 Tao Cai
Tao Cai Jieting Huang
Jieting Huang Xiuwei Ma
Xiuwei Ma Siqi Hu
Siqi Hu Lina Zhu
Lina Zhu Jinwen Zhu
Jinwen Zhu Zhichun Feng
Zhichun Feng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 11 July 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.892940
This article is part of the Research Topic Advancing Genomics for Rare Disease Diagnosis and Therapy Development Vol II View all 42 articles
Background: Genetic causes in most affected children with intellectual disability and/or development delay remain unknown.
Methods: To identify potential variants responsible for these disorders, we recruited 161 affected families and performed whole-exome sequencing and associated bioinformatics analysis.
Results: In the present study, we report the identification of variants in the ALG13 gene in two of the families. In family 1, a known pathogenic missense variant (c.23T > C; p.V8A) of ALG13 was identified in a boy and his mother. In family 2, a novel missense variant (c.862C > G; p.L288V) of the same gene was identified in the affected boy and his phenotypically normal mother. Genotype–phenotype correlation analysis by comparing reported 28 different variants (HGMD) showed that three major phenotypes, including various seizures/epilepsy, intellectual disability, and development delay (such as growth, speech, motor, etc.), are present in most affected individuals. However, other phenotypes, such as strabismus and absence of seizure in our second patient, are not reported if any, which may represent a unique case of X-linked recessive nonsyndromic disorder caused by a mutation in ALG13.
Conclusion: We identified two missense variants in ALG13 in a cohort of 161 families with affected individuals diagnosed as intellectual disability and/or development delay. A novel c.862C > G mutation may represent a case of X-linked recessive.
The ALG13 gene encodes a subunit of a bipartite UDP-N-acetylglucosamine transferase that regulates protein folding and stability, which is mapped to Xq23 and widely expressed in human tissues, such as brain, liver, and kidney (MIM: 300776). The first ALG13 mutation with de novo origin was identified in a male infant diagnosed with congenital disorders of glycosylation type I with refractory epilepsy, microcephaly, extrapyramidal, and pyramidal symptoms (Timal, et al., 2012).
Many of the affected individuals were diagnosed with developmental and epileptic encephalopathy 36 (DEE36, MIM: 300884), which is caused by heterozygous or hemizygous mutation in ALG13. DEE36 is characterized by the onset of seizures at a mean age of 6.5 months. Most patients present with infantile spasms associated with hypsarrhythmia on EEG, consistent with a clinical diagnosis of West syndrome.
To date, a total of 28 different mutations in ALG13 (HGMD) have been identified in affected individuals or families with epileptic encephalopathies (Epi, et al., 2013; Moller, et al., 2016), intellectual disability (Bissar-Tadmouri, et al., 2014), infantile spasms (Michaud, et al., 2014), West syndrome (Hino-Fukuyo, et al., 2015) or Lennox–Gastaut syndrome (Zhou, et al., 2018; Stranneheim, et al., 2021), congenital disorder of glycosylation (Alsharhan, et al., 2021), and several rare conditions such as left ventricular obstruction (Jin, et al., 2017) and fetal alcohol syndrome (de la Morena-Barrio, et al., 2018).
In the present study, we report the identification of two variants of ALG13 from two affected males with development delay and seizures or intellectual disability binocular strabismus, including a novel missense variant (c.862C > G; p.L288V) and a previously reported variant (c.23T > C; p.V8A). For a better understanding of this extremely rare disease, we present a detailed phenotype–genotype correlation analysis and a brief literature review.
Informed consents were obtained from all participants and in the case of minors, from their parents. This study was approved by the Seventh Medical Center of PLA General Hospital Ethics Committee at Beijing (no. 2022-37). A total of 503 individuals in 161 families, including 175 diagnosed as intellectual disability and/or development delay and 328 unaffected individuals, were recruited for genetic analysis. In the current study, three affected individuals with development defects in two families were presented.
