 Jiangyan Xu1
Jiangyan Xu1 Jian Wu
Jian Wu Yuan Wang
Yuan Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet., 26 April 2022
Sec. Applied Genetic Epidemiology
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.880359
Tumor is one of the important factors affecting human life and health in today’s world, and scientists have studied it extensively and deeply, among which autophagy and JAK/STAT3 signaling pathway are two important research directions. The JAK/STAT3 axis is a classical intracellular signaling pathway that assumes a key role in the regulation of cell proliferation, apoptosis, and vascular neogenesis, and its abnormal cell signaling and regulation are closely related to the occurrence and development of tumors. Therefore, the JAK/STAT3 pathway in tumor cells and various stromal cells in their microenvironment is often considered as an effective target for tumor therapy. Autophagy is a process that degrades cytoplasmic proteins and organelles through the lysosomal pathway. It is a fundamental metabolic mechanism for intracellular degradation. The mechanism of action of autophagy is complex and may play different roles at various stages of tumor development. Altered STAT3 expression has been found to be accompanied by the abnormal autophagy activity in many oncological studies, and the two may play a synergistic or antagonistic role in promoting or inhibiting the occurrence and development of tumors. This article reviews the recent advances in autophagy and its interaction with JAK/STAT3 signaling pathway in the pathogenesis, prevention, diagnosis, and treatment of tumors.
Tumor is an ancient disease, which occurs not only in humans but also in plants and animals, and human records of tumors can be traced back thousands of years. The Chinese character “Liu (tumor)" was already found in the oracle bone inscriptions of Yin Ruins in China, and the ancient book “Zhou Li” (The Rites of Zhou), dating 2,400 years ago, recorded that there were doctors specializing in treating swelling and ulcers during the Zhou Dynasty, known as “ulcer doctors”. In the West, there were records of tumors almost at the commence of medical history. In the ancient Egyptian papyrus era, an ointment made of arsenic was applied to treat tumors with ulcers. However, cancer remains a highly fatal disease which has not been conquered by the mankind up to date. According to the Global Cancer Report released by the International Agency for Research on Cancer (IARC), there were 18.1 million new cases of cancer and 9.6 million cancer-related deaths in 2018. The numbers for China were 3.80 million new cases and 2.3 million deaths, ranking first worldwide. Therefore, it is particularly important to investigate the mechanisms, prevention and treatment strategies, and prognoses of tumors.
Tumor had long been regarded as a systemic disease, and it was not until the development of cytopathology that the understanding of tumor histogenesis has been enhanced dramatically. The past century has witnessed the increasingly in-depth research on cancer mechanisms, owing to the development of basic disciplines such as pathology, immunology, and cell biology. The roles of cellular autophagy and JAK/STAT3 signaling pathway in the pathogenesis, development, and treatment of tumors have become hot research topics in recent years (Hu et al., 2020a; Jacquet et al., 2021). Related studies have important significance for the development and prevention of tumor drugs (Khan et al., 2020; Muñoz-Guardiola et al., 2021). This article reviews the recent advances in autophagy and its interaction with JAK/STAT3 signaling pathway in the pathogenesis, prevention, diagnosis, and treatment of tumors.
JAK-STAT signal pathway is a cytokine-stimulated signal transduction pathway and is involved in many important biological processes such as cell proliferation, differentiation, apoptosis, and immune regulation. Compared with other signal pathways, JAK-STAT signal pathway is relatively simple. It is composed of three major components: receptor tyrosine kinase (RTK), Janus kinase (JAK), and signal transducer and activator of transcription (STAT) (Figure 1)
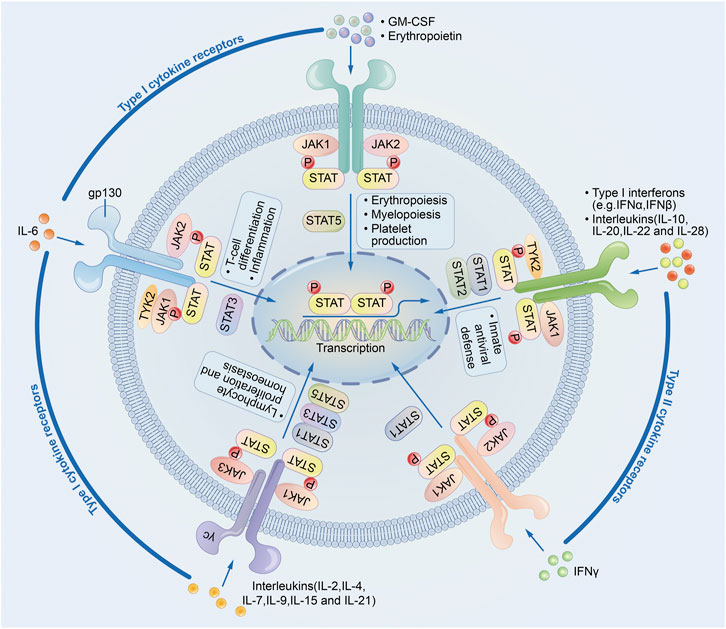
FIGURE 1. JAK-STAT signaling pathway.
JAK has four family members: JAK1, JAK2, JAK3, and Tyk2, which are non-receptor protein tyrosine kinases (PTKs) that play an important role in the signal transduction of cytokine receptor superfamily members (Rodig et al., 1998; Wu et al., 2015). JAKs are the upstream kinases of STAT3, and the phosphorylation of STAT3 is key for its biological activity, so JAKs transmit signals generated by cytokines through the JAK/STAT signaling pathway. Blocking the activity of JAKs can inhibit tumor and inflammation. In a model of acute inflammation of intestinal epithelial cells, JAK-specific siRNAs can significantly inhibit the activity of STAT3, downregulate the expression of IL-1β and TNF-α, inhibit cellular inflammation (Hammarén et al., 2019).
STAT belongs to a unique family of proteins that can bind to DNA. The STAT family includes seven structurally- and functionally-related proteins including STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6. STAT family members usually consist of 750–900 amino acids. Although the protein structures are similar among these seven members, each STAT protein is encoded by separate genes within the cells (O’shea et al., 2004; Sanda et al., 2013; Furtek et al., 2016; Yu et al., 2009). As a transcription factor, STAT3 plays a key role in signaling and transcriptional activation. Unactivated STAT3 is present in the cytoplasm (Singh et al., 1996); when cells are stimulated, STAT3 binds to and is phosphorylated by specific peptides containing phosphorylated tyrosines through its SH2 and SH3 structural domains (Levy and Lee, 2002). Tyrosine phosphorylation is the classical STAT3 activation pathway and is mainly activated by phosphorylation of a specific tyrosine residue (Tyr705) (Wakahara et al., 2012). STAT3 can be directly catalyzed by either receptor tyrosine kinases (RTKs) (e.g., EGFR, KDR, and MET) or non-receptor tyrosine kinases (e.g., JAKs) (Butti et al., 2018). Once activated, STAT3 polymerizes in the form of activated transcription factors, and these transcriptional activators enter the nucleus to promote transcription of target genes (Kisseleva et al., 2002; Steelman et al., 2004; Wu et al., 2017a).
