 Lihong Fan
Lihong Fan Pengzhen Jin2
Pengzhen Jin2 Yeqing Qian
Yeqing Qian Minyue Dong
Minyue Dong- 1Center of Prenatal Diagnosis, Huzhou Maternity & Child Health Care Hospital, Huzhou, China
- 2Women’s Hospital, School of Medicine Zhejiang University, Hangzhou, China
- 3Key Laboratory of Reproductive Genetics (Zhejiang University), Ministry of Education, Hangzhou, China
Postaxial polydactyly is a common congenital malformation which involves complex genetic factors. This retrospective study analyzed the cytogenetic and molecular results of a Chinese fetus diagnosed with postaxial polydactyly of all four limbs. Fetal karyotyping and chromosomal microarray analysis (CMA) did not find any abnormality while trio whole-exome sequencing (trio-WES) identified bi-allelic variants in smoothened (SMO) and (NM_005631.5: c.1219C > G, NP_005622.1: p. Pro407Ala, and NM_005631.5: c.1619C > T, NP_005622.1: p. Ala540Val). Sanger sequencing validated these variants. The mutations are highly conserved across multiple species. In-depth bioinformatics analysis and familial co-segregation implied the compound heterozygous variants as the likely cause of postaxial polydactyly in this fetus. Our findings provided the basis for genetic counseling and will contribute to a better understanding of the complex genetic mechanism that underlies postaxial polydactyly.
Introduction
Postaxial polydactyly (PAP), characterized by an extra digit at the fifth finger or toe, is one of the most common congenital malformations (Umair et al., 2018). It can be a marker of a wide variety of neurological and systemic abnormities and usually occurs in syndromic types (Verma and El-Harouni, 2015; Chandra et al., 2017). Limb growth in vertebrates is regulated by a complex network of intercellular communication and gene expression. The Sonic hedgehog protein (SHH), one of three mammalian hedgehog (HH) proteins, is expressed in the zone of polarizing activity (ZPA) of the limb bud and those of the notochord and floor plate in the neural tube (Riddle et al., 1993; Johnson et al., 1994; Jessell, 2000; Xavier et al., 2016). There is compelling evidence that the SHH signaling pathway plays an important role in regulating the patterning and growth of the developing limb (Hill et al., 2003; Tickle and Barker, 2013; Chinnaiya et al., 2014; Choudhry et al., 2014). The major components of the SHH pathway include smoothened (SMO), PTCH, and GLI transcription factors. In the absence of ligand binding of SHH, PTCH binds to SMO and inhibits its activity. Here, SUFU binds to the GLI transcription factors, which are subsequently phosphorylated by PKA, CK1, and GSK3β and degraded by the proteasome. Under this condition, GLI is converted to GLIr with the C-terminal domain truncated and enters the nucleus, inhibiting the transcription of downstream target genes (Figure 1A). When SHH is present, it binds to the extracellular domain of PTCH and releases its repressive effects on SMO. Freed of PTCH1-mediated suppression, SMO relieves the sequestration by SUFU and phosphorylation from PKA, CK1, and GSK3β, thus stabilizing the GLI proteins in their full-length transcriptional activator form. The activated GLI (GLIa) translocates into the nucleus and promotes the transcription of SHH target genes (Figure 1B) (Briscoe and Thérond, 2013; Niida et al., 2021; Sigafoos et al., 2021). Here, we observed that the SHH signaling pathway is a highly regulated cascade of extracellular ligands, receptor proteins, cytoplasmic signaling molecules, transcription factors, coregulators, and target genes. Mutations in SHH, PTCH, SMO, and GLI can lead to the downregulation of the SHH pathway and ultimately to malformations (Johnson et al., 2014; Le et al., 2020; Yi et al., 2020). Most studies on PAP-related mutations have focused on GLI, while few studies, on SMO (Zou et al., 2019; Cao et al., 2020; Xiang et al., 2020; Patel et al., 2021). To the best of our knowledge, no previous study on prenatal diagnosis of SMO mutations has been available so far. The present study reported novel bi-allelic variants in SMO likely causing the PAP in a fetus.