Genomic DNAs were isolated from peripheral blood leukocytes. The captured exome by a SureSelect Human All Exon Kit (Agilent, Santa Clara, CA) was sequenced by HiSeq2000 sequencer (Illumina, San Diego, CA) and analyzed as previously described (Yu, et al., 2016; Zhu, et al., 2019; Li, et al., 2021). The reads were aligned to hg19, and the variants were identified through the GATK pipeline. An average sequence depth of coverage was 149× for exome sequences. Potential pathogenic variants were selected for further bioinformatics analysis. Primers (forward: TCACAGAAGGCAGTCACT; reverse: CGGAATAATGGGAAGAGGAA) were used for Sanger sequencing confirmation of the c.23T > C variant in the ALG13 gene (NM_001099922). Primer sequences for the confirmation of the second variant (c.862C > G) in ALG13 include the forward ACCATAATTGTTGAGCTGAGCA and reverse TTGGATTCAACACAGCTGGC.
The ALG13 protein motif was predicted by the SmartMotif (http://smart.embl-heidelberg.de/). Three-dimensional structure of the AlphaFold ALG13 prediction was obtained from UniProt (https://www.uniprot.org; model ID: AF-Q9NP73-F1). Its associated figures were produced using the program PyMOL (https://pymol.org/2/).
In family 1, the affected boy (Figure 1A) was first referred to the clinic when he was 2 years old due to intellectual disability, speech and motor development delay, and seizures. Seizures were first observed when the affected boy was 9 months old, which occurred 1–2 times per week and could be controlled by levetiracetam. He could only call “mom” but no other words at 3 years of age. His walking was unstable and could easily fall down. His height and head circumference were in normal ranges. His mother was also diagnosed with intellectual disability, but with no seizures. She could not read or count. His father was normal. An EEK examination at nearly 2 years of age of the body showed abnormal slow-wave activities in bilateral brain. His MRI showed protruding temporal angle of the left lateral ventricle and slightly wider bilateral frontotemporal extracerebral space.
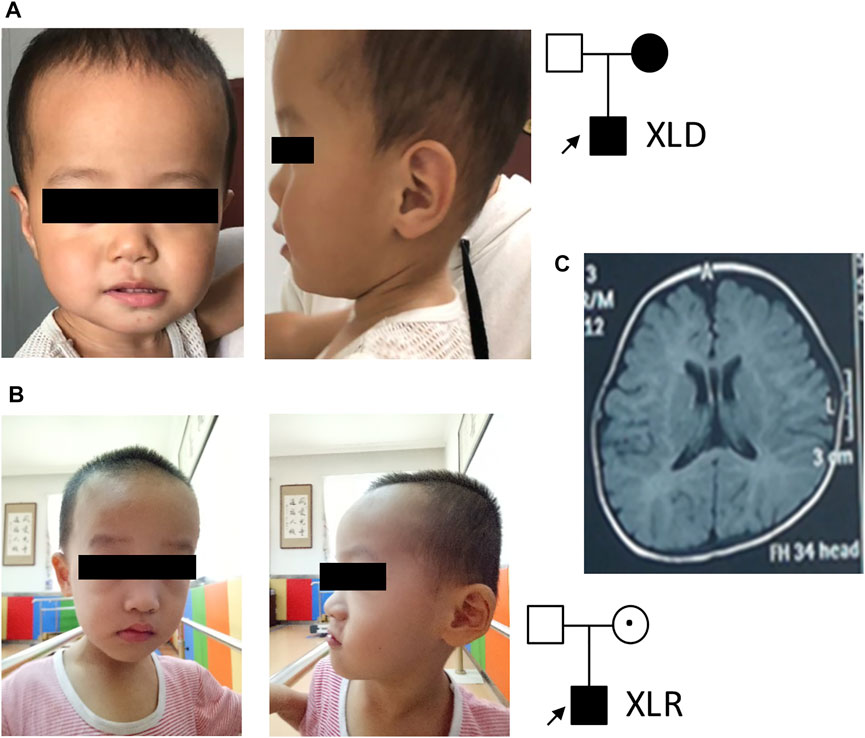
FIGURE 1. Patient pedigrees and radiographic findings. (A) Patient 1 photo and pedigree. (B) Patient 2 photo and pedigree. (C) Representative MRI image of patient 2.