The JAK/STAT3 axis is a classical intracellular signaling pathway that assumes a key role in the regulation of cell proliferation, apoptosis, and vascular neogenesis, and its abnormal cell signaling and regulation are closely related to the occurrence and development of tumors (Xu and Neamati, 2013; Xiong et al., 2014). It was initially believed that STAT3 was an acute-phase gene transcription enhancer activated by IL-6 (Akira et al., 1994). In subsequent studies, STAT3 was confirmed to regulate many functional genes of cells and bodies, showing inducing or suppressing effects. Its downstream signaling pathways regulate many key molecules (e.g., cyclin D1, survivin Bcl-2, and MMP2) related to cell growth/apoptosis, angiogenesis, and tumor metastasis. For example, STAT3 can stimulate cell proliferation by upregulating Bcl-2; in contrast, STAT3 can also lead to downregulation of MYC and MYB, which ultimately results in cell differentiation and growth arrest (Talbot et al., 2014; Wang et al., 2014; Klupp et al., 2015; Wu et al., 2017b). Many studies have demonstrated that STAT3 is not only involved in the various physiological processes mentioned above but also plays a role in malignant transformation of cells (Yu et al., 2009; Bonetto et al., 2012; Min et al., 2019). Excessive activation of STAT3 has been observed in a variety of tumor cells and tissues. When activated, STAT3 is a point of convergence for many oncogenic tyrosine kinase signaling pathways and displays oncogenicity by promoting cell proliferation and blocking apoptosis through multiple pathways (e.g., EGFR, IL-6/JAK, and Src).
In addition, JAK/STAT3 is also associated with tumor metastasis and blood vessel formation. Hu, F et al. confirmed that in colorectal cancer metastasis cells, BECN1 downregulation increased the phosphorylation of STAT3, and that activation of JAK/STAT3 signaling promoted colorectal cancer metastasis (Hu et al., 2020b). In cisplatin-resistant ovarian cancer cells, JAK/STAT3 signaling is hyperactive, with enhanced cell colony-forming capacity and metastasis capacity, which can be reduced by STAT3 inhibitors (Xia et al., 2022). In lung adenocarcinoma cells, highly expressed membrane progesterone receptor α (mPR) activation enhances JAK/STAT3 signaling, increases VEGF secretion in the tumor microenvironment, and promotes tumor blood vessel formation (Xia et al., 2022).
The JAK/STAT3 signaling pathway, as a signaling hub, plays an important role in the tumor epithelial-mesenchymal transformation (EMT). Unno J, et al. showed that activated STAT3 binds to the endoplasmic reticulum (ER), induces LIV-1 expression, enhances EMT in prostate cancer (Unno et al., 2009). In cisplatin-resistant lung cancer cells, Ataxia Telangiectasia Mutated (ATM) upregulates the EMT and metastatic capacity of cancer cells by activating the JAK/STAT signaling pathway, thus mediating cell resistance to cisplatin; inhibition of the JAK/STAT pathway by SiRNA significantly reduced the EMT and invasiveness of the resistant cells. The results indicate that JAK/STAT signaling is a mediator of ATM promoting EMT and metastasis in lung cancer cells (Shen et al., 2019).
The role of JAK/STAT signaling pathway in cancer stem cells (CSC) formation is also important. It was found that IL 8-mediated production of IL 6 and TGF 1 promotes the binding of STAT3 to the AUF 1 promoter, reducing p16, p21 and p53 levels, leading to activation of mammary stromal fibroblasts and inducing CSC formation in breast cancer (Al-Khalaf et al., 2019). Studies have found that chemotherapy resistance in myxoid liposarcoma is associated with the number of cells in CSC subsets, and JAK-STAT signaling can control the number of cells with CSC properties. Researchers use the JAK inhibitor ruxolitinib and doxorubicin, which can overcome chemoresistance resistance in MLS patients (Dolatabadi et al., 2019).
Therefore, the JAK/STAT3 pathway in tumor cells and various stromal cells in their microenvironment is often considered as an effective target for tumor therapy. The inhibitors targeting STAT3 are the focus of current research.
The STAT3 inhibitors are usually divided into the following five types. ①: STAT3 DNA binding domain inhibitor: The regulation of gene expression by STAT3 requires the interaction of STAT3 and DNA; therefore, the inhibition of STAT3 DNA binding domain may reduce the activity of STAT3 and intervene in the regulation of gene expression by STAT3. DBD-1 is a small molecule peptide that specifically recognizes the STAT3 DNA binding domain and causes significant increased apoptosis in murine melanoma B16 cells (Nagel-Wolfrum et al., 2004). It confirms that the STAT3 DNA binding domain may be a novel target for anticancer therapy. ②: STAT3 N-terminal domain inhibitors: The STAT3 N-terminal domain has eight helices, which are involved in STAT3 dimerization, assembly of transcriptional complexes, and binding to promoter. It has been demonstrated that STAT3 inhibits the transcription of pro-apoptotic genes in tumor cells through its N-terminal domain (Timofeeva et al., 2013). Thus, inhibitors targeting the STAT3 N-terminal domain may also inhibit tumorigenesis. ③: STAT3 SH2 domain inhibitor: The STAT3 SH2 domain plays an important role in mediating the interaction between receptor tyrosine residues and its dimerization process (Johnston and Grandis, 2011). Thus, inhibition of the SH2 domain not only interferes with the activation of this transcription factor, but also affects its dimerization. For example, garcinol, a polyisoprenylated benzophenone, was reported that can directly bind to the SH2 domain of STAT3 and inhibit its dimerization and acetylation, thus affecting its binding to DNA (Sethi et al., 2014). ④: Inhibition of the STAT3-importin interaction: Importin is a member of a family of nuclear transport receptors. Importin helps in the nuclear translocation of activated STAT3. In colorectal cancer cells, inhibition of the STAT3-importin interaction interferes with cytoplasmic-nuclear displacement, leading to increase in apoptosis (Souissi et al., 2011). Therefore, the elimination of STAT3 nuclear translocation by pharmacological inhibitors could be used as a promising strategy to prevent and treat tumors. ⑤: Blocking the activity of upstream kinases: The biological activity of STAT3 depends on upstream kinase phosphorylation, so the inhibition of upstream kinase activity is a reasonable way to prevent and treat tumos. For example, inhibition of JAK with synthetic compounds such as AZD1480 ultimately inhibits the growth of human melanoma cells and Hodgkin’s lymphoma cells (Scuto et al., 2011). In addition, compounds of natural origin have been shown to have low toxicity profiles; meanwhile, they have synergistic effects with anticancer drugs and may potentially reverse chemotherapy resistance (Cort and Ozben, 2015). Several small-molecule natural STAT3 inhibitors are currently under investigations (Subramaniam et al., 2013; Siveen et al., 2014a; Siveen et al., 2014b). They have shown significant effects in inhibiting STAT3 in a variety of tumor cell lines and preclinical cancer models and may be used as potential anticancer therapeutic agents.