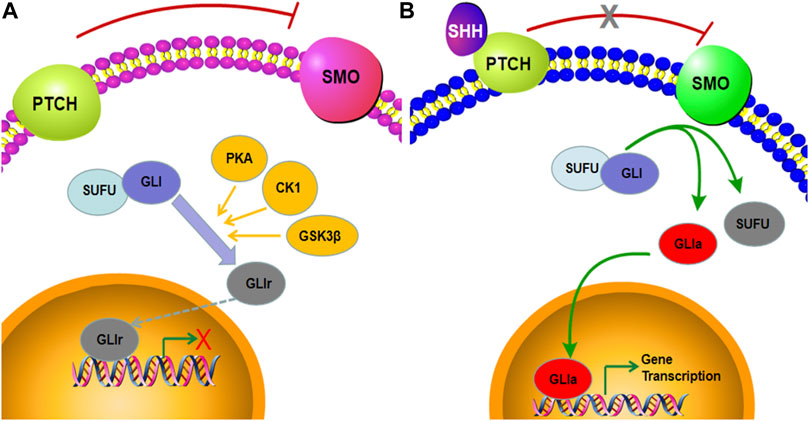
FIGURE 1. Graphical representation of active and inactive SHH signaling pathways. (A) When SHH is absent, the full-length GLI is phosphorylated by PKA, CK1, and GSK3β and proteolytic cleavaged into the GLI repressor, which subsequently suppresses the expression of SHH target genes. (B) In the presence of the SHH ligand, SMO inhibits the sequestration by SUFU and phosphorylation by PKA, CK1, and GSK3β, leading to the formation of the GLI activator and ultimately to induction of target gene transcription.
Patients and Methods
Case Presentation
A 29-year-old primigravida woman was referred to our center for further evaluation at 23+2 weeks of gestation. First-trimester screening and second-trimester screening for Down syndrome indicated a low risk. Routine prenatal ultrasound scans (US) at 22+1 weeks of gestation suggested the PAP of all four limbs (Figure 2A). MRI scans subsequently confirmed the result of fetal PAP (Figure 2B). The mother denied being exposed to teratogenic agents or irradiation or using nicotine or alcohol during the pregnancy. No family history of neurological disease or congenital malformations was recorded.
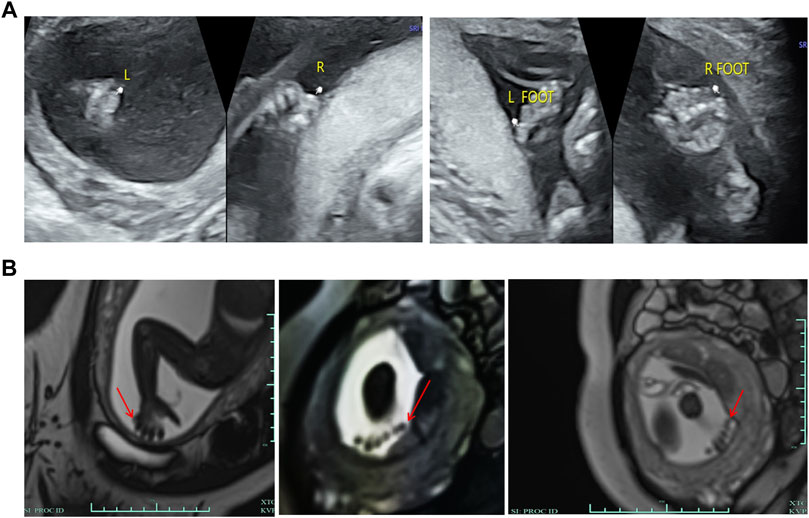
FIGURE 2. Clinical features of the fetus. (A) Ultrasonography showed that there were echoes of the sixth toe on the lateral side of the little toe of both feet and the lateral side of the little finger of both hands, with a size of about 0.6*0.4 cm, and no obvious bony structure in them. (B) MRI showed a finger-like signal shadow on the outside of the right little finger, while the number of fingers on the left hand was not clear due to the position of the fetus. Six toe-like signal shadows were seen on both feet.
The current investigation was approved by the Ethics Committee of Women’s Hospital, School of Medicine Zhejiang University. All participants provided their written informed consent.