In family 2, a 3-year-old boy (Figure 1B) with intellectual disability and speech and motor development delay was recruited for genetic analysis. His speech was delayed. He could call “mom” and stand up after 2 years of age. His hand movements were not coordinated. His walk was not stable and fell down easily. No seizure was observed. Physical examination showed binocular strabismus and abnormal finger-nose test (FNT). His height and head circumference were in normal ranges. Both of his parents were phenotypically normal. MRI images (Figure 1C) showed delayed myelination and widening of bilateral frontotemporal extracerebral space. His EEK report was normal. A combined analysis of DDST, Gesell, and Bayley tests when he was about 3 years old revealed low levels in his language, social behavior, movement, adaptability, and development quotient.
Trio-WES analysis for family 1 identified a known pathogenic missense variant (c.23T > C; p.V8A) in the ALG13 gene (NM_001099922.3) and further confirmed by Sanger sequencing (Figure 2A) from both the affected boy and his mother. The same mutation as a de novo allele was previously detected in a female patient (Datta, et al., 2021), who showed mild developmental delay and seizures starting from the second year of life (Table 1). The p.V8 residue is located in the Glyco_tran_28_C domain (amino acids 3–133) at the N-terminal region of the encoded protein (Figures 2C,D), which involves monogalactosyldiacylglycerol synthase and UDP-N-acetylglucosamine transferase (Pfam, SmartMotif).
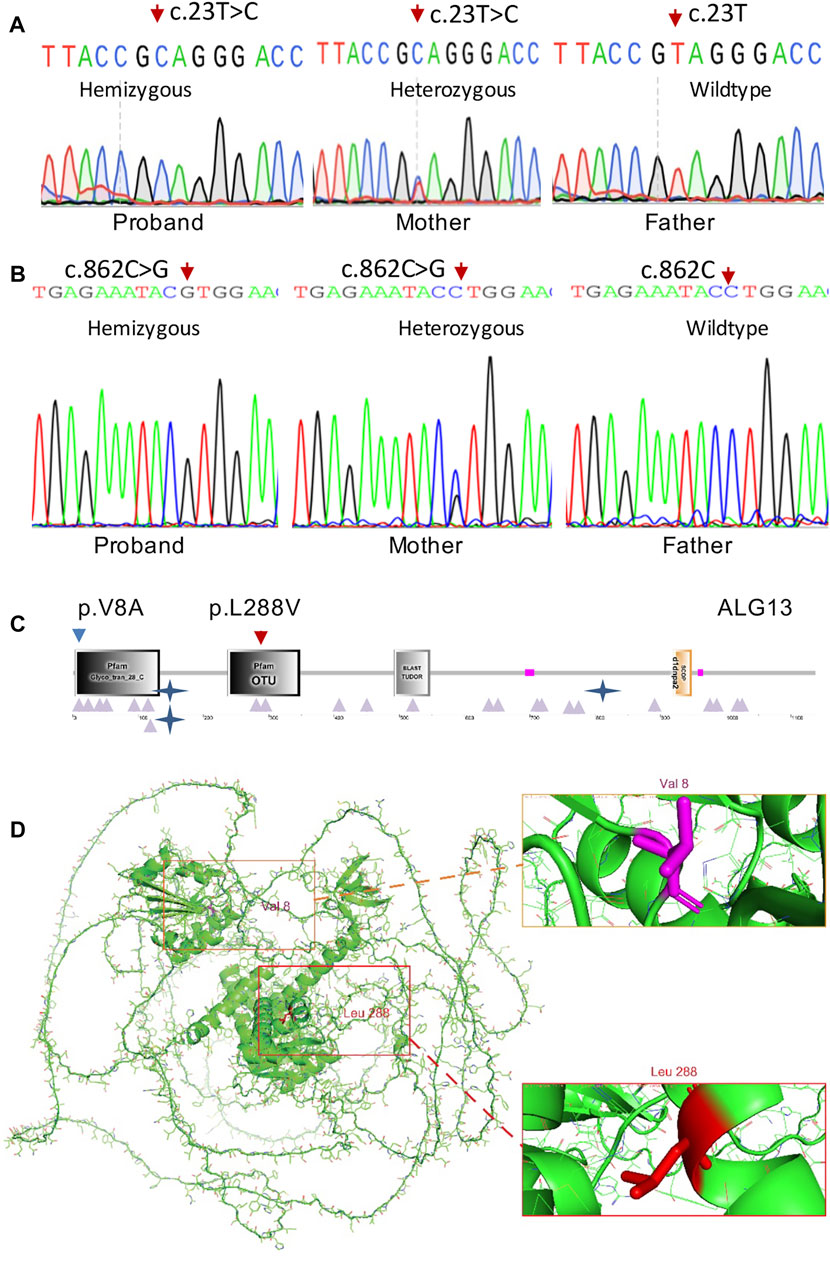
FIGURE 2. ALG13 mutations and expression. (A) Sanger sequencing confirmation of the c.23T > C mutation in case 1 and his mother. (B) Sanger sequencing confirmation of the c.862C > G mutation in case 2 and his mother. (C) Schematic representations of functional domains of ALG13. The p.V8A mutation of family 1 is located in the Glyco_tans_28 domain at N-terminus (amino acid 3–133). The p.L288V mutation of family 2 is mapped in the OUT domain (amino acid 237–348). (D) Residue p.V8 and p.L288 positions are indicated in red in the 3-dimensional structure of the ALG13 protein.