Autophagy was first observed in rat hepatocytes by Porter and his student Ashford using electron microscopy in 1962 (Shim et al., 2003); Christian de Duve coined the name “autophagy” in 1963 (Kabeya et al., 2000; Zhou, 2016). Autophagy is a process that degrades cytoplasmic proteins and organelles through the lysosomal pathway. It is evolutionarily highly-conserved and is a fundamental metabolic mechanism for intracellular degradation (Mohan et al., 2000). Physiologically, it maintains the dynamic balances between intracellular protein synthesis and degradation and between organelle synthesis and clearance. Therefore, autophagy is often referred to as a recycling process (Cosway and Lovat, 2016). Under pathological conditions, abnormal autophagy often leads to dysfunctional cellular clearance, which in turn triggers the development of a variety of diseases (including neurodegenerative diseases, tumors, muscle diseases, cardiovascular diseases, autoimmune diseases, and tissue fibrosis) by inducing biological effects such as inflammation (Okura and Nakamura, 2012).
The autophagic process is participated by several autophagy-associated proteins (ATGs), which mainly include ULK1/Atg1, ATG5, BECN1/Beclin 1/ATG6, ATG7, LC3, and ATG9A (Mizushima et al., 2011). LC3 exists in two forms, namely cytosolic (LC3-I) or membrane bound (LC3-II). During autophagy, LC3-I in the cytoplasm enzymatically cleaves a small segment of polypeptide and converts them into LC3-II. Therefore, the expression of LC3-II is positively correlated with the level of autophagy and thus can be used to mark the occurrence of autophagy (Klionsky, 2008; Agrotis et al., 2019).
The mechanism of action of autophagy is complex and may play different roles at various stages of tumor development. For example, enhanced autophagic activity may play a role in inhibiting cancer cell proliferation and metastasis during tumorigenesis by clearing the damaged organelles and stabilizing genome; with the progression of the disease, autophagy may promote cancer cell survival and proliferation via the following mechanism: the cancer cells initiate the protective autophagy in a hostile environment to degrade nonessential organelles and proteins, thus offering essential energy support for cell survival (Abraham et al., 2018; Akkoç and Gözüaçık, 2018; Bishop and Bradshaw, 2018; Limpert et al., 2018; Wang et al., 2019; Zamame Ramirez et al., 2020). In mice, loss of the ATG gene and other autophagy regulators makes the animals more prone to tumorigenesis (Mariño et al., 2007); in addition, it has been observed that spontaneous tumors are more likely to occur in Becn1 heterozygous mice, which may be related to their reduced autophagy levels in vivo (Qu et al., 2003); a similar situation was observed in immortalized epithelial cells lacking Becn1 or ATG5, with increased DNA damage, gene amplification, aneuploidy, and enhanced cell tumorigenicity (Mathew et al., 2009). Similarly, Becn1 allele deletion has also been found in two thirds of patients with breast, ovarian, or prostate cancers (Morikawa et al., 2015).
However, activation of autophagy can help the advanced cancers to cope with intracellular and environmental stresses (e.g. hypoxia, nutritional deficiencies, and cancer treatment), thus favoring tumor progression (Macintosh and Ryan, 2013). Yoshihara et al. detected the expression levels of autophagy-related markers (e.g. LC3, p62) in 50 cutaneous SCC specimens, calculated the percentage of positive cells in each low-magnification microscopic field, and assessed their correlation to clinicopathological factors (Yoshihara et al., 2014). The results showed that autophagy was activated during disease progression. As the number of autophagosomes increased, LC3 expression was increased, while p62 expression was decreased. In addition, Sivridis et al. applied IHC technology to measure the thickness and LC3 expression of different skin squamous cell carcinoma (SCC) tissue samples. The results showed that LC3 expression was more in the high thickness group (>6 mm), which was significantly different from SCC in the moderate thickness group (2.1–6 mm) and the mild thickness group (<2 mm), This study suggested that high LC3 expression could be used as a marker of tumor invasive ability in SCC tissue (Sivridis et al., 2011).
Although, autophagy provides an important path for cancer cell survival in cases of nutrient deficiency. However, the induction of autophagy depends on the interaction between the concentration of diet-derived nutrients and their respective nutrient sensors. Studies have shown that diet-derived nutrients concentration plays an important role in regulating autophagy induction, and the concentration signal of growth factors, amino acids and low glucose controls the induction of autophagy through ULK1, ATG13 and RB1CC1 protein (Mack et al., 2012; Saxton and Sabatini, 2017).
Membrane type 1 matrix metalloproteinase (MT1-MMP) functions to degrade extracellular proteins and transduce intracellular signals. A series of studies have elucidated the mechanism by which MT1-MMP promotes autophagy and induced autophagic death in tumor cells. MT1-MMP expression was increased in ConA-activated glioblastoma cells, and the expression of autophagy-related markers Bcl-2, ATG9, and LC3 was also enhanced, with autophagosome formation; after silencing of the MT1-MMP genes by SiRNA, the expression of these genes was decreased and autophagy was inhibited (Pratt et al., 2012; Pratt et al., 2016; Desjarlais and Annabi, 2019).
Many studies have demonstrated that the altered STAT3 expression is accompanied by the abnormal autophagy activity, and they may promote or inhibit the occurrence and development of tumors in a synergistic or antagonistic manner in different tumor stages (Tables 1, 2).

TABLE 1. Regulation of tumor autophagy by STAT3.

TABLE 2. Regulation of STAT3 by autophagy.
Several studies have shown that STAT3 plays an important role in the regulation of autophagy (Yokoyama et al., 2007; Kroemer et al., 2010; Du Toit, 2012; Shen et al., 2012; Maycotte et al., 2014). Shen et al. have found that chemical inhibition of STAT3 induces autophagy, while high STAT3 expression strongly inhibits autophagy both in vitro and in vivo. Notably, different subcellular localization patterns of STAT3 affect autophagy in various ways. For example, activation of nuclear STAT3 is followed by upregulation of Bcl-2 expression, which disrupts the formation of Beclin 1/Vps34 complex and ultimately inhibits autophagy; in contrast, inactivated STAT3 usually induces upregulation of autophagy (Liu et al., 2018). Cytoplasmic STAT3 inhibits autophagy by binding its SH2 domain to the C-terminal (aa259-552) of protein kinase R (PKR), thereby preventing the phosphorylation of eukaryotic initiation factor 2a (EIF2a), which is necessary for the induction of autophagy initiated by the LC3b and ATG5 cascades (Rouschop et al., 2010). In addition, mitochondrial STAT3 inhibits oxidative stress-induced autophagy and drastically protects mitochondria from mitotic degradation (You et al., 2015a).
Many studies have shown that STAT3 phosphorylation negatively regulates autophagy. Siegelin et al. evaluated the in vitro and in vivo efficacy of sorafenib, a multikinase inhibitor, on glioblastoma cells and found that treatment of patient-derived glioblastoma cells with low-concentration sorafenib significantly inhibited cell proliferation and STAT3 phosphorylation and induced apoptosis and autophagy, resulting in significant inhibition of intracranial glioma growth (Siegelin et al., 2010). You et al. found in a study on drug resistance mechanisms in lung cancer that crizotinib induced cytoprotective autophagy by inhibiting STAT3 expression in lung cancer cells, which led to the development of drug resistance (You et al., 2015b). Therefore, when crizotinib is targeted in treating lung cancer patients, inhibition of autophagic activity could improve the efficacy of therapy. Chen et al. demonstrated the antagonistic regulation of autophagy by cyclic HIPK3 (circHIPK3) and linear HIPK3 in lung cancer, in which the deletion of cycHIPK3 induced autophagy through the MIR124-3p-STAT3-PRKAA/AMPKa axis and significantly inhibited cell proliferation, migration, and invasion (Chen et al., 2020). In research on hepatocellular carcinoma (HCC), Tai et al. found that sorafenib and the novel sorafenib derivative SC-59 activated autophagy and inhibited tumor cell growth in a dose- and time-dependent manner in several HCC cell lines via a mechanism that led to disruption of the Beclin 1-Mcl-1 complex through downregulation of p-STAT3, which in turn decreased Mcl-1 expression; however, sorafenib did not affect the amount of Beclin 1 (Tai et al., 2013). Su et al. showed that overexpression of STAT3 in HCC cell lines inhibited autophagy; however, after further dephosphorylation of STAT3 using SC-2001, the release of Beclin 1 increased and the level of autophagy increased significantly (Su et al., 2014).
Real et al. found in estrogen receptor-negative metastatic breast cancer cell lines that activation of the STAT3-Bcl-2 pathway inhibited the autophagy and apoptosis of these cells, increased their survival benefits, and contributed to the development of drug resistance (Real et al., 2018). Qin et al. found that the adding of IL-6 to starvation-induced lymphoma U937 cells significantly increased the phosphorylation level of STAT3, while the level of autophagy was significantly downregulated, suggesting that IL-6 inhibits cellular autophagy through activation of the STAT3 signaling pathway (Qin et al., 2022). Kim et al. reported that an herbal formulation SH003 activated autophagy by decreasing the phosphorylation level of STAT3 and could be used as a therapeutic strategy for hypoxia-mediated chemotherapy resistance (Kim et al., 2020). CYT997 is a novel microtubule-disrupting agent that is expected to be an antitumor candidate for gastric cancer treatment. It has been shown to induce autophagy and apoptosis in gastric cancer cells by activating mitochondrial ROS accumulation and silencing the JAK2/STAT3 pathway (Cao et al., 2020). Blessing et al. have been looking for drugs that induce autophagy in cancer cells to eliminate residual ovarian cancer cells after conventional surgery and chemotherapy; they found that crizotinib induced autophagy and mediated apoptosis in cancer cells by reducing the phosphorylation of STAT3 and Bcl-2 expression when treating ovarian cancer, which provided a new idea for ovarian cancer treatment (Blessing et al., 2020). Chen et al. found that autophagy was induced by knockdown of STAT3 in STK11 mutant lung cancer cell lines (A549 and H838), indicating that circular RNA may be a potential therapeutic target for lung cancer (Chen et al., 2020). In addition to activated STAT3, non-phosphorylated STAT3 in the cytoplasm can also inhibit cellular autophagy (You et al., 2015a). Moreover, in ovarian (Chu et al., 2020), gastric (Nie and Yang, 2017), and colorectal cancers (Zhang et al., 2019), it has been shown that downregulation of p-STAT3 activity increases autophagy of these tumor cells, and the occurrence and progression of these tumors are also suppressed.
However, some studies have also concluded the opposite: STAT3 positively regulates autophagy. Yamada et al. found that the activated IL-6/STAT3 pathway upregulated autophagy levels in pancreatic cancer cells (Yamada et al., 2012). Pratt et al. found in glioblastoma U87 cells that ConA induced overexpression of MT1-MMP gene and protein, followed by increased STAT3 phosphorylation, which finally led to increased autophagy (upregulated expressions of biomarker BNIP3 gene and protein) (Pratt and Annabi, 2014). According to Yang, under glucose-deficient conditions in vitro and in vivo, the kinase activator MOB1A, which plays an important role in many diseases and cancers, plays a key role in the development of gallbladder cancer (GBC) by promoting autophagy through activation of the IL6/STAT3 signaling pathway and modulation of gemcitabine chemosensitivity (Yang et al., 2020).
Chemotherapy resistance is one of the main reasons for the low survival rate of tumor patients, and it has been shown that formation of chemoresistance in cancer cells is associated with its altered autophagic activity. Meng et al. further showed that STAT3 could promote the transcription of activating transcription factor 6 (ATF6), which induced endoplasmic reticulum (ER) stress to enhance cellular autophagy activity. Finally, the cancer cells became resistant to both cisplatin and paclitaxel treatments. This study revealed that chemoresistance in cancer cells may be mediated through STAT3/ATF6-induced autophagy (Meng et al., 2020). A recent study has shown that prostate cancer stem cells (PCSCs) have an enhanced tendency to be chemoresistant and may be a prognostic factor for prostate cancer recurrence; the activated STAT3 modulates chemoresistance in PCSCs by protecting autophagy and regulating MDR1 on the surface of PCSCs, which sheds new light on the selective targeted therapy against PCSCs (Talukdar et al., 2020).
Researches has shown that autophagy mainly shows a positive regulation of STAT3. Inhibition of autophagy by 3-methyladenine (3-MA), a specific autophagy inhibitor, effectively blocks the phosphorylation of STAT3 and NF-κB, suppresses the infiltration of macrophages and lymphocytes, and suppresses the release of multiple pro-fibrotic cytokines/chemokines (Shi et al., 2020). Yeo et al. found that downregulating the autophagy regulator FIP200 in mouse breast cancer models impaired STAT3 or TGFβ/Smad pathway, respectively, and diminished the tumor-initiating properties of both ALDH+ and CD29hiCD61 + breast cancer stem cells, thereby limiting tumor growth and reducing the number of breast cancer stem cells (Yeo et al., 2016). According to Maycotte et al. (Maycotte et al., 2014; Maycotte et al., 2015) and Romero et al. (Romero et al., 2019), different breast cancer cell lines can be either autophagy-dependent or non-autophagy-dependent. For some breast cancer cells, autophagy is essential for survival even in the absence of any nutritional stress. Such a phenotype is even more abundant in triple-negative breast cancer (TNBC) cell lines because the TNBC cell lines are more dependent on STAT3 phosphorylation, whereas autophagy can upregulate p-STAT3; when autophagy is inhibited, p-STAT3 is downregulated and tumor cell growth is inhibited in autophagy-dependent breast cancer cell lines. In contrast, no such phenomenon has been observed in autophagy-independent breast tumors. Therefore, autophagy inhibitors and other drugs may be synergistic when used in combination for autophagy-dependent TNBC, and inhibition of autophagy may be a potential therapeutic strategy for TNBC that currently lacks effective targeted therapy. Similar results have obtained in some studies on pancreatic cancer (Kang et al., 2012; Gong et al., 2014): autophagy activated by RAGE promoted IL-6-induced STAT3 activation; in addition, downregulation of autophagic activity in RAGE-targeted knockout KC mice inhibited STAT3 activation and ATP generation in mitochondria and delayed tumor development.
As a degradation and reuse process of damaged proteins or organelles in eukaryotic cells, autophagy maintains the intracellular environment stable in response to a range of extracellular damages. However, these damages may trigger STAT3 signaling pathway, another key downstream signaling pathway in the stress responses, which is closely related to autophagy.
It has been shown that STAT3 and autophagy are associated with the development of various tumors. STAT3 negatively regulates autophagy in most studies, although some studies showed the opposite. The different results may be explained by the differences in cell lines and/or STAT3 phosphorylation sites. For example, the phosphorylation site used was Ser727 in a pancreatic cancer experiment (Yamada et al., 2012) but was Tyr705 in the U937 cell experiment (Qin et al., 2022013). Although pancreatic cancer cells were used in both experiments, the phosphorylation site played an important role in the localization of p-STAT3 in mitochondria. In addition, different intercellular localization of STAT3 also has different effects on autophagy. Furthermore, they may form a feedback loop, which may yield different results at different nodes of the assay (Jonchère et al., 2013; Myojin et al., 2019). In addition, autophagy can also influence tumor progression by regulating the phosphorylation level of STAT3, although it has only been investigated in a limited number of studies.
Recent studies have shown that autophagy inhibition has promising clinical applications in cancer therapy. Combination of hydroxychloroquine (an autophagy inhibitor) with temsirolimus (an inhibitor of mammalian target of rapamycin, mTOR) (Rangwala et al., 2014) or bortezomib (a proteasome inhibitor) (Vogl et al., 2014) has been found to be tolerable and effective in phase I clinical trials. Thus, autophagy inhibition may be a powerful complement to STAT3-targeted therapies. However, the overall inhibition of autophagy poses a new problem. That is, normal cells have reduced tolerance to harsh environments. Autophagy is a defense mechanism in normal cells, and activation of autophagy has been reported to protect hepatocytes from chemically-induced hepatotoxicity (Ni et al., 2012). Thus, research and development of more targeted drugs or delivery methods remains a challenge for clinical application.
In summary, STAT3 interacts with autophagy in a very complex manner, involving various factors such as phosphorylation sites, mode of action, and subcellular localization. The specific mechanisms warrant further investigations. A series of targeted drugs exert their antitumor effects by blocking STAT3 signaling, which inevitably affects the autophagic pathway (Xia et al., 2018). Therefore, a clearer understanding of the regulatory mechanisms between STAT3 and autophagy will help the R&D of new drugs targeting the STAT3-autophagic pathway and shed new light on tumor prevention and treatment.
The original contributions presented in the study are included in the article. Further inquiries can be directed to the corresponding authors.
YW had the idea for the article; JX and JZ performed the literature search and data analysis; JW and Q-FM drafted and critically revised the work.
This research was supported by Zhejiang Provincial Natural Science Foundation (grant No. LY19H290005 to YW), the science and technology project of Yancheng (grant No. YK2019091 to JZ), and Zhejiang traditional Chinese medicine science and technology plan project (grant No. 2019ZA031 to Q-FM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ATG, autophagy related gene; ATF, activating transcription factor; EGFR, epidermal growth factor receptor; GBC, gallbladder cancer; IARC, The international agency for research on cancer; JAK, janus kinase; KDR, kinase-insert domain-containing receptor; MET, mesenchymmal—epithelial transition Factor; MT1-MMP, membrane-type 1 matrix metalloproteinase; mTOR, mammalian target of rapamycin; mPRα, membrane progesterone receptor α; PCSCs, prostate cancer stem cells; PKR, protein kinase R; RTKs, receptor tyrosine kinases; RAGE, receptor for advanced glycation endproducts; RB1CC1, RB1-inducible coiled-coil 1; STAT, signal transducer and activator of transcription; SLS, stone-like structures; TNBC, triple-negative breast cancer; ULK1, unc-51-like kinase 1; VEGF, vascular endothelial growth factor.
Abraham, N., Kirubel, M., and Abraham, D. (2018). Autophagy as a Possible Target for Cancer Therapy. J. Orthop. Oncol. 4, 1–9. doi:10.4172/2472-016X.1000124
Agrotis, A., Pengo, N., Burden, J. J., and Ketteler, R. (2019). Redundancy of Human ATG4 Protease Isoforms in Autophagy and LC3/GABARAP Processing Revealed in Cells. Autophagy 15 (6), 976–997. doi:10.1080/15548627.2019.1569925
Akira, S., Nishio, Y., Inoue, M., Wang, X.-J., We, S., Matsusaka, T., et al. (1994). Molecular Cloning of APRF, a Novel IFN-Stimulated Gene Factor 3 P91-Related Transcription Factor Involved in the Gp130-Mediated Signaling Pathway. Cell 77 (1), 63–71. doi:10.1016/0092-8674(94)90235-6
Akkoç, Y., and Gözüaçık, D. (2018). Autophagy and Liver Cancer. Turkish J. Gastroenterol. 29 (3), 270. doi:10.5152/tjg.2018.150318
Al-Khalaf, H. H., Ghebeh, H., Inass, R., and Aboussekhra, A. (2019). Senescent Breast Luminal Cells Promote Carcinogenesis through Interleukin-8-dependent Activation of Stromal Fibroblasts. Mol. Cel Biol 39 (2), e00359–18. doi:10.1128/MCB.00359-18
Bishop, E., and Bradshaw, T. D. (2018). Autophagy Modulation: a Prudent Approach in Cancer Treatment? Cancer Chemother. Pharmacol. 82 (6), 913–922. doi:10.1007/s00280-018-3669-6
Blessing, A. M., Santiago‐O'Farrill, J. M., Mao, W., Pang, L., Ning, J., Pak, D., et al. (2020). Elimination of Dormant, Autophagic Ovarian Cancer Cells and Xenografts through Enhanced Sensitivity to Anaplastic Lymphoma Kinase Inhibition. Cancer 126 (15), 3579–3592. doi:10.1002/cncr.32985
Bonetto, A., Aydogdu, T., Jin, X., Zhang, Z., Zhan, R., Puzis, L., et al. (2012). JAK/STAT3 Pathway Inhibition Blocks Skeletal Muscle Wasting Downstream of IL-6 and in Experimental Cancer Cachexia. Am. J. Physiology-Endocrinology Metab. 303 (3), E410–E421. doi:10.1152/ajpendo.00039.2012
Butti, R., Das, S., Gunasekaran, V. P., Yadav, A. S., Kumar, D., and Kundu, G. C. (2018). Receptor Tyrosine Kinases (RTKs) in Breast Cancer: Signaling, Therapeutic Implications and Challenges. Mol. Cancer 17 (1), 34–18. doi:10.1186/s12943-018-0797-x
Cao, Y., Wang, J., Tian, H., and Fu, G. H. (2020). Mitochondrial ROS Accumulation Inhibiting JAK2/STAT3 Pathway Is a Critical Modulator of CYT997-Induced Autophagy and Apoptosis in Gastric Cancer. J. Exp. Clin. Cancer Res. 39 (1), 119–215. doi:10.1186/s13046-020-01621-y
Chen, X., Mao, R., Su, W., Yang, X., Geng, Q., Guo, C., et al. (2020). Circular RNA circHIPK3 Modulates Autophagy via MIR124-3p-STAT3-Prkaa/ampkα Signaling in STK11 Mutant Lung Cancer. Autophagy 16 (4), 659–671. doi:10.1080/15548627.2019.1634945
Chu, Y., Wang, Y., Li, K., Liu, M., Zhang, Y., Li, Y., et al. (2020). Human Omental Adipose-Derived Mesenchymal Stem Cells Enhance Autophagy in Ovarian Carcinoma Cells through the STAT3 Signalling Pathway. Cell Signal. 69, 109549. doi:10.1016/j.cellsig.2020.109549
Cort, A., and Ozben, T. (2015). Natural Product Modulators to Overcome Multidrug Resistance in Cancer. Nutr. Cancer 67 (3), 411–423. doi:10.1080/01635581.2015.1002624
Cosway, B., and Lovat, P. (2016). The Role of Autophagy in Squamous Cell Carcinoma of the Head and Neck. Oral Oncol. 54, 1–6. doi:10.1016/j.oraloncology.2015.12.007
Desjarlais, M., and Annabi, B. (2019). Dual Functions of ARP101 in Targeting Membrane Type-1 Matrix Metalloproteinase: Impact on U87 Glioblastoma Cell Invasion and Autophagy Signaling. Chem. Biol. Drug Des. 93 (3), 272–282. doi:10.1111/cbdd.13410
Dolatabadi, S., Jonasson, E., Lindén, M., Fereydouni, B., Bäcksten, K., Nilsson, M., et al. (2019). JAK-STAT Signalling Controls Cancer Stem Cell Properties Including Chemotherapy Resistance in Myxoid Liposarcoma. Int. J. Cancer 145 (2), 435–449. doi:10.1002/ijc.32123
Du Toit, A. (2012). STAT3 Maintains Order. Nat. Rev. Mol. Cel Biol 13 (12), 754. doi:10.1038/nrm3472
Furtek, S. L., Backos, D. S., Matheson, C. J., and Reigan, P. (2016). Strategies and Approaches of Targeting STAT3 for Cancer Treatment. ACS Chem. Biol. 11 (2), 308–318. doi:10.1021/acschembio.5b00945
Gong, J., Muñoz, A. R., Chan, D., Ghosh, R., and Kumar, A. P. (2014). STAT3 Down Regulates LC3 to Inhibit Autophagy and Pancreatic Cancer Cell Growth. Oncotarget 5 (9), 2529–2541. doi:10.18632/oncotarget.1810
Hammarén, H. M., Virtanen, A. T., Raivola, J., and Silvennoinen, O. (2019). The Regulation of JAKs in Cytokine Signaling and its Breakdown in Disease. Cytokine 118, 48–63. doi:10.1016/j.cyto.2018.03.041
Hu, F., Li, G., Huang, C., Hou, Z., Yang, X., Luo, X., et al. (2020). The Autophagy-independent Role of BECN1 in Colorectal Cancer Metastasis through Regulating STAT3 Signaling Pathway Activation. Cell Death Dis 11 (5), 304–313. doi:10.1038/s41419-020-2467-3
Hu, H., Zhang, Q., Chen, W., Wu, T., Liu, S., Li, X., et al. (2020). MicroRNA-301a Promotes Pancreatic Cancer Invasion and Metastasis through the JAK/STAT3 Signaling Pathway by Targeting SOCS5. Carcinogenesis 41 (4), 502–514. doi:10.1093/carcin/bgz121
Jacquet, M., Guittaut, M., Fraichard, A., and Despouy, G. (2021). The Functions of Atg8-Family Proteins in Autophagy and Cancer: Linked or Unrelated? Autophagy 17 (3), 599–611. doi:10.1080/15548627.2020.1749367
Johnston, P. A., and Grandis, J. R. (2011). STAT3 Signaling: Anticancer Strategies and Challenges. Mol. interventions 11 (1), 18–26. doi:10.1124/mi.11.1.4
Jonchère, B., Bélanger, A., Guette, C., Barré, B., and Coqueret, O. (2013). STAT3 as a New Autophagy Regulator. Jak-Stat 2 (3), 667–680. doi:10.4161/jkst.24353
Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda, T., et al. (2000). LC3, a Mammalian Homologue of Yeast Apg8p, Is Localized in Autophagosome Membranes after Processing. EMBO J. 19 (21), 5720–5728. doi:10.1093/emboj/19.21.5720
Kang, R., Loux, T., Tang, D., Schapiro, N. E., Vernon, P., Livesey, K. M., et al. (2012). The Expression of the Receptor for Advanced Glycation Endproducts (RAGE) Is Permissive for Early Pancreatic Neoplasia. Proc. Natl. Acad. Sci. U.S.A. 109 (18), 7031–7036. doi:10.1073/pnas.1113865109
Khan, A. Q., Ahmed, E. I., Elareer, N., Fathima, H., Prabhu, K. S., Siveen, K. S., et al. (2020). Curcumin-mediated Apoptotic Cell Death in Papillary Thyroid Cancer and Cancer Stem-like Cells through Targeting of the JAK/STAT3 Signaling Pathway. Ijms 21 (2), 438. doi:10.3390/ijms21020438
Kim, T. W., Cheon, C., and Ko, S. G. (2020). SH003 Activates Autophagic Cell Death by Activating ATF4 and Inhibiting G9a under Hypoxia in Gastric Cancer Cells. Cel Death Dis 11 (8), 717–814. doi:10.1038/s41419-020-02924-w
Kisseleva, T., Bhattacharya, S., Braunstein, J., and Schindler, C. W. (2002). Signaling through the JAK/STAT Pathway, Recent Advances and Future Challenges. Gene 285 (1-2), 1–24. doi:10.1016/s0378-1119(02)00398-0
Klionsky, D. J. (2008). Autophagy revisited: a conversation with Christian de Duve. Autophagy 4 (6), 740–743. doi:10.4161/auto.6398
Klupp, F., Diers, J., Kahlert, C., Neumann, L., Halama, N., Franz, C., et al. (2015). Expressional STAT3/STAT5 Ratio Is an Independent Prognostic Marker in colon Carcinoma. Ann. Surg. Oncol. 22 Suppl 3 (3), S1548–S1555. doi:10.1245/s10434-015-4485-4
Kroemer, G., Mariño, G., and Levine, B. (2010). Autophagy and the Integrated Stress Response. Mol. Cel. 40 (2), 280–293. doi:10.1016/j.molcel.2010.09.023
Levy, D. E., and Lee, C.-k. (2002). What Does Stat3 Do? J. Clin. Invest. 109 (9), 1143–1148. doi:10.1172/jci0215650
Limpert, A. S., Lambert, L. J., Bakas, N. A., Bata, N., Brun, S. N., Shaw, R. J., et al. (2018). Autophagy in Cancer: Regulation by Small Molecules. Trends Pharmacological Sciences 39 (12), 1021–1032. doi:10.1016/j.tips.2018.10.004
Liu, D., Lin, J., Su, J., Chen, X., Jiang, P., and Huang, K. (2018). Glutamine Deficiency Promotes PCV2 Infection through Induction of Autophagy via Activation of ROS-Mediated JAK2/STAT3 Signaling Pathway. J. Agric. Food Chem. 66 (44), 11757–11766. doi:10.1021/acs.jafc.8b04704
Mack, H. I. D., Zheng, B., Asara, J. M., and Thomas, S. M. (2012). AMPK-dependent Phosphorylation of ULK1 Regulates ATG9 Localization. Autophagy 8 (8), 1197–1214. doi:10.4161/auto.20586
Mariño, G., Salvador-Montoliu, N., Fueyo, A., Knecht, E., Mizushima, N., and López-Otín, C. (2007). Tissue-specific Autophagy Alterations and Increased Tumorigenesis in Mice Deficient in Atg4C/autophagin-3. J. Biol. Chem. 282 (25), 18573–18583. doi:10.1074/jbc.M701194200
Mathew, R., Karp, C. M., Beaudoin, B., Vuong, N., Chen, G., Chen, H.-Y., et al. (2009). Autophagy Suppresses Tumorigenesis through Elimination of P62. Cell 137 (6), 1062–1075. doi:10.1016/j.cell.2009.03.048
Maycotte, P., Gearheart, C. M., Barnard, R., Aryal, S., Mulcahy Levy, J. M., Fosmire, S. P., et al. (2014). STAT3-mediated Autophagy Dependence Identifies Subtypes of Breast Cancer where Autophagy Inhibition Can Be Efficacious. Cancer Res. 74 (9), 2579–2590. doi:10.1158/0008-5472.can-13-3470
Maycotte, P., Jones, K. L., Goodall, M. L., Thorburn, J., and Thorburn, A. (2015). Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Mol. Cancer Res. 13 (4), 651–658. doi:10.1158/1541-7786.mcr-14-0487
Meng, J., Liu, K., Shao, Y., Feng, X., Ji, Z., Chang, B., et al. (2020). ID1 Confers Cancer Cell Chemoresistance through STAT3/ATF6-Mediated Induction of Autophagy. Cel Death Dis 11 (2), 137–216. doi:10.1038/s41419-020-2327-1
Min, T. R., Park, H. J., Ha, K. T., Chi, G. Y., Choi, Y. H., and Park, S. H. (2019). Suppression of EGFR/STAT3 Activity by Lupeol Contributes to the Induction of the Apoptosis of Human Non-small C-ell L-ung C-ancer C-ells. Int. J. Oncol. 55 (1), 320–330. doi:10.3892/ijo.2019.4799
Mizushima, N., Yoshimori, T., and Ohsumi, Y. (2011). The Role of Atg Proteins in Autophagosome Formation. Annu. Rev. Cel Dev. Biol. 27, 107–132. doi:10.1146/annurev-cellbio-092910-154005
Mohan, R., Sivak, J., Ashton, P., Russo, L. A., Pham, B. Q., Kasahara, N., et al. (2000). Curcuminoids Inhibit the Angiogenic Response Stimulated by Fibroblast Growth Factor-2, Including Expression of Matrix Metalloproteinase Gelatinase B. J. Biol. Chem. 275 (14), 10405–10412. doi:10.1074/jbc.275.14.10405
Morikawa, A., Takeuchi, T., Kito, Y., Saigo, C., Sakuratani, T., Futamura, M., et al. (2015). Expression of Beclin-1 in the Microenvironment of Invasive Ductal Carcinoma of the Breast: Correlation with Prognosis and the Cancer-Stromal Interaction. PLoS One 10 (5), e0125762. doi:10.1371/journal.pone.0125762
Muñoz-Guardiola, P., Casas, J., Megías-Roda, E., Solé, S., Perez-Montoyo, H., Yeste-Velasco, M., et al. (2021). The Anti-cancer Drug ABTL0812 Induces ER Stress-Mediated Cytotoxic Autophagy by Increasing Dihydroceramide Levels in Cancer Cells. Autophagy 17 (6), 1349–1366. doi:10.1080/15548627.2020.1761651
Myojin, Y., Hikita, H., Kodama, T., Makino, Y., Yamada, R., Saito, Y., et al. (2019). PS-045-HCC Promotes Autophagy in Hepatic Stellate Cells, Leading to HCC Progression via IL-6/STAT3 Signaling. J. Hepatol. 70 (1), e28. doi:10.1016/s0618-8278(19)30051-9
Nagel-Wolfrum, K., Buerger, C., Wittig, I., Butz, K., Hoppe-Seyler, F., and Groner, B. (2004). The Interaction of Specific Peptide Aptamers with the DNA Binding Domain and the Dimerization Domain of the Transcription Factor Stat3 Inhibits Transactivation and Induces Apoptosis in Tumor Cells. Mol. Cancer Res. 2 (3), 170–182. doi:10.1016/j.nimb.2009.01.080
Ni, H.-M., Bockus, A., Boggess, N., Jaeschke, H., and Ding, W.-X. (2012). Activation of Autophagy Protects against Acetaminophen-Induced Hepatotoxicity. Hepatology 55 (1), 222–232. doi:10.1002/hep.24690
Nie, G. Q. W. X., and Yang, M. Y. (2017). E804 a Derivative of Indirubin,promotes Autophagy of Gastric Cancer Cells through Stat3 Signaling Pathway. World Chin. J. Digestology 25, 3184–3190. doi:10.11569/wcjd.v25.i36.3184
O'shea, J. J., Pesu, M., Borie, D. C., and Changelian, P. S. (2004). A New Modality for Immunosuppression: Targeting the JAK/STAT Pathway. Nat. Rev. Drug Discov. 3 (7), 555–564. doi:10.1038/nrd1441
Okura, R., and Nakamura, M. (2012). Overexpression of Autophagy-Related Beclin-1 in Cutaneous Squamous Cell Carcinoma with Lymph-Node Metastasis. Eur. J. Dermatol. 21 (6), 1002–1003. doi:10.1684/ejd.2011.1516
Pratt, J., and Annabi, B. (2014). Induction of Autophagy Biomarker BNIP3 Requires a JAK2/STAT3 and MT1-MMP Signaling Interplay in Concanavalin-A-Activated U87 Glioblastoma Cells. Cell Signal. 26 (5), 917–924. doi:10.1016/j.cellsig.2014.01.012
Pratt, J., Iddir, M., Bourgault, S., and Annabi, B. (2016). Evidence of MTCBP-1 Interaction with the Cytoplasmic Domain of MT1-MMP: Implications in the Autophagy Cell index of High-Grade Glioblastoma. Mol. Carcinog. 55 (2), 148–160. doi:10.1002/mc.22264
Pratt, J., Roy, R., and Annabi, B. (2012). Concanavalin-A-induced Autophagy Biomarkers Requires Membrane Type-1 Matrix Metalloproteinase Intracellular Signaling in Glioblastoma Cells. Glycobiology 22 (9), 1245–1255. doi:10.1093/glycob/cws093
Qin, B., He, J., and Ding, S. (2022). IL-6 Inhibited Starvation-Induced Autophagy through STAT3/Bcl2/Beclin1 Pathway. Cytokine 63 (3), 291. doi:10.1016/j.cyto.2013.06.205
Qu, X., Yu, J., Bhagat, G., Furuya, N., Hibshoosh, H., Troxel, A., et al. (2003). Promotion of Tumorigenesis by Heterozygous Disruption of the Beclin 1 Autophagy Gene. J. Clin. Invest. 112 (12), 1809–1820. doi:10.1172/jci20039
Rangwala, R., Chang, Y. C., Hu, J., Algazy, K. M., Evans, T. L., Fecher, L. A., et al. (2014). Combined MTOR and Autophagy Inhibition. Autophagy 10 (8), 1391–1402. doi:10.4161/auto.29119
Real, S. A. S., Parveen, F., Rehman, A. U., Khan, M. A., Deo, S. V. S., Shukla, N. K., et al. (2018). Aberrant Promoter Methylation of YAP Gene and its Subsequent Downregulation in Indian Breast Cancer Patients. BMC cancer 18 (1), 711–715. doi:10.1186/s12885-018-4627-8
R. L. Macintosh, and K. M. Ryan (Editors) (2013). “Autophagy in Tumour Cell Death,” Seminars in Cancer Biology (Elsevier).
Rodig, S. J., Meraz, M. A., White, J. M., Lampe, P. A., Riley, J. K., Arthur, C. D., et al. (1998). Disruption of the Jak1 Gene Demonstrates Obligatory and Nonredundant Roles of the Jaks in Cytokine-Induced Biologic Responses. Cell 93 (3), 373–383. doi:10.1016/s0092-8674(00)81166-6
Romero, M. A., Bayraktar Ekmekcigil, O., Bagca, B. G., Avci, C. B., Sabitaliyevich, U. Y., Zhenisovna, T. G., et al. (2019). Role of Autophagy in Breast Cancer Development and Progression: Opposite Sides of the Same coin. Breast Cancer Metastasis Drug Resist. 10, 65–73. doi:10.1007/978-3-030-20301-6_5
Rouschop, K. M. A., van den Beucken, T., Dubois, L., Niessen, H., Bussink, J., Savelkouls, K., et al. (2010). The Unfolded Protein Response Protects Human Tumor Cells during Hypoxia through Regulation of the Autophagy Genes MAP1LC3B and ATG5. J. Clin. Invest. 120 (1), 127–141. doi:10.1172/jci40027
Sanda, T., Tyner, J. W., Gutierrez, A., Ngo, V. N., Glover, J., Chang, B. H., et al. (2013). TYK2-STAT1-BCL2 Pathway Dependence in T-Cell Acute Lymphoblastic Leukemia. Cancer Discov. 3 (5), 564–577. doi:10.1158/2159-8290.cd-12-0504
Saxton, R. A., and Sabatini, D. M. (2017). mTOR Signaling in Growth, Metabolism, and Disease. Cell 168 (6), 960–976. doi:10.1016/j.cell.2017.02.004
Scuto, A., Krejci, P., Popplewell, L., Wu, J., Wang, Y., Kujawski, M., et al. (2011). The Novel JAK Inhibitor AZD1480 Blocks STAT3 and FGFR3 Signaling, Resulting in Suppression of Human Myeloma Cell Growth and Survival. Leukemia 25 (3), 538–550. doi:10.1038/leu.2010.289
Sethi, G., Chatterjee, S., Rajendran, P., Li, F., Shanmugam, M. K., Wong, K. F., et al. (2014). Inhibition of STAT3 Dimerization and Acetylation by Garcinol Suppresses the Growth of Human Hepatocellular Carcinoma In Vitro and In Vivo. Mol. Cancer 13 (1), 66–14. doi:10.1186/1476-4598-13-66
Shen, M., Xu, Z., Xu, W., Jiang, K., Zhang, F., Ding, Q., et al. (2019). Inhibition of ATM Reverses EMT and Decreases Metastatic Potential of Cisplatin-Resistant Lung Cancer Cells through JAK/STAT3/PD-L1 Pathway. J. Exp. Clin. Cancer Res. 38 (1), 149–214. doi:10.1186/s13046-019-1161-8
Shen, S., Niso-Santano, M., Adjemian, S., Takehara, T., Malik, S. A., Minoux, H., et al. (2012). Cytoplasmic STAT3 Represses Autophagy by Inhibiting PKR Activity. Mol. Cel. 48 (5), 667–680. doi:10.1016/j.molcel.2012.09.013
Shi, Y., Tao, M., Ma, X., Hu, Y., Huang, G., Qiu, A., et al. (2020). Delayed Treatment with an Autophagy Inhibitor 3-MA Alleviates the Progression of Hyperuricemic Nephropathy. Cel Death Dis 11 (6), 467–516. doi:10.1038/s41419-020-2673-z
Shim, J. S., Kim, J. H., Cho, H. Y., Yum, Y. N., Kim, S. H., Park, H.-J., et al. (2003). Irreversible Inhibition of CD13/aminopeptidase N by the Antiangiogenic Agent Curcumin. Chem. Biol. 10 (8), 695–704. doi:10.1016/s1074-5521(03)00169-8
Siegelin, M. D., