015). Universal screening for familial hypercholesterolemia in children. J. Am. Coll. Cardiol. 66 (11), 1250–1257. doi:10.1016/j.jacc.2015.07.017
Klitzman, R., Chung, W., Marder, K., Shanmugham, A., Chin, L. J., Stark, M., et al. (2013). Attitudes and practices among internists concerning genetic testing. J. Genet. Couns. 22 (1), 90–100. doi:10.1007/s10897-012-9504-z
Kriege, M., Brekelmans, C. T., Boetes, C., Besnard, P. E., Zonderland, H. M., Obdeijn, I. M., et al. (2004). Efficacy of MRI and mammography for breast-cancer screening in women with a familial or genetic predisposition. N. Engl. J. Med. 351 (5), 427–437. doi:10.1056/NEJMoa031759
Kuchenbaecker, K. B., Hopper, J. L., Barnes, D. R., Phillips, K. A., Mooij, T. M., Roos-Blom, M. J., et al. (2017). Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 317 (23), 2402–2416. doi:10.1001/jama.2017.7112
Kuhl, C. K., Schrading, S., Leutner, C. C., Morakkabati-Spitz, N., Wardelmann, E., Fimmers, R., et al. (2005). Mammography, breast ultrasound, and magnetic resonance imaging for surveillance of women at high familial risk for breast cancer. J. Clin. Oncol. 23 (33), 8469–8476. doi:10.1200/JCO.2004.00.4960
Kunnackal John, G., Das Villgran, V., Caufield-Noll, C., and Giardiello, F. (2021). Worldwide variation in lynch syndrome screening: Case for universal screening in low colorectal cancer prevalence areas. Fam. Cancer 20 (2), 145–156. doi:10.1007/s10689-020-00206-0
Lacaze, P., Tiller, J., and Winship, I. (2019). Healthcare system-funded preventive genomic screening: Challenges for Australia and other single-payer systems. Public Health Genomics 22 (3-4), 140–144. doi:10.1159/000502917
Lacaze, P. A., Tiller, J., Winship, I., and Group, D. S. I. (2022). Population DNA screening for medically actionable disease risk in adults. Med. J. Aust. 216 (6), 278–280. doi:10.5694/mja2.51454
Ladabaum, U., Ford, J. M., Martel, M., and Barkun, A. N. (2015). American gastroenterological association technical review on the diagnosis and management of lynch syndrome. Gastroenterology 149 (3), 783–813. e20. doi:10.1053/j.gastro.2015.07.037
Laforest, F., Kirkegaard, P., Mann, B., and Edwards, A. (2019). Genetic cancer risk assessment in general practice: Systematic review of tools available, clinician attitudes, and patient outcomes. Br. J. Gen. Pract. 69 (679), e97–e105. doi:10.3399/bjgp18X700265
Lahtinen, A. M., Havulinna, A. S., Jula, A., Salomaa, V., and Kontula, K. (2015). Prevalence and clinical correlates of familial hypercholesterolemia founder mutations in the general population. Atherosclerosis 238 (1), 64–69. doi:10.1016/j.atherosclerosis.2014.11.015
Landry, L. G., Ali, N., Williams, D. R., Rehm, H. L., and Bonham, V. L. (2018). Lack of diversity in genomic databases is A barrier to translating precision medicine research into practice. Health Aff. 37 (5), 780–785. doi:10.1377/hlthaff.2017.1595
Landry, L. G., and Rehm, H. L. (2018). Association of racial/ethnic categories with the ability of genetic tests to detect a cause of cardiomyopathy. JAMA Cardiol. 3 (4), 341–345. doi:10.1001/jamacardio.2017.5333
Lázaro, P., Pérez de Isla, L., Watts, G. F., Alonso, R., Norman, R., Muniz, O., et al. (20172017). Cost-effectiveness of a cascade screening program for the early detection of familial hypercholesterolemia. J. Clin. Lipidol. 11 (1), 260–271. doi:10.1016/j.jacl.2017.01.002
Leach, M. O., Boggis, C. R., Dixon, A. K., Easton, D. F., Eeles, R. A., Evans, D. G. R., et al. (20052005). Screening with magnetic resonance imaging and mammography of a UK population at high familial risk of breast cancer: A prospective multicentre cohort study (MARIBS). Lancet 365 (9473), 1769–1778. doi:10.1016/S0140-6736(05)66481-1
Lebo, M. S., Zakoor, K. R., Chun, K., Speevak, M. D., Waye, J. S., McCready, E., et al. (2018). Data sharing as a national quality improvement program: Reporting on BRCA1 and BRCA2 variant-interpretation comparisons through the Canadian open genetics Repository (COGR). Genet. Med. 20 (3), 294–302. doi:10.1038/gim.2017.80
Lee, C., Elsekaily, O., Kochan, D. C., Alhalabi, L., Faizee, F., Sharp, R., et al. (2021). Penetrance and outcomes at 1-year following return of actionable variants identified by genome sequencing. Genet. Med. 23 (7), 1192–1201. doi:10.1038/s41436-021-01142-9
Lee, C., Rivera-Valerio, M., Bangash, H., Prokop, L., and Kullo, I. J. (2019). New case detection by cascade testing in familial hypercholesterolemia: A systematic review of the literature. Circ. Genom. Precis. Med. 1112 (11), e002723. doi:10.1161/CIRCGEN.119.002723
Lee, C. Y., Yen, H. Y., Zhong, A. W., and Gao, H. (2021). Resolving misalignment interference for NGS-based clinical diagnostics. Hum. Genet. 140 (3), 477–492. doi:10.1007/s00439-020-02216-5
Lee, W., Shickh, S., Assamad, D., Luca, S., Clausen, M., Somerville, C., et al. (2022). Patient-facing digital tools for delivering genetic services: a systematic review. J. Med. Genet.. doi:10.1136/jmg-2022-108653
Lehman, C. D., Lee, J. M., DeMartini, W. B., Hippe, D. S., Rendi, M. H., Kalish, G., et al. (2016). Screening MRI in women with a personal history of breast cancer. J. Natl. Cancer Inst. 108 (3), djv349. doi:10.1093/jnci/djv349