TABLE 1. Genotype–phenotype correlation analyses for affected individuals with ALG13 variants. The bold presents the phenotype in this study or a special phenotype related to this study.
Trio-WES analysis for family 2 identified a previously undescribed potentially pathogenic missense variant (c.862C > G) from the affected boy and his phenotypically normal mother. Sanger sequencing further confirmed this variant (Figure 2B). Bioinformatics analysis revealed that this variant is not present in ExAC or the in-house database and predicted to be disease-causing by MutationTaster and Polyphen2. Smart Motif analysis revealed that the mutation p.L288 is mapped to the OTU domain (amino acids 237–348) in the ALG13 protein (Figures 2C,D), which is OTU-like cysteine protease motif (Makarova, et al., 2000).
As shown in Table 1, we summarized 28 different mutations that are listed in HGMD and two mutations identified in the present study. Affected individuals and/or families were either in X-linked dominant (XLD) or X-linked recessive (XLR) pattern or with de novo mutation (DNM) origins. Family 1 in our case is in the XLD form, while family 2 is in the XLR pattern. Most of the mutations in Table 1 are missense variants (27/30); three of them are splicing and deletion mutations (3/30). Three major phenotypes, including various seizures/epilepsy, intellectual disability, and development delay (such as growth, speech, motor, etc.), are observed in most of the cases. Less frequently observed phenotypes include strabismus, optic nerve atrophy, left ventricular obstruction, and ataxia.
In the present study, we identified two variants in the ALG13 gene in patients with either typical phenotypes in family 1 with XLD inheritance form (seizures, intellectual disability, speech, and motor development delay) or atypical phenotypes in family 2 with XLR inheritance pattern (mild intellectual disability, speech and motor development delay, mild ataxia, and binocular strabismus, but no seizures). Previously, only 28 different variants were reported (HGMD). Three of them (c.845G > A; p.G282E, c.1233G > C; p.K411N and c.384-5C > T) were identified in Chinese populations (Wei, et al., 2018; Zhou, et al., 2018; Jiao, et al., 2019). Our findings expanded the ALG-13-related mutation spectrum and ALG-13-associated clinic manifestations.
Genotype–phenotype correlation analysis indicates that the mild clinical manifestations of the patient in family 2 is resulted from a mild pathogenic mutation (c.862C > G; p.L288V). In fact, both leucine and valine in the p.L288V allele are alpha-amino acids, which implies that they contain an alpha-amino group, an alpha-carboxylic acid group, and a side chain isobutyl group/isopropyl group. Based on the AlphaFold predicted structure model (Figure 2D), the Leu288 residue is located in α-helix.
In contrast, alanine in the p.V8A variant in family 1 is a simple amino acid, which has just a methyl as its side chain. Based on the AlphaFold predicted structure model (Figure 2D), Val8 residue is located in the loop region involving glycosyltransferase activity (amino acids 1–125), thereby causing more severe clinic phenotypes as we described earlier.