Amniocentesis
Amniocentesis was performed aseptically under the guidance of ultrasonography at 23+2 weeks of gestation. Thirty milliliters of amniotic fluid were obtained, of which 15 mL was for cell culture, while the remaining 15 mL was for DNA extraction.
Karyotype Analysis
Karyotype analysis included the culture of amniocytes in appropriate culture media and G-banded karyotyping at the 320–400 band level.
Chromosomal Microarray Analysis
Genomic DNA was extracted from the amniotic fluid and the peripheral blood of the parents. Then, fetal DNA was analyzed using the CytoScan™ HD whole-genome SNP array (Affymetrix, United States), and data were analyzed by Chromosome Analysis Suite 4.2, according to manufacturers’ instructions.
Trio Whole-Exome Sequencing and Sanger Sequencing
Genomic DNA extracted from the amniotic fluid and the peripheral blood of the parents was used for Trio-WES. Exome capture was performed using the SureSelect Human All Exon V4 kit (Agilent Technologies, Santa Clara, CA, United States), followed by sequencing using an Illumina HiSeq2500 system (Illumina, San Diego, CA, United States). After sequencing and filtering out low-quality reads, high-quality reads were compared to the GRCh37/hg19 reference human genome using Sentieon BWA (Sentieon, United States) with the MEM align method, and only the variants located in the coding sequence or splice site regions would be retained. Variant calling was performed using the Genome Analysis Tool Kit (GATK v4.0). Then, the candidate variants, including single-nucleotide variants (SNVs) and indels, were filtered by frequencies on specific databases, including the Human Gene Mutation Database (HGMD), ClinVar database, 1000 Genomes Project, Exome Aggregation Consortium (ExAC), Exome Sequencing Project 6500 (ESP6500), database of single-nucleotide polymorphisms (dbSNP), and Genome Aggregation Database (gnomAD). Mutation sites, known as polymorphic sites, were excluded, and the variants with allele frequency≤1% were retained. We used PolyPhen2, SIFT, Condel, MutationTaster, and phyloP to predict the effect of variants. The interpretation of sequence variants was performed according to the American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015). Online Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) was used to analyze the evolutionarily conserved sequences among species (humans, chimpanzees, macaques, mice, rats, Sus scrofa, horses, and Bos taurus). Finally, the identified variants were confirmed with Sanger sequencing. Sequences containing the two potential variants were amplified. The primer sequences were designed by Primer3 (http://primer3.ut.ee/), and all the primers are shown in Supplementary Table S1. Then, the PCR products were sequenced on the ABI 3500DX, followed by analysis by DNASTAR 5.0 software.
Results
CMA and karyotype analysis did not reveal any abnormalities. However, the trio-WES study identified a compound heterozygote in SMO (chr7:128846383, NM_005631.5: c.1219C > G, NP_005622.1: p. Pro407Ala; chr7:128850356, NM_005631.5: c.1619C > T, NP_005622.1: p. Ala540Val) inherited from the mother and father, respectively. Sanger sequencing confirmed the variants and showed that the two compound heterozygous mutations in SMO co-segregate with the disorder in this family (Figure 3A). The two mutations were absent in the 1000 Genomes, ESP6500, dbSNP144, and ExAC databases, and only the mutation c.1619C > T had a low allele frequency (0.00001989, all heterozygote) observed in gnomAD. The alignment of the amino acid sequences indicates that the two mutant residues were 100% conserved among many species (Figure 3B). Moreover, both variants were predicted to be pathogenic by the SIFT, PROVEAN, and MutationTaster tools (Table 1). Based on the consistency of genotype–phenotype correlation and the effect of the mutations, we speculated that the compound heterozygous mutations in SMO might be responsible for the limb abnormalities in this Chinese family.

FIGURE 3. Genetic variations identified in this case. (A) Two variants detected in the fetus and parents. (B) Conservation of the mutated amino acids (Pro407 and Ala540) across different species.

TABLE 1. In silico analysis of the SMO variants c.1219C > G and c.1619C > T.
Finally, the couple chose to terminate the pregnancy after carefully considering the abnormal ultrasonography findings and trio-WES results.