Lehmann, L. S., Weeks, J. C., Klar, N., and Garber, J. E. (20022002). A population-based study of Ashkenazi Jewish women's attitudes toward genetic discrimination and BRCA1/2 testing. Genet. Med. 4 (5), 346–352. doi:10.1097/00125817-200209000-00005
Lemke, A. A., Wolf, W. A., Hebert-Beirne, J., and Smith, M. E. (2010). Public and biobank participant attitudes toward genetic research participation and data sharing. Public Health Genomics 13 (6), 368–377. doi:10.1159/000276767
Levine, D. A., Argenta, P. A., Yee, C. J., Marshall, D. S., Olvera, N., Bogomolniy, F., et al. (2003). Fallopian tube and primary peritoneal carcinomas associated with BRCA mutations. J. Clin. Oncol. 21 (22), 4222–4227. doi:10.1200/JCO.2003.04.131
Li, J., Dai, H., Feng, Y., Tang, J., Chen, S., Tian, X., et al. (2015). A comprehensive strategy for accurate mutation detection of the highly homologous PMS2. J. Mol. Diagn. 17 (5), 545–553. doi:10.1016/j.jmoldx.2015.04.001
Li, X., You, R., Wang, X., Liu, C., Xu, Z., Zhou, J., et al. (2016). Effectiveness of prophylactic surgeries in BRCA1 or BRCA2 mutation carriers: A meta-analysis and systematic review. Clin. Cancer Res. 22 (15), 3971–3981. doi:10.1158/1078-0432.CCR-15-1465
Lieberman, S., Lahad, A., Tomer, A., Cohen, C., Levy-Lahad, E., and Raz, A. (2017). Population screening for BRCA1/BRCA2 mutations: Lessons from qualitative analysis of the screening experience. Genet. Med. 19 (6), 628–634. doi:10.1038/gim.2016.175
Lieberman, S., Tomer, A., Ben-Chetrit, A., Olsha, O., Strano, S., Beeri, R., et al. (2017). Population screening for BRCA1/BRCA2 founder mutations in Ashkenazi Jews: Proactive recruitment compared with self-referral. Genet. Med. 19 (7), 754–762. doi:10.1038/gim.2016.182
Lin, J. S., Perdue, L. A., Henrikson, N. B., Bean, S. I., and Blasi, P. R. (2021). Screening for colorectal cancer: Updated evidence report and systematic review for the US preventive services task force. JAMA 325 (19), 1978–1998. doi:10.1001/jama.2021.4417
Linderman, M. D., Nielsen, D. E., and Green, R. C. (2016). Personal genome sequencing in Ostensibly Healthy individuals and the PeopleSeq Consortium. J. Pers. Med. 6 (2), E14. doi:10.3390/jpm6020014
Lindor, N. M., McMaster, M. L., Lindor, C. J., and Greene, M. H. (2008). Concise handbook of familial cancer susceptibility syndromes - second edition. JNCI Monogr. (38), 3–93. doi:10.1093/jncimonographs/lgn001
Lindor, N. M., Petersen, G. M., Hadley, D. W., Kinney, A. Y., Miesfeldt, S., Lu, K. H., et al. (2006). Recommendations for the care of individuals with an inherited predisposition to lynch syndrome: A systematic review. JAMA. 296 (12), 1507–1517. doi:10.1001/jama.296.12.1507
Lowry, K. P., Lee, J. M., Kong, C. Y., McMahon, P. M., Gilmore, M. E., Cott Chubiz, J. E., et al. (2012). Annual screening strategies in BRCA1 and BRCA2 gene mutation carriers: A comparative effectiveness analysis. Cancer 118 (8), 2021–2030. doi:10.1002/cncr.26424
Lozano, P., Henrikson, N. B., and Dunn, J. (2016). Lipid screening in childhood and adolescence for detection of familial hypercholesterolemia: A systematic evidence review for the U.S. Preventive services task force.
Lozano, P., Henrikson, N. B., Dunn, J., Morrison, C. C., Nguyen, M., Blasi, P. R., et al. (2016). Lipid screening in childhood and adolescence for detection of familial hypercholesterolemia: Evidence report and systematic review for the US preventive services task force. JAMA. 316 (6), 645–655. doi:10.1001/jama.2016.6176
Lu, J. T., Ferber, M., Hagenkord, J., Levin, E., South, S., Kang, H. P., et al. (2019). Evaluation for genetic Disorders in the absence of a clinical indication for testing: Elective genomic testing. J. Mol. Diagn. 21 (1), 3–12. doi:10.1016/j.jmoldx.2018.09.006
Lynch, H. T., Casey, M. J., Snyder, C. L., Bewtra, C., Lynch, J. F., Butts, M., et al. (2009). Hereditary ovarian carcinoma: Heterogeneity, molecular genetics, pathology, and management. Mol. Oncol. 3 (2), 97–137. doi:10.1016/j.molonc.2009.02.004
Macklin, S., Durand, N., Atwal, P., and Hines, S. (2018). Observed frequency and challenges of variant reclassification in a hereditary cancer clinic. Genet. Med. 20 (3), 346–350. doi:10.1038/gim.2017.207
Maddison, A. R., Asada, Y., and Urquhart, R. (2011). Inequity in access to cancer care: A review of the Canadian literature. Cancer Causes Control 22 (3), 359–366. doi:10.1007/s10552-010-9722-3
Manchanda, R., Burnell, M., Gaba, F., Desai, R., Wardle, J., GeSSler, S., et al. (2020). Randomised trial of population-based BRCA testing in Ashkenazi jews: Long-term outcomes. BJOG. 127 (3), 364–375. doi:10.1111/1471-0528.15905
Manchanda, R., Burnell, M., Gaba, F., SanderSon, S., Loggenberg, K., GeSSler, S., et al. (2019). Attitude towards and factors affecting uptake of population-based BRCA testing in the Ashkenazi jewish population: A cohort study. BJOG. 126 (6), 784–794. doi:10.1111/1471-0528.15654