Based on several commonly used gene expression databases, such as BioGPS and human brain transcriptome, the human ALG13 gene is widely expressed in many tissues, including neurons in developing and adult brain tissues (Supplementary Figure S1). Brain-associated clinical manifestations, such as seizures and intellectual disability, are apparently correlated with cortical and central nervous dysfunctions in the affected individuals with ALG13 variants. Additional rare phenotypes, such as ataxia, nystagmus, and strabismus, are potentially associated with developmental defects or dysfunctions of the cerebellum and brain stem tissues.
In the Alg13 knockout mouse model, Alg13 deficiency resulted in an increased seizure and susceptibility in the Alg13−/− mice (Gao, et al., 2019). Previous studies also explored the possible mechanisms of Alg13-involved epilepsy by showing hyperactive mTOR signaling pathways in the cortex and hippocampus of Alg13−/− mice (Gao, et al., 2019; Huo, et al., 2020). Further studies using patch-clamp recordings demonstrated that Alg13−/− mice show a marked decrease in the gamma-aminobutyric acid A receptor (GABAAR)–mediated inhibitory synaptic transmission (Huo, et al., 2020). At the human level, a majority of variants are missense, which are linked to either X-linked dominant phenotypes due to stronger pathogenic variants (such as the variant in family 1) or X-linked recessive phenotypes due to mild pathogenetic variants (such as the variant in family 2).
Taken together, we provided clinical and bioinformatics evidences that two ALG13 variants are pathogenic for the affected individuals with ALG13-associated phenotypes. However, the underlying mechanism remains to be explored in further studies.
The original datasets presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the Seventh Medical Center of PLA General Hospital Ethics Committee. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s) and minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
TC and ZF conceived and designed the experiment and wrote the manuscript. JH, SH, and JZ provided WES and bioinformatics analysis and performed the experiments. XM and LZ provided clinical information.
The authors are grateful to the patients and family members for their participation in this study.
Author JZ was employed by Angen Gene Medicine Technology, Beijing, China.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.892940/full#supplementary-material
Alsharhan, H., He, M., Edmondson, A. C., Daniel, E. J. P., Chen, J., Donald, T., et al. (2021). ALG13 X‐linked Intellectual Disability: New Variants, Glycosylation Analysis, and Expanded Phenotypes. J Inher Metab Disea 44 (4), 1001–1012. doi:10.1002/jimd.12378
Amadori, E., Scala, M., Cereda, G. S., Vari, M. S., Marchese, F., Di Pisa, V., et al. (2020). Targeted Re-sequencing for Early Diagnosis of Genetic Causes of Childhood Epilepsy: the Italian Experience from the 'beyond Epilepsy' Project. Ital. J. Pediatr. 46 (1), 92. doi:10.1186/s13052-020-00860-1
Bissar-Tadmouri, N., Donahue, W. L., Al-Gazali, L., Nelson, S. F., Bayrak-Toydemir, P., and Kantarci, S. (2014). X Chromosome Exome Sequencing Reveals a novelALG13mutation in a Nonsyndromic Intellectual Disability Family with Multiple Affected Male Siblings. Am. J. Med. Genet. 164 (1), 164–169. doi:10.1002/ajmg.a.36233
Datta, A. N., Bahi‐Buisson, N., Bienvenu, T., Buerki, S. E., Gardiner, F., Cross, J. H., et al. (2021). The Phenotypic Spectrum of X‐linked, Infantile Onset ALG13 ‐related Developmental and Epileptic Encephalopathy. Epilepsia 62 (2), 325–334. doi:10.1111/epi.16761
de la Morena-Barrio, M. E., Ballesta-Martínez, M. J., López-Gálvez, R., Antón, A. I., López-González, V., Martínez-Ribot, L., et al. (2018). Genetic Predisposition to Fetal Alcohol Syndrome: Association with Congenital Disorders of N-Glycosylation. Pediatr. Res. 83 (1-1), 119–127. doi:10.1038/pr.2017.201
Epi, K. C., Epilepsy Phenome/Genome, P., Allen, A. S., Berkovic, S. F., Cossette, P., Delanty, N., et al. (2013). De Novo mutations in Epileptic Encephalopathies. Nature 501 (7466), 217–221. doi:10.1038/nature12439
Farwell, K. D., Shahmirzadi, L., El-Khechen, D., Powis, Z., Chao, E. C., Tippin Davis, B., et al. (2015). Enhanced Utility of Family-Centered Diagnostic Exome Sequencing with Inheritance Model-Based Analysis: Results from 500 Unselected Families with Undiagnosed Genetic Conditions. Genet. Med. 17 (7), 578–586. doi:10.1038/gim.2014.154
Gadomski, T. E., Bolton, M., Alfadhel, M., Dvorak, C., Ogunsakin, O. A., Nelson, S. L., et al. (2017). ALG13‐CDG in a Male with Seizures, Normal Cognitive Development, and Normal Transferrin Isoelectric Focusing. Am. J. Med. Genet. 173 (10), 2772–2775. doi:10.1002/ajmg.a.38377
Gao, P., Wang, F., Huo, J., Wan, D., Zhang, J., Niu, J., et al. (2019). ALG13 Deficiency Associated with Increased Seizure Susceptibility and Severity. Neuroscience 409, 204–221. doi:10.1016/j.neuroscience.2019.03.009
Geisheker, M. R., Heymann, G., Wang, T., Coe, B. P., Turner, T. N., Stessman, H. A. F., et al. (2017). Hotspots of Missense Mutation Identify Neurodevelopmental Disorder Genes and Functional Domains. Nat. Neurosci. 20 (8), 1043–1051. doi:10.1038/nn.4589
Harripaul, R., Vasli, N., Mikhailov, A., Rafiq, M. A., Mittal, K., Windpassinger, C., et al. (2018). Mapping Autosomal Recessive Intellectual Disability: Combined Microarray and Exome Sequencing Identifies 26 Novel Candidate Genes in 192 Consanguineous Families. Mol. Psychiatry 23 (4), 973–984. doi:10.1038/mp.2017.60
Hesse, A. N., Bevilacqua, J., Shankar, K., and Reddi, H. V. (2018). Retrospective Genotype-Phenotype Analysis in a 305 Patient Cohort Referred for Testing of a Targeted Epilepsy Panel. Epilepsy Res. 144, 53–61. doi:10.1016/j.eplepsyres.2018.05.004
Hino-Fukuyo, N., Kikuchi, A., Arai-Ichinoi, N., Niihori, T., Sato, R., Suzuki, T., et al. (2015). Genomic Analysis Identifies Candidate Pathogenic Variants in 9 of 18 Patients with Unexplained West Syndrome. Hum. Genet. 134 (6), 649–658. doi:10.1007/s00439-015-1553-6
Huo, J., Ren, S., Gao, P., Wan, D., Rong, S., Li, X., et al. (2020). ALG13 Participates in Epileptogenesis via Regulation of GABAA Receptors in Mouse Models. Cell Death Discov. 6 (1), 87. doi:10.1038/s41420-020-00319-6
Ji, J., Shen, L., Bootwalla, M., Quindipan, C., Tatarinova, T., Maglinte, D. T., et al. (2019). A Semiautomated Whole-Exome Sequencing Workflow Leads to Increased Diagnostic Yield and Identification of Novel Candidate Variants. Cold Spring Harb. Mol. Case Stud. 5 (2), a003756. doi:10.1101/mcs.a003756
Jiao, Q., Sun, H., Zhang, H., Wang, R., Li, S., Sun, D., et al. (2019). The Combination of Whole‐exome Sequencing and Copy Number Variation Sequencing Enables the Diagnosis of Rare Neurological Disorders. Clin. Genet. 96 (2), 140–150. doi:10.1111/cge.13548
Jin, S. C., Homsy, J., Zaidi, S., Lu, Q., Morton, S., DePalma, S. R., et al. (2017). Contribution of Rare Inherited and De Novo Variants in 2,871 Congenital Heart Disease Probands. Nat. Genet. 49 (11), 1593–1601. doi:10.1038/ng.3970
Li, Y., Xiong, J., Zhang, Y., Xu, L., Liu, J., and Cai, T. (2021). Case Report: Exome Sequencing Identified Variants in Three Candidate Genes from Two Families with Hearing Loss, Onychodystrophy, and Epilepsy. Front. Genet. 12, 728020. doi:10.3389/fgene.2021.728020
Makarova, K. S., Aravind, L., and Koonin, E. V. (2000). A Novel Superfamily of Predicted Cysteine Proteases from Eukaryotes, Viruses and Chlamydia Pneumoniae. Trends Biochem. Sci. 25 (2), 50–52. doi:10.1016/s0968-0004(99)01530-3
Michaud, J. L., Lachance, M., Hamdan, F. F., Carmant, L., Lortie, A., Diadori, P., et al. (2014). The Genetic Landscape of Infantile Spasms. Hum. Mol. Genet. 23 (18), 4846–4858. doi:10.1093/hmg/ddu199
Møller, R. S., Larsen, L. H. G., Johannesen, K. M., Talvik, I., Talvik, T., Vaher, U., et al. (2016). Gene Panel Testing in Epileptic Encephalopathies and Familial Epilepsies. Mol. Syndromol. 7 (4), 210–219. doi:10.1159/000448369
Monies, D., Abouelhoda, M., Assoum, M., Moghrabi, N., Rafiullah, R., Almontashiri, N., et al. (2019). Lessons Learned from Large-Scale, First-Tier Clinical Exome Sequencing in a Highly Consanguineous Population. Am. J. Hum. Genet. 105 (6), 879–201. doi:10.1016/j.ajhg.2019.04.011
Paprocka, J., Jezela-Stanek, A., Boguszewicz, Ł., Sokół, M., Lipiński, P., Jamroz, E., et al. (2021). The First Metabolome Analysis in Children with Epilepsy and ALG13-CDG Resulting from c.320A>G Variant. Children 8 (3), 251. doi:10.3390/children8030251
Stranneheim, H., Lagerstedt-Robinson, K., Magnusson, M., Kvarnung, M., Nilsson, D., Lesko, N., et al. (2021). Integration of Whole Genome Sequencing into a Healthcare Setting: High Diagnostic Rates across Multiple Clinical Entities in 3219 Rare Disease Patients. Genome Med. 13 (1), 40. doi:10.1186/s13073-021-00855-5
Timal, S., Hoischen, A., Lehle, L., Adamowicz, M., Huijben, K., Sykut-Cegielska, J., et al. (2012). Gene Identification in the Congenital Disorders of Glycosylation Type I by Whole-Exome Sequencing. Hum. Mol. Genet. 21 (19), 4151–4161. doi:10.1093/hmg/dds123
Wei, C. M., Xia, G. Z., and Ren, R. N. (2018). Gene Mutations in Unexplained Infantile Epileptic Encephalopathy: an Analysis of 47 Cases. Zhongguo Dang Dai Er Ke Za Zhi 20 (2), 125–129.
Yu, P., Yang, W., Han, D., Wang, X., Guo, S., Li, J., et al. (2016). Mutations in WNT10B Are Identified in Individuals with Oligodontia. Am. J. Hum. Genet. 99 (1), 195–201. doi:10.1016/j.ajhg.2016.05.012
Zhou, P., He, N., Zhang, J.-W., Lin, Z.-J., Wang, J., Yan, L.-M., et al. (2018). Novel Mutations and Phenotypes of Epilepsy-Associated Genes in Epileptic Encephalopathies. Genes, Brain Behav. 17 (8), e12456. doi:10.1111/gbb.12456
Keywords: ALG13, mutation, whole-exome sequencing, development delay, X-linked
Citation: Cai T, Huang J, Ma X, Hu S, Zhu L, Zhu J and Feng Z (2022) Case Report: Identification of Two Variants of ALG13 in Families With or Without Seizure and Binocular Strabismus: Phenotypic Spectrum Analysis. Front. Genet. 13:892940. doi: 10.3389/fgene.2022.892940
Received: 09 March 2022; Accepted: 07 June 2022;
Published: 11 July 2022.
Edited by:
Tieliu Shi, Hunan University of Arts and Science, ChinaReviewed by:
Ning Wang, Jiangnan University, ChinaCopyright © 2022 Cai, Huang, Ma, Hu, Zhu, Zhu and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Cai, Z2VuZmF4MUBnbWFpbC5jb20=; Zhichun Feng, emhqZmVuZ3pjQDEyNi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.