Discussion
The genetic mechanism underlying polydactyly is highly complex. The SHH pathway is the essential evolutionarily conserved pathway related to the growth and patterning of the limbs (Patterson et al., 2009; Rahi and Mehan, 2020). SMO, a seven-transmembrane, encoded by SMO, is required for active SHH signaling (Zhang et al., 2001; Le et al., 2020). A previous study showed that removing SMO from the apical ectodermal ridge resulted in the loss of functional SHH signaling, ultimately leading to disruption of digit patterns and the formation of additional postaxial cartilage contraction (Bouldin et al., 2010).
In the present study, we identified bi-allelic variants in SMO, c.1219C > G (p. Pro407Ala), and c.1619C > T (p. Ala540Val) in a Chinese family. The second variant was previously reported in a Han Chinese boy, also in a state of compound heterozygosity (Zhang et al., 2019). It was predicted that this mutation affects the signaling ability of SMO as the alanine residue at position 540 is quite close to the seventh TM domain, which is considered to be the key site for signaling transduction. In addition, the previous study also performed a gene expression analysis and showed a significant decrease in the SMO expression in the patient compared with healthy controls. Back to our study, although the mRNA and protein level in the fetus cannot be detected due to the unavailability of the fetus’ sample, the pathogenic effect of Pro407Ala and Ala540Val substitution in SMO may be supported by the following points: 1) both loci were quite conserved among different species; 2) in silico bioinformatics applications predicted that both mutations are deleterious; 3) SMO belongs to recessive genes, and the two mutations co-segregated with the disorder in the family; 4) no other pathogenic variants were detected in the known polydactyly or limb development genes, such as SHH, PTCH, and GLI; and 5) the phenotype of PAP found in the fetus could be explained by SMO defects.
We searched the PubMed database to find relevant literature on SMO mutations. As a result, we found that the previous studies on SMO were limited and focused on activating somatic mutations that caused diseases such as sporadic basal cell carcinoma (BCC), medulloblastoma (MB), and Curry–Jones syndrome (CRJS) (Kool et al., 2014; Twigg et al., 2016; Bisceglia et al., 2020). Germline mutations, leading to developmental disorders, have not been reported until recent years. According to the previous reports, there were, in total, 10 patients with a series of congenital developmental abnormalities caused by SMO mutations (Rubino et al., 2018; Zhang et al., 2019; Le et al., 2020; Green et al., 2022). The fact that all the patients had compound heterozygous variants, while their parents were in a heterozygous state and had no associated phenotype, confirmed that SMO belongs to recessive genes. PAP can be observed in nine out of 10 patients, and 100% (9/9) of the patients with PAP also had other symptoms, such as thalamic hamartoma, cystic epilepsy, atrioventricular septal defect (AVSD), and aganglionosis. The most severe case was one who presented AVSD and died at 3 months of age (Le et al., 2020). In addition, all nine cases mentioned previously were diagnosed after birth, and our report on the fetus is to date the only prenatal genetic diagnosis of SMO mutations. We speculated that PAP may be the earliest prenatal manifestation and that the fetus may present other related symptoms after birth.
As one of the most common congenital malformations, PAP often appears as a key feature of malformation syndromes, such as Meckel–Gruber syndrome (Turkyilmaz et al., 2021), Bardet–Biedl syndrome (Ullah et al., 2017), and Pallister–Hall syndrome (Yang et al., 2021). According to previous studies, about 8% of bilateral PAP cases are related to a set of other congenital syndromic defects. As is well known, many syndromes have postnatal features that cannot be detected prenatally on ultrasound scans. The fetal phenotype is useful but sometimes limited source of information for the diagnosis of many Mendelian diseases. In prenatal cases with a less severe phenotype or an isolated anomaly, it is always quite difficult for parents to make a decision on continuing or terminating the pregnancy. We believe that prenatal WES is particularly valuable in detecting genetic variants of certain genes that may be critical to human development and helps us predict the postnatal phenotypes associated with the detected genes.
The most important limitation to our study lies in the fact that there is a lack of postpartum assessment and validation of associated malformations, such as thalamic hamartoma, cystic epilepsy, atrioventricular septal defect, and aganglionosis, due to our failure to get the permission for an autopsy. Therefore, it is still uncertain whether the fetus’ polydactyly was isolated or syndromic, although no abnormality in organs such as the heart and the brain was indicated by multiple prenatal ultrasounds and MRI. Given the very scarce reporting of occasional mutations, more studies in patients with related phenotypes are urgently needed to describe a full spectrum of SMO mutations. Detailed functional experiments are also needed to confirm how these mutations work.
In conclusion, we identified a novel compound heterozygous mutation (c.1219C > G, p. Pro407Ala and c.1619C > T, p. Ala540Val) in the gene SMO. There is convincing evidence, although no definite proof, that this mutation is causative for the PAP in the present family. The genetic diagnosis provided evidence for assessing the risk of recurrence and was invaluable for genetic counseling of the couple contemplating future pregnancies.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Women’s Hospital, School of Medicine Zhejiang University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
MD designed the study and reviewed the manuscript. LF analyzed the data and wrote the manuscript. XS and GS performed the experiments. YQ and PJ collected the data. All authors have read and approved the manuscript.
Funding
The work was supported by the Zhejiang medical and health technology program (2021KY1085) and the Huzhou science and technology program (2020GY26).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the family for their participation in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.887082/full#supplementary-material
References
Bisceglia, M., Panniello, G., Galliani, C. A., Centola, M., D’Errico, M. M., Minenna, E., et al. (2020). Metastatic Basal Cell Carcinoma of the Skin: A Comprehensive Literature Review, Including Advances in Molecular Therapeutics. Adv. Anat. Pathol. 27 (5), 331–353. doi:10.1097/pap.0000000000000267
Bouldin, C. M., Gritli-Linde, A., Ahn, S., and Harfe, B. D. (2010). Shh Pathway Activation Is Present and Required within the Vertebrate Limb Bud Apical Ectodermal Ridge for Normal Autopod Patterning. Proc. Natl. Acad. Sci. U.S.A. 107 (12), 5489–5494. doi:10.1073/pnas.0912818107
Briscoe, J., and Thérond, P. P. (2013). The Mechanisms of Hedgehog Signalling and its Roles in Development and Disease. Nat. Rev. Mol. Cell Biol. 14 (7), 416–429. doi:10.1038/nrm3598
Cao, R., Liu, S., Chai, W., and Shen, P. (2020). Polydactyly Patient Carried a Mutation of PTCH1 Which Has Been Identified in Nevoid Basal Cell Nevus Syndrome. DNA Cell Biol. 39 (10), 1754–1759. doi:10.1089/dna.2019.5236
Chandra, S., Daryappa, M., Mukheem Mudabbir, M., Pooja, M., and Arivazhagan, A. (2017). Pallister-Hall Syndrome. J. Pediatr. Neurosci. 12 (3), 276–279. doi:10.4103/jpn.JPN_101_17
Chinnaiya, K., Tickle, C., and Towers, M. (2014). Sonic Hedgehog-Expressing Cells in the Developing Limb Measure Time by an Intrinsic Cell Cycle Clock. Nat. Commun. 5, 4230. doi:10.1038/ncomms5230
Choudhry, Z., Rikani, A. A., Choudhry, A. M., Tariq, S., Zakaria, F., Asghar, M. W., et al. (2014). Sonic Hedgehog Signalling Pathway: a Complex Network. Ans 21 (1), 28–31. doi:10.5214/ans.0972.7531.210109
Green, T. E., Schimmel, M., Schubert, S., Lemke, J. R., Bennett, M. F., Hildebrand, M. S., et al. (2022). Bi-allelic SMO Variants in Hypothalamic Hamartoma: a Recessive Cause of Pallister-Hall Syndrome. Eur. J. Hum. Genet. 30, 384–388. doi:10.1038/s41431-021-01023-4
Hill, R. E., Heaney, S. J. H., and Lettice, L. A. (2003). Sonic Hedgehog: Restricted Expression and Limb Dysmorphologies. J. Anat. 202 (1), 13–20. doi:10.1046/j.1469-7580.2003.00148.x
Jessell, T. M. (2000). Neuronal Specification in the Spinal Cord: Inductive Signals and Transcriptional Codes. Nat. Rev. Genet. 1 (1), 20–29. doi:10.1038/35049541
Johnson, E. J., Neely, D. M., Dunn, I. C., and Davey, M. G. (2014). Direct Functional Consequences of ZRS Enhancer Mutation Combine with Secondary Long Range SHH Signalling Effects to Cause Preaxial Polydactyly. Dev. Biol. 392 (2), 209–220. doi:10.1016/j.ydbio.2014.05.025
Johnson, R. L., Laufer, E., Riddle, R. D., and Tabin, C. (1994). Ectopic Expression of Sonic Hedgehog Alters Dorsal-Ventral Patterning of Somites. Cell 79 (7), 1165–1173. doi:10.1016/0092-8674(94)90008-6
Kool, M., Jones, D. T. W., Jäger, N., Northcott, P. A., Pugh, T. J., Hovestadt, V., et al. (2014). Genome Sequencing of SHH Medulloblastoma Predicts Genotype-Related Response to Smoothened Inhibition. Cancer Cell 25 (3), 393–405. doi:10.1016/j.ccr.2014.02.004
Le, T.-L., Sribudiani, Y., Dong, X., Huber, C., Kois, C., Baujat, G., et al. (2020). Bi-allelic Variations of SMO in Humans Cause a Broad Spectrum of Developmental Anomalies Due to Abnormal Hedgehog Signaling. Am. J. Hum. Genet. 106 (6), 779–792. doi:10.1016/j.ajhg.2020.04.010
Niida, Y., Togi, S., and Ura, H. (2021). Molecular Bases of Human Malformation Syndromes Involving the SHH Pathway: GLIA/R Balance and Cardinal Phenotypes. Ijms 22 (23), 13060. doi:10.3390/ijms222313060
Patel, R., Singh, S. K., Bhattacharya, V., and Ali, A. (2021). Novel GLI3 Pathogenic Variants in Complex Pre- and Postaxial Polysyndactyly and Greig Cephalopolysyndactyly Syndrome. Am. J. Med. Genet. 185 (1), 97–104. doi:10.1002/ajmg.a.61919
Patterson, V. L., Damrau, C., Paudyal, A., Reeve, B., Grimes, D. T., Stewart, M. E., et al. (2009). Mouse Hitchhiker Mutants Have Spina Bifida, Dorso-Ventral Patterning Defects and Polydactyly: Identification of Tulp3 as a Novel Negative Regulator of the Sonic Hedgehog Pathway. Hum. Mol. Genet. 18 (10), 1719–1739. doi:10.1093/hmg/ddp075
Rahi, S., and Mehan, S. (2020). Understanding Abnormal SMO-SHH Signaling in Autism Spectrum Disorder: Potential Drug Target and Therapeutic Goals. Cell Mol. Neurobiol. 42, 931–953. doi:10.1007/s10571-020-01010-1
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Riddle, R. D., Johnson, R. L., Laufer, E., and Tabin, C. (1993). Sonic Hedgehog Mediates the Polarizing Activity of the ZPA. Cell 75 (7), 1401–1416. doi:10.1016/0092-8674(93)90626-2
Rubino, S., Qian, J., Pinheiro-Neto, C. D., Kenning, T. J., and Adamo, M. A. (2019). A Familial Syndrome of Hypothalamic Hamartomas, Polydactyly, and SMO Mutations: a Clinical Report of 2 Cases. J. Neurosurg. Pediatr. 23 (1), 98–103. doi:10.3171/2018.7.Peds18292
Sigafoos, A. N., Paradise, B. D., and Fernandez-Zapico, M. E. (2021). Hedgehog/GLI Signaling Pathway: Transduction, Regulation, and Implications for Disease. Cancers 13 (14), 3410. doi:10.3390/cancers13143410
Tickle, C., and Barker, H. (2013). The Sonic Hedgehog Gradient in the Developing Limb. WIREs Dev. Biol. 2 (2), 275–290. doi:10.1002/wdev.70
Turkyilmaz, A., Geckinli, B. B., Alavanda, C., Arslan Ates, E., Buyukbayrak, E. E., Eren, S. F., et al. (2021). Meckel-Gruber Syndrome: Clinical and Molecular Genetic Profiles in Two Fetuses and Review of the Current Literature. Genet. Test. Mol. Biomarkers 25 (6), 445–451. doi:10.1089/gtmb.2020.0311
Twigg, S. R. F., Hufnagel, R. B., Miller, K. A., Zhou, Y., McGowan, S. J., Taylor, J., et al. (2016). A Recurrent Mosaic Mutation in SMO , Encoding the Hedgehog Signal Transducer Smoothened, Is the Major Cause of Curry-Jones Syndrome. Am. J. Hum. Genet. 98 (6), 1256–1265. doi:10.1016/j.ajhg.2016.04.007
Ullah, A., Umair, M., Yousaf, M., Khan, S. A., Nazim-Ud-Din, M., Shah, K., et al. (2017). Sequence Variants in Four Genes Underlying Bardet-Biedl Syndrome in Consanguineous Families. Mol. Vis. 23, 482–494.
Umair, M., Ahmad, F., Bilal, M., Ahmad, W., and Alfadhel, M. (2018). Clinical Genetics of Polydactyly: An Updated Review. Front. Genet. 9, 447. doi:10.3389/fgene.2018.00447
Verma, P. K., and El-Harouni, A. A. (2015). Review of Literature: Genes Related to Postaxial Polydactyly. Front. Pediatr. 3, 8. doi:10.3389/fped.2015.00008
Xavier, G. M., Seppala, M., Barrell, W., Birjandi, A. A., Geoghegan, F., and Cobourne, M. T. (2016). Hedgehog Receptor Function during Craniofacial Development. Dev. Biol. 415 (2), 198–215. doi:10.1016/j.ydbio.2016.02.009
Xiang, Y., Li, X., Zhan, Z., Feng, J., Cai, H., Li, Y., et al. (2020). A Novel Nonsense GLI3 Variant Is Associated with Polydactyly and Syndactyly in a Family by Blocking the Sonic Hedgehog Signaling Pathway. Front. Genet. 11, 542004. doi:10.3389/fgene.2020.542004
Yang, Y., Shen, F., Jing, X.-P., Zhang, N., Xu, S.-Y., Li, D.-D., et al. (2021). Case Report: Whole-Exome Sequencing of Hypothalamic Hamartoma from an Infant with Pallister-Hall Syndrome Revealed Novel De Novo Mutation in the GLI3. Front. Surg. 8, 734757. doi:10.3389/fsurg.2021.734757
Yi, X., Yuan, X., Xie, H., Chen, X., and Zhu, Y. (2020). A Familial Sonic Hedgehog (SHH) Stop-Gain Mutation Associated with Agenesis of the Corpus Callosum, Mild Intellectual Disability and Facial Dysmorphism. Brain Dev. 42 (10), 771–774. doi:10.1016/j.braindev.2020.07.004
Zhang, J., Li, Y., Fan, Y., Wu, D., and Xu, J. (2019). Compound Heterozygous Mutations in SMO Associated with Anterior Segment Dysgenesis and Morning Glory Syndrome. Gene 713, 143973. doi:10.1016/j.gene.2019.143973
Zhang, X. M., Ramalho-Santos, M., and McMahon, A. P. (2001). Smoothened Mutants Reveal Redundant Roles for Shh and Ihh Signaling Including Regulation of L/R Asymmetry by the Mouse Node. Cell 105 (6), 781–792. doi:10.1016/s0092-8674(01)00385-3
Keywords: postaxial polydactyly, whole-exome sequencing, bi-allelic variants, smoothened, genetic counseling
Citation: Fan L, Jin P, Qian Y, Shen G, Shen X and Dong M (2022) Case Report: Prenatal Diagnosis of Postaxial Polydactyly With Bi-Allelic Variants in Smoothened (SMO). Front. Genet. 13:887082. doi: 10.3389/fgene.2022.887082
Received: 01 March 2022; Accepted: 13 May 2022;
Published: 22 June 2022.
Edited by:
Long Guo, RIKEN Center for Integrative Medical Sciences, JapanReviewed by:
Asmat Ullah, University of Copenhagen, DenmarkSajid Malik, Quaid-i-Azam University, Pakistan
Copyright © 2022 Fan, Jin, Qian, Shen, Shen and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Minyue Dong, ZG9uZ215QHpqdS5lZHUuY24=