Manchanda, R., Burnell, M., Loggenberg, K., Desai, R., Wardle, J., Sanderson, S. C., et al. (2016). Cluster-randomised non-inferiority trial comparing DVD-assisted and traditional genetic counselling in systematic population testing for BRCA1/2 mutations. J. Med. Genet. 53 (7), 472–480. doi:10.1136/jmedgenet-2015-103740
Manchanda, R., Gaba, F., Talaulikar, V., Pundir, J., Gessler, S., Davies, M., et al. (2022). Risk-reducing salpingo-oophorectomy and the use of hormone replacement therapy below the age of natural menopause: Scientific impact Paper No. 66 october 2021: Scientific impact paper No. 66. BJOG 129 (1), e16–e34. doi:10.1111/1471-0528.16896
Manchanda, R., Legood, R., Burnell, M., McGuire, A., Raikou, M., Loggenberg, K., et al. (2015). Cost-effectiveness of population screening for BRCA mutations in Ashkenazi jewish women compared with family history-based testing. J. Natl. Cancer Inst. 107 (1), 380. doi:10.1093/jnci/dju380
Manchanda, R., Lieberman, S., Gaba, F., Lahad, A., and Levy-Lahad, E. (2020). Population screening for inherited predisposition to breast and ovarian cancer. Annu. Rev. Genomics Hum. Genet. 3121, 373–412. doi:10.1146/annurev-genom-083118-015253
Manchanda, R., Loggenberg, K., Sanderson, S., Burnell, M., Wardle, J., Gessler, S., et al. (2015). Population testing for cancer predisposing BRCA1/BRCA2 mutations in the ashkenazi-jewish community: A randomized controlled trial. J. Natl. Cancer Inst. 107 (1), 379. doi:10.1093/jnci/dju379
Manchanda, R., Patel, S., Antoniou, A. C., Levy-Lahad, E., Turnbull, C., Evans, D. G., et al. (2017). Cost-effectiveness of population based BRCA testing with varying Ashkenazi Jewish ancestry. Am. J. Obstet. Gynecol. 11217 (5), e1–e1578. e12. doi:10.1016/j.ajog.2017.06.038
Manchanda, R., Patel, S., Gordeev, V. S., Antoniou, A. C., Smith, S., Lee, A., et al. (2018). Cost-effectiveness of population-based BRCA1, BRCA2, RAD51C, RAD51D, BRIP1, PALB2 mutation testing in unselected general population women. J. Natl. Cancer Inst. 110 (7), 714–725. doi:10.1093/jnci/djx265
Manchanda, R., Sun, L., Patel, S., Evans, O., Wilschut, J., De Freitas Lopes, A. C., et al. (2020). Economic evaluation of population-based BRCA1/BRCA2 mutation testing across multiple countries and health systems. Cancers (Basel) 12 (7), E1929. doi:10.3390/cancers12071929
Manickam, K., Buchanan, A. H., Schwartz, M. L. B., Hallquist, M. L. G., Williams, J. L., Rahm, A. K., et al. (2018). Exome sequencing-based screening for BRCA1/2 expected pathogenic variants among adult biobank participants. JAMA Netw. Open 1 (5), e182140. doi:10.1001/jamanetworkopen.2018.2140
Manickam, K., McClain, M. R., Demmer, L. A., Biswas, S., Kearney, H. M., Malinowski, J., et al. (2021). Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: An evidence-based clinical guideline of the American College of medical genetics and genomics (ACMG). Genet. Med. 1123 (11), 2029–2037. doi:10.1038/s41436-021-01242-6
Mann, R. M., Kuhl, C. K., and Moy, L. (2019). Contrast-enhanced MRI for breast cancer screening. J. Magn. Reson. Imaging 50 (2), 377–390. doi:10.1002/jmri.26654
Manrai, A. K., Funke, B. H., Rehm, H. L., Olesen, M. S., Maron, B. A., Szolovits, P., et al. (2016). Genetic misdiagnoses and the potential for health disparities. N. Engl. J. Med. 375 (7), 655–665. doi:10.1056/NEJMsa1507092
Marks, D., Wonderling, D., Thorogood, M., Lambert, H., Humphries, S. E., and Neil, H. A. (2002). Cost effectiveness analysis of different approaches of screening for familial hypercholesterolaemia. BMJ 324 (7349), 1303. doi:10.1136/bmj.324.7349.1303
Marquina, C., Lacaze, P., Tiller, J., Riaz, M., Sturm, A. C., Nelson, M. R., et al. (2021). Population genomic screening of young adults for familial hypercholesterolaemia: A cost-effectiveness analysis. Eur. Heart J. 11, 3243–3254. doi:10.1093/eurheartj/ehab770
Marshall, D. A., Deal, K., Bombard, Y., Leighl, N., MacDonald, K. V., and Trudeau, M. (2016). How do women trade-off benefits and risks in chemotherapy treatment decisions based on gene expression profiling for early-stage breast cancer? A discrete choice experiment. BMJ Open 6 (6), e010981. doi:10.1136/bmjopen-2015-010981
Matsunaga, K., Mizobuchi, A., Fu, H. Y., Ishikawa, S., Tada, H., Kawashiri, M. A., et al. (2021). Universal screening for familial hypercholesterolemia in children in kagawa, Japan. J. Atheroscler. Thromb. 29, 839–849. doi:10.5551/jat.62780
Mavaddat, N., Peock, S., Frost, D., Ellis, S., Platte, R., Fineberg, E., et al. (2013). Cancer risks for BRCA1 and BRCA2 mutation carriers: Results from prospective analysis of EMBRACE. J. Natl. Cancer Inst. 105 (11), 812–822. doi:10.1093/jnci/djt095
Maynard, A. (2013). Health care rationing: Doing it better in public and private health care systems. J. Health Polit. Policy Law 38 (6), 1103–1127. doi:10.1215/03616878-2373157
McCuaig, J. M., Thain, E., Malcolmson, J., Keshavarzi, S., Armel, S. R., and Kim, R. H. (2021). A comparison of patient-reported outcomes following consent for genetic testing using an oncologist- or genetic counselor-mediated model of care. Curr. Oncol. 28 (2), 1459–1471. doi:10.3390/curroncol28020138
McKay, S., Humphris, J., Johns, A., Gill, A., and Tucker, K. (2016). Inherited pancreatic cancer. Cancer Forum 40 (1).
Meisel, S. F., Rahman, B., Side, L., Fraser, L., Gessler, S., Lanceley, A., et al. (2016). Genetic testing and personalized ovarian cancer screening: A survey of public attitudes. BMC Womens Health 16, 46. doi:10.1186/s12905-016-0325-3 26
Menon, U., Gentry-Maharaj, A., Burnell, M., Singh, N., Ryan, A., Karpinskyj, C., et al. (2021). Ovarian cancer population screening and mortality after long-term follow-up in the UK collaborative trial of ovarian cancer screening (UKCTOCS): A randomised controlled trial. Lancet 397 (10290), 2182–2193. doi:10.1016/S0140-6736(21)00731-5
Menon, U., Gentry-Maharaj, A., Hallett, R., Ryan, A., Burnell, M., Sharma, A., et al. (2009). Sensitivity and specificity of multimodal and ultrasound screening for ovarian cancer, and stage distribution of detected cancers: Results of the prevalence screen of the UK collaborative trial of ovarian cancer screening (UKCTOCS). Lancet. Oncol. 10 (4), 327–340. doi:10.1016/S1470-2045(09)70026-9
Menzin, A. W., Anderson, B. L., Williams, S. B., and Schulkin, J. (2010). Education and experience with breast health maintenance and breast cancer care: A study of obstetricians and gynecologists. J. Cancer Educ. 25 (1), 87–91. doi:10.1007/s13187-009-0019-8
Mersch, J., Brown, N., Pirzadeh-Miller, S., Mundt, E., Cox, H. C., Brown, K., et al. (2018). Prevalence of variant reclassification following hereditary cancer genetic testing. Jama 320 (12), 1266–1274. doi:10.1001/jama.2018.13152
Meshkani, Z., Aboutorabi, A., Moradi, N., Langarizadeh, M., and Motlagh, A. G. (2021). Population or family history based BRCA gene tests of breast cancer? A systematic review of economic evaluations. Hered. Cancer Clin. Pract. 19 (1), 35. doi:10.1186/s13053-021-00191-0
Metcalfe, K. A., Mian, N., Enmore, M., Poll, A., Llacuachaqui, M., Nanda, S., et al. (2012). Long-term follow-up of Jewish women with a BRCA1 and BRCA2 mutation who underwent population genetic screening. Breast Cancer Res. Treat. 133 (2), 735–740. doi:10.1007/s10549-011-1941-0
Metcalfe, K. A., Poll, A., Llacuachaqui, M., TulmAn, A., MiaNN., , et al. (2010). Patient satisfaction and cancer-related distress among unselected Jewish women undergoing genetic testing for BRCA1 and BRCA2. Clin. Genet. 78 (5), 411–417. doi:10.1111/j.1399-0004.2010.01499.x
Metcalfe, K. A., Poll, A., Royer, R., Llacuachaqui, M., Tulman, A., Sun, P., et al. (2010). Screening for founder mutations in BRCA1 and BRCA2 in unselected Jewish women. J. Clin. Oncol. 28 (3), 387–391. doi:10.1200/JCO.2009.25.0712
Metcalfe, K. A., Poll, A., Royer, R., LlacuachaquiM., , Sun, P., et al. (2013). A comparison of the detection of BRCA mutation carriers through the provision of Jewish population-based genetic testing compared with clinic-based genetic testing. Br. J. Cancer 109 (3), 777–779. doi:10.1038/bjc.2013.309
Metcalfe, K. A., Price, M. A., Mansfield, C., Hallett, D. C., Lindeman, G. J., Fairchild, A., et al. (2020). Predictors of long-term cancer-related distress among female BRCA1 and BRCA2 mutation carriers without a cancer diagnosis: An international analysis. Br. J. Cancer 123 (2), 268–274. doi:10.1038/s41416-020-0861-3
Mighton, C., Charames, G., Wang, M., Zakoor, K. R., Wong, A., Shickh, S., et al. (2019). Variant classification changes over time in BRCA1 and BRCA2. Genet. Med. 21 (10), 2248–2254. doi:10.1038/s41436-019-0493-2
Mighton, C., Clausen, M., Sebastian, A., Muir, S. M., Shickh, S., Baxter, N. N., et al. (2021). Patient and public preferences for being recontacted with updated genomic results: A mixed methods study. Hum. Genet. 140, 1695–1708. doi:10.1007/s00439-021-02366-0
Mighton, C., Shickh, S., Uleryk, E., Pechlivanoglou, P., and Bombard, Y. (2021). Clinical and psychological outcomes of receiving a variant of uncertain significance from multigene panel testing or genomic sequencing: A systematic review and meta-analysis. Genet. Med. 23 (1), 22–33. doi:10.1038/s41436-020-00957-2
Mighton, C., Smith, A. C., Mayers, J., Tomaszewski, R., Taylor, S., Hume, S., et al. (2021). Data sharing to improve concordance in variant interpretation across laboratories: Results from the Canadian open genetics repository. J. Med. Genet. 59, 571–578. doi:10.1136/jmedgenet-2021-107738
Miller, F. A., Hayeems, R. Z., Bombard, Y., Cressman, C., Barg, C. J., Carroll, J. C., et al. (2015). Public perceptions of the benefits and risks of newborn screening. Pediatrics 136 (2), e413–e423. doi:10.1542/peds.2015-0518
Moran, A., O'Hara, C., Khan, S., Woodward, E., Maher, E. R., et al. (2012). Risk of cancer other than breast or ovarian in individuals with BRCA1 and BRCA2 mutations. Fam. Cancer 11 (2), 235–242. doi:10.1007/s10689-011-9506-2
Morgan, K. M., Hamilton, J. G., Symecko, H., Kamara, D., Jenkins, C., Lester, J., et al. (2021). Targeted BRCA1/2 population screening among Ashkenazi Jewish individuals using a web-enabled medical model: An observational cohort study. Genet. Med. 24, 564–575. doi:10.1016/j.gim.2021.10.016
Muller, C., Lee, S. M., Barge, W., Siddique, S. M., Berera, S., Wideroff, G., et al. (2018). Low referral rate for genetic testing in racially and ethnically diverse patients despite universal colorectal cancer screening. Clin. Gastroenterol. Hepatol. 1216 (12), 1911–1918. e2. doi:10.1016/j.cgh.2018.08.038
Murray, M. F., Giovanni, M. A., Doyle, D. L., Harrison, S. M., Lyon, E., Manickam, K., et al. (2021). DNA-Based screening and population health: A points to consider statement for programs and sponsoring organizations from the American College of medical genetics and genomics (ACMG). Genet. Med. 23 (6), 989–995. doi:10.1038/s41436-020-01082-w
Murray, M. F. (2016). Your DNA is not your diagnosis: Getting diagnoses right following secondary genomic findings. Genet. Med. 18 (8), 765–767. doi:10.1038/gim.2015.134
Narod, S. A., Brunet, J. S., Ghadirian, P., RobsonM., , Heimdal, K., Neuhausen, S. L., et al. (2000). Tamoxifen and risk of contralateral breast cancer in BRCA1 and BRCA2 mutation carriers: A case-control study. Hereditary breast cancer clinical study group. Lancet 356 (9245), 1876–1881. doi:10.1016/s0140-6736(00)03258-x
National Academies of Sciences Engineering and Medicine (2017). Health and medicine Division, board on health care services, board on the health of Select populations, Committee on the evidence base for genetic testing. An evidence framework for genetic testing. Washington, DC: National Academies Press (US).
National Comprehensive Cancer Network (NCCN) (2021). NCCN clinical practice guidelines in Oncology. Genetic/familial high-risk Assessment: Breast, ovarian, and pancreatic. National Comprehensive Cancer Network NCCN. Version 1.2022.
National Comprehensive Cancer Network (NCCN) (2021). NCCN clinical practice guidelines in oncology. Genetic/familial high-risk assessment: Colorectal. Version 1.2021. National Comprehensive Cancer Network NCCN.
Ndugga-Kabuye, M. K., and Issaka, R. B. (2019). Inequities in multi-gene hereditary cancer testing: Lower diagnostic yield and higher VUS rate in individuals who identify as hispanic, african or asian and pacific islander as compared to European. Fam. Cancer 18 (4), 465–469. doi:10.1007/s10689-019-00144-6
Nelson, H. D., Pappas, M., Cantor, A., Griffin, J., Daeges, M., and Humphrey, L. (2016). Harms of breast cancer screening: Systematic review to update the 2009 U.S. Preventive services task force recommendation. Ann. Intern. Med. 164 (4), 256–267. doi:10.7326/M15-0970
Nguyen-Pham, S., Leung, J., and McLaughlin, D. (2014). Disparities in breast cancer stage at diagnosis in urban and rural adult women: A systematic review and meta-analysis. Ann. Epidemiol. 24 (3), 228–235. doi:10.1016/j.annepidem.2013.12.002
Nherera, L., Marks, D., Minhas, R., Thorogood, M., and Humphries, S. E. (2011). Probabilistic cost-effectiveness analysis of cascade screening for familial hypercholesterolaemia using alternative diagnostic and identification strategies. Heart 97 (14), 1175–1181. doi:10.1136/hrt.2010.213975
O'Shea, R., Taylor, N., Crook, A., Jacobs, C., Jung Kang, Y., Lewis, S., et al. (2021). Health system interventions to integrate genetic testing in routine oncology services: A systematic review. PLoS One 16 (5), e0250379. doi:10.1371/journal.pone.0250379
Office of the Auditor General (2017). Annual report: Section 3.07 laboratory services in the health sector. Toronto, ON: Ontario Ministry of Health and Long-term Care.
Oliva, J., López-Bastida, J., Moreno, S. G., Mata, P., and Alonso, R. (2009). Cost-effectiveness analysis of a genetic screening program in the close relatives of Spanish patients with familial hypercholesterolemia. Rev. Espanola Cardiol. 62 (1), 57–65. doi:10.1016/s1885-5857(09)71514-2
Ontario Ministry of Health and Long-Term Care (2018). The Ontario public health standards: Requirements for programs, services, and accountability are published as the public health standards for the provision of mandatory health programs and services by the minister of health and long-term care, pursuant to section 7 of the health protection and promotion act.. Toronto, ON: Ontario Ministry of Health and Long-Term Care.
Otten, E., Plantinga, M., Birnie, E., Verkerk, M. A., Lucassen, A. M., Ranchor, A. V., et al. (2015). Is there a duty to recontact in light of new genetic technologies? A systematic review of the literature. Genet. Med. 17 (8), 668–678. doi:10.1038/gim.2014.173
Pande, M., Peterson, S., and Lynch, P. M. (2022). Development and evaluation of an online, patient-driven, family outreach intervention to facilitate sharing of genetic risk information in families with Lynch syndrome. J. Med. Genet. 59 (6), 589–596. doi:10.1136/jmedgenet-2020-107615
Pang, J., Sullivan, D. R., Brett, T., Kostner, K. M., Hare, D. L., and Watts, G. F. (2020). Familial hypercholesterolaemia in 2020: A leading tier 1 genomic application. Heart Lung Circ. 29 (4), 619–633. doi:10.1016/j.hlc.2019.12.002
Pelczarska, A., Jakubczyk, M., Jakubiak-Lasocka, J., Banach, M., Mysliwiec, M., Gruchala, M., et al. (2018). The cost-effectiveness of screening strategies for familial hypercholesterolaemia in Poland. Atherosclerosis 270, 132–138. doi:10.1016/j.atherosclerosis.2018.01.036
Peterson, J. M., Pepin, A., Thomas, R., Biagi, T., Stark, E., Sparks, A. D., et al. (2020). Racial disparities in breast cancer hereditary risk assessment referrals. J. Genet. Couns. 29 (4), 587–593. doi:10.1002/jgc4.1250
Petrucelli, N., Daly, M. B., and Pal, T. (1998). “BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer,” in GeneReviews® [Internet]. Editors M. P. Adam, D. B. Everman, and G. M. Mirzaa (Seattle, WA: University of Washington, Seattle), 1993–2022. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1247/
Petrucelli, N., Daly, M., and Pal, T. (2022). Chap BRCA1- and BRCA2-associated hereditary breast and ovarian cancer. GeneReviews.
Piedimonte, S., Power, J., Foulkes, W. D., Weber, E., Palma, L., Schiavi, A., et al. (2020). BRCA testing in women with high-grade serous ovarian cancer: Gynecologic oncologist-initiated testing compared with genetics referral. Int. J. Gynecol. Cancer 1130 (11), 1757–1761. doi:10.1136/ijgc-2020-001261
Pijpe, A., Andrieu, N., Easton, D. F., Kesminiene, A., Cardis, E., Nogues, C., et al. (2012). Exposure to diagnostic radiation and risk of breast cancer among carriers of BRCA1/2 mutations: Retrospective cohort study (GENE-RAD-RISK). BMJ 345, e5660. doi:10.1136/bmj.e5660
Pitini, E., D'Andrea, E., De Vito, C., Rosso, A., Unim, B., Marzuillo, C., et al. (2019). A proposal of a new evaluation framework towards implementation of genetic tests. PLoS One 14 (8), e0219755. doi:10.1371/journal.pone.0219755
Ponti, G., Castellsagué, E., Ruini, C., Percesepe, A., and Tomasi, A. (2015). Mismatch repair genes founder mutations and cancer susceptibility in Lynch syndrome. Clin. Genet. 87 (6), 507–516. doi:10.1111/cge.12529
Popejoy, A. B., Crooks, K. R., Fullerton, S. M., Hindorff, L. A., Hooker, G. W., Koenig, B. A., et al. (2020). Clinical genetics lacks standard definitions and protocols for the collection and use of diversity measures. Am. J. Hum. Genet. 107 (1), 72–82. doi:10.1016/j.ajhg.2020.05.005
Popejoy, A. B., and Fullerton, S. M. (2016). Genomics is failing on diversity. Nature 538 (7624), 161–164. doi:10.1038/538161a
Purificacion, S. J., French, J. G., and d'Agincourt-Canning, L. (2015). Inequities in access to cancer care in Canada: An ethical perspective. Healthc. Manage. Forum 28 (6), 265–269. doi:10.1177/0840470415599136
Ramsey, M. L., Tomlinson, J., Pearlman, R., Abushahin, L., Aeilts, A., Chen, H. Z., et al. (2022). Mainstreaming germline genetic testing for patients with pancreatic cancer increases uptake. Fam. Cancer. doi:10.1007/s10689-022-00300-5
Reed Johnson, F., Lancsar, E., Marshall, D., Kilambi, V., Muhlbacher, A., Regier, D. A., et al. (20132013). Constructing experimental designs for discrete-choice experiments: Report of the ISPOR conjoint analysis experimental design good research practices task force. Value Health 16 (1), 3–13. doi:10.1016/j.jval.2012.08.2223
Reisel, D., Burnell, M., Side, L., Loggenberg, K., Gessler, S., Desai, R., et al. (2022). Jewish cultural and religious factors and uptake of population-based BRCA testing across denominations: A cohort study. BJOG. 129 (6), 959–968. doi:10.1111/1471-0528.16994
Rew, L., Kaur, M., McMillan, A., Mackert, M., and Bonevac, D. (2010). Systematic review of psychosocial benefits and harms of genetic testing. Issues Ment. Health Nurs. 31 (10), 631–645. doi:10.3109/01612840.2010.510618
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Ridic, G., Gleason, S., and Ridic, O. (2012). Comparisons of health care systems in the United States, Germany and Canada. Mater. Sociomed. 24 (2), 112–120. doi:10.5455/msm.2012.24.112-120
Riedl, C. C., Ponhold, L., Flöry, D., Weber, M., Kroiss, R., Wagner, T., et al. (2007). Magnetic resonance imaging of the breast improves detection of invasive cancer, preinvasive cancer, and premalignant lesions during surveillance of women at high risk for breast cancer. Clin. Cancer Res. 13 (20), 6144–6152. doi:10.1158/1078-0432.CCR-07-1270
Roa, B. B., Boyd, A. A., Volcik, K., and Richards, C. S. (1996). Ashkenazi Jewish population frequencies for common mutations in BRCA1 and BRCA2. Nat. Genet. 14 (2), 185–187. doi:10.1038/ng1096-185
Rolle, L., Zayhowski, K., Koeller, D., Chiluiza, D., and Carmichael, N. (2021). Transgender patients' perspectives on their cancer genetic counseling experiences. J. Genet. Couns. 31, 781–791. doi:10.1002/jgc4.1544
Rosenthal, E., Moyes, K., Arnell, C., Evans, B., and Wenstrup, R. J. (2015). Incidence of BRCA1 and BRCA2 non-founder mutations in patients of Ashkenazi Jewish ancestry. Breast Cancer Res. Treat. 149 (1), 223–227. doi:10.1007/s10549-014-3218-x
Rowley, S. M., Mascarenhas, L., Devereux, L., Li, N., Amarasinghe, K. C., Zethoven, M., et al. (2019). Population-based genetic testing of asymptomatic women for breast and ovarian cancer susceptibility. Genet. Med. 21 (4), 913–922. doi:10.1038/s41436-018-0277-0
Rubenstein, J. H., Enns, R., Heidelbaugh, J., Barkun, A., and Committee, C. G. (2015). American Gastroenterological association Institute guideline on the diagnosis and management of lynch syndrome. Gastroenterology. Gastroenterology 149 (3), 777–782. quiz e16-7. doi:10.1053/j.gastro.2015.07.036
Rubinsak, L. A., Kleinman, A., Quillin, J., Gordon, S. W., Sullivan, S. A., Sutton, A. L., et al. (2019). Awareness and acceptability of population-based screening for pathogenic BRCA variants: Do race and ethnicity matter? Gynecol. Oncol. 154 (2), 383–387. doi:10.1016/j.ygyno.2019.06.009
Sanderson, S., Zimmern, R., Kroese, M., Higgins, J., Patch, C., and Emery, J. (2005). How can the evaluation of genetic tests be enhanced? Lessons learned from the ACCE framework and evaluating genetic tests in the United Kingdom. Genet. Med. 7 (7), 495–500. doi:10.1097/01.gim.0000179941.44494.73
Sardanelli, F., Podo, F., D'Agnolo, G., Verdecchia, A., Santaquilani, M., Musumeci, R., et al. (2007). Multicenter comparative multimodality surveillance of women at genetic-familial high risk for breast cancer (HIBCRIT study): Interim results. Radiology 242 (3), 698–715. doi:10.1148/radiol.2423051965
Savatt, J. M., Ortiz, N. M., Thone, G. M., McDonald, W. S., Kelly, M. A., Berry, A. S. F., et al. (2022). Observational study of population genomic screening for variants associated with endocrine tumor syndromes in a large, healthcare-based cohort. BMC Med. 20 (1), 205. doi:10.1186/s12916-022-02375-4
Scheinberg, T., Young, A., Woo, H., Goodwin, A., Mahon, K. L., and Horvath, L. G. (2021). Mainstream consent programs for genetic counseling in cancer patients: A systematic review. Asia. Pac. J. Clin. Oncol. 17 (3), 163–177. doi:10.1111/ajco.13334
Schmeler, K. M., Lynch, H. T., Chen, L. M., Munsell, M. F., Soliman, P. T., Clark, M. B., et al. (2006). Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N. Engl. J. Med. 354 (3), 261–269. doi:10.1056/NEJMoa052627
Schofield, L., Grieu, F., Amanuel, B., Carrello, A., Spagnolo, D., Kiraly, C., et al. (2014). Population-based screening for lynch syndrome in Western Australia. Int. J. Cancer 135 (5), 1085–1091. doi:10.1002/ijc.28744
Schwartz, M. L. B., McCormick, C. Z., Lazzeri, A. L., Lindbuchler, D. M., Hallquist, M. L. G., Manickam, K., et al. (2018). A model for genome-first care: Returning secondary genomic findings to participants and their healthcare providers in a large research cohort. Am. J. Hum. Genet. 103 (3), 328–337. doi:10.1016/j.ajhg.2018.07.009
Screening programmes: a short guide (2020). Increase effectiveness, maximize benefits and minimize harm. Copenhagen: WHO Regional Office for Europe.
Senter, L., Clendenning, M., Sotamaa, K., Hampel, H., Green, J., Potter, J. D., et al. (2008). The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology 135 (2), 419–428. doi:10.1053/j.gastro.2008.04.026
Shickh, S., Rafferty, S. A., Clausen, M., Kodida, R., Mighton, C., Panchal, S., et al. (2021). The role of digital tools in the delivery of genomic medicine: Enhancing patient-centered care. Genet. Med. 23 (6), 1086–1094. doi:10.1038/s41436-021-01112-1
Shkedi-Rafid, S., Ofer-Bialer, G., Meiner, V., and Calderon-Margalit, R. (2013). Clinicians' attitudes toward general screening of the Ashkenazi-Jewish population for prevalent founder BRCA1/2 and LRRK2 mutations. Public Health Genomics 16 (4), 174–183. doi:10.1159/000351592
Siegel, J. E., Heeringa, J. W., and Carman, K. L. (2013). Public deliberation in decisions about health research. Virtual Mentor 15 (1), 56–64. doi:10.1001/virtualmentor.2013.15.1.pfor2-1301
Sirchia, F., Carrieri, D., Dheensa, S., Benjamin, C., Kayserili, H., Cordier, C., et al. (2018). Recontacting or not recontacting? A survey of current practices in clinical genetics centres in Europe. Eur. J. Hum. Genet. 26 (7), 946–954. doi:10.1038/s41431-018-0131-5
Slavin, T. P., Manjarrez, S., Pritchard, C. C., Gray, S., and Weitzel, J. N. (2019). The effects of genomic germline variant reclassification on clinical cancer care. Oncotarget 10 (4), 417–423. doi:10.18632/oncotarget.26501
Slavin, T. P., Van Tongeren, L. R., Behrendt, C. E., Solomon, I., Rybak, C., Nehoray, B., et al. (2018). Prospective study of cancer genetic variants: Variation in rate of reclassification by ancestry. J. Natl. Cancer Inst. 110, 1059–1066. doi:10.1093/jnci/djy027
Smith, A. J., Turner, E. L., and Kinra, S. (20162016). Universal cholesterol screening in childhood: A systematic review. Acad. Pediatr. 16 (8), 716–725. doi:10.1016/j.acap.2016.06.005
Solano, A. R., Liria, N. C., Jalil, F. S., Faggionato, D. M., Mele, P. G., Mampel, A., et al. (2018). BRCA1 and BRCA2 mutations other than the founder alleles among Ashkenazi jewish in the population of Argentina. Front. Oncol. 8, 323. doi:10.3389/fonc.2018.00323
Solomon, I. B., McGraw, S., Shen, J., Albayrak, A., Alterovitz, G., Davies, M., et al. (2020). Engaging patients in precision oncology: Development and usability of a web-based patient-facing genomic sequencing report. JCO Precis. Oncol. 4doi, 307–318. doi:10.1200/PO.19.00195
Soper, E. R., Suckiel, S. A., Braganza, G. T., Kontorovich, A. R., Kenny, E. E., and Abul-Husn, N. S. (2021). Genomic screening identifies individuals at high risk for hereditary transthyretin amyloidosis. J. Pers. Med. 11 (1), 49. doi:10.3390/jpm11010049
Stjepanovic, N., Moreira, L., Carneiro, F., BalaguerF., , CervAntes, A., Balmana, J., et al. (2019). Hereditary gastrointestinal cancers: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 30 (10), 1558–1571. doi:10.1093/annonc/mdz233
Stoll, K., Kubendran, S., and Cohen, S. A. (2018). The past, present and future of service delivery in genetic counseling: Keeping up in the era of precision medicine. Am. J. Med. Genet. C Semin. Med. Genet. 178 (1), 24–37. doi:10.1002/ajmg.c.31602
Suther, S., and Kiros, G. E. (2009). Barriers to the use of genetic testing: A study of racial and ethnic disparities. Genet. Med. 11 (9), 655–662. doi:10.1097/GIM.0b013e3181ab22aa
Ten Haaf, K., Jeon, J., Tammemägi, M. C., Han, S. S., Kong, C. Y., Plevritis, S. K., et al. (2017). Risk prediction models for selection of lung cancer screening candidates: A retrospective validation study. PLoS Med. 14 (4), e1002277. doi:10.1371/journal.pmed.1002277
Terris-Prestholt, F., Neke, N., Grund, J. M., Plotkin, M., Kuringe, E., Osaki, H., et al. (2019). Using discrete choice experiments to inform the design of complex interventions. Trials 20 (1), 157. doi:10.1186/s13063-019-3186-x
Teutsch, S. M., Bradley, L. A., Palomaki, G. E., Haddow, J. E., Piper, M., Calonge, N., et al. (2009). The evaluation of genomic applications in practice and prevention (EGAPP) initiative: Methods of the EGAPP working group. Genet. Med. 11 (1), 3–14. doi:10.1097/GIM.0b013e318184137c
The Breast Cancer Linkage Consortium (1999). Cancer risks in BRCA2 mutation carriers. J. Natl. Cancer Inst. 91 (15), 1310–1316. doi:10.1093/jnci/91.15.1310
Tischkowitz, M., Balmaña, J., Foulkes, W. D., James, P., Ngeow, J., Schmutzler, R., et al. (2021). Management of individuals with germline variants in PALB2: A clinical practice resource of the American college of medical genetics and genomics (ACMG). Genet. Med. 23 (8), 1416–1423. doi:10.1038/s41436-021-01151-8
Tognetto, A., Michelazzo, M. B., Calabró, G. E., Unim, B., Di Marco, M., Ricciardi, W., et al. (2017). A systematic review on the existing screening pathways for lynch syndrome identification. Front. Public Health 5, 243. doi:10.3389/fpubh.2017.00243
Toland, A. E., Forman, A., Couch, F. J., Culver, J. O., Eccles, D. M., Foulkes, W. D., et al. (2018). Clinical testing of BRCA1 and BRCA2: A worldwide snapshot of technological practices. NPJ Genom. Med. 3, 7. doi:10.1038/s41525-018-0046-7
Turner, S. A., Rao, S. K., Morgan, R. H., Vnencak-Jones, C. L., and Wiesner, G. L. (2018). The impact of variant classification on the clinical management of hereditary cancer syndromes. Genet. Med. 21, 426–430. doi:10.1038/s41436-018-0063-z
Villegas, C., and Haga, S. B. (2019). Access to genetic counselors in the Southern United States. J. Pers. Med. 9 (3), E33. doi:10.3390/jpm9030033
Vohnout, B., Gabcova, D., Huckova, M., Klimes, I., Gasperikova, D., and Raslova, K. (2016). Genetic testing of familial hypercholesterolemia in a real clinical setting. Wien. Klin. Wochenschr. 128 (23-24), 916–921. doi:10.1007/s00508-016-1053-2
Wade, C. H. (2019). What is the psychosocial impact of providing genetic and genomic health information to individuals? An overview of systematic reviews. Hastings Cent. Rep. 49 (1), S88–S96. doi:10.1002/hast.1021
Wade, C. H., Wilfond, B. S., and McBride, C. M. (2010). Effects of genetic risk information on children's psychosocial wellbeing: A systematic review of the literature. Genet. Med. 12 (6), 317–326. doi:10.1097/GIM.0b013e3181de695c
Wald, D. S., Bestwick, J. P., Morris, J. K., Whyte, K., Jenkins, L., and Wald, N. J. (2016). Child-parent familial hypercholesterolemia screening in primary care. N. Engl. J. Med. 10375 (17), 1628–1637. doi:10.1056/NEJMoa1602777 27
Walsh, R., Bezzina, C., and Wilde, A. A. M. (2022). First steps of population genomic medicine in the arrhythmia world: Pros and Cons. Circulation 145 (12), 892–895. doi:10.1161/CIRCULATIONAHA.122.058738
Walsh, T., Mandell, J. B., Norquist, B. M., Casadei, S., Gulsuner, S., Lee, M. K., et al. (2017). Genetic predisposition to breast cancer due to mutations other than BRCA1 and BRCA2 founder alleles among Ashkenazi jewish women. JAMA Oncol. 3 (12), 1647–1653. doi:10.1001/jamaoncol.2017.1996
Warner, E., Messersmith, H., Causer, P., Eisen, A., Shumak, R., and Plewes, D. (2008). Systematic review: Using magnetic resonance imaging to screen women at high risk for breast cancer. Ann. Intern. Med. 148 (9), 671–679. doi:10.7326/0003-4819-148-9-200805060-00007
Warner, E., Plewes, D. B., Hill, K. A., Causer, P. A., Zubovits, J. T., Jong, R. A., et al. (2004). Surveillance of BRCA1 and BRCA2 mutation carriers with magnetic resonance imaging, ultrasound, mammography, and clinical breast examination. JAMA. 292 (11), 1317–1325. doi:10.1001/jama.292.11.1317
Warner, E., Zhu, S., Plewes, D. B., Hill, K., Ramsay, E. A., Causer, P. A., et al. (2020). Breast cancer mortality among women with a BRCA1 or BRCA2 mutation in a magnetic resonance imaging Plus mammography screening program. Cancers 23 (11), E3479. doi:10.3390/cancers12113479
Watkins, K. E., Way, C. Y., Fiander, J. J., Meadus, R. J., Esplen, M. J., Green, J. S., et al. (2011). Lynch syndrome: Barriers to and facilitators of screening and disease management. Hered. Cancer Clin. Pract. 9, 8. doi:10.1186/1897-4287-9-8
Watts, G. F., Sullivan, D. R., Hare, D. L., Kostner, K. M., Horton, A. E., Bell, D. A., et al. (2021). Essentials of a new clinical practice guidance on familial hypercholesterolaemia for physicians. Intern. Med. J. 51 (5), 769–779. doi:10.1111/imj.15327
Wauters, A., and Van Hoyweghen, I. (2016). Global trends on fears and concerns of genetic discrimination: A systematic literature review. J. Hum. Genet. 61 (4), 275–282. doi:10.1038/jhg.2015.151
Wilson, J. M., and Jungner, Y. G. (1968). Principles and practice of mass screening for disease. Bol. Oficina Sanit. Panam. 65 (4), 281–393.
Wonderling, D., Umans-Eckenhausen, M. A., Marks, D., Defesche, J. C., Kastelein, J. J., and Thorogood, M. (2004). Cost-effectiveness analysis of the genetic screening program for familial hypercholesterolemia in The Netherlands. Semin. Vasc. Med. 4 (1), 97–104. doi:10.1055/s-2004-822992
Yoon, S. Y., Wong, S. W., Lim, J., Ahmad, S., Mariapun, S., Padmanabhan, H., et al. (2021). Oncologist-led BRCA counselling improves access to cancer genetic testing in middle-income Asian country, with no significant impact on psychosocial outcomes. J. Med. Genet. 59, 220–229. doi:10.1136/jmedgenet-2020-107416
Yorkshire Cancer Research (2022). Yorkshire Cancer Research announces £7.3 million in funding for new research. Harrogate, United Kingdom: Yorkshire Cancer Research. Availlable at: https://yorkshirecancerresearch.org.uk/news/7-3-million-in-funding-for-new-research.
Yurgelun, M. B., Hiller, E., and Garber, J. E. (2015). Population-Wide screening for germline BRCA1 and BRCA2 mutations: Too much of a good thing? J. Clin. Oncol. 33 (28), 3092–3095. doi:10.1200/JCO.2015.60.8596
Zhang, L., Bao, Y., Riaz, M., Tiller, J., Liew, D., Zhuang, X., et al. (2019). Population genomic screening of all young adults in a health-care system: A cost-effectiveness analysis. Genet. Med. 21 (9), 1958–1968. doi:10.1038/s41436-019-0457-6
Keywords: population screening, tier 1 conditions, hereditary breast and ovarian cancer (HBOC), lynch syndrome, familial hypercholestelemia, genetic testing
Citation: Mighton C, Shickh S, Aguda V, Krishnapillai S, Adi-Wauran E and Bombard Y (2022) From the patient to the population: Use of genomics for population screening. Front. Genet. 13:893832. doi: 10.3389/fgene.2022.893832
Received: 10 March 2022; Accepted: 26 September 2022;
Published: 24 October 2022.
Edited by:
Yann Joly, McGill University, CanadaReviewed by:
Lisa Dive, University of Technology Sydney, AustraliaRanjit Manchanda, Queen Mary University of London, United Kingdom
Copyright © 2022 Mighton, Shickh, Aguda, Krishnapillai, Adi-Wauran and Bombard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yvonne Bombard, eXZvbm5lLmJvbWJhcmRAdXRvcm9udG8uY2E=
†These authors have contributed equally to this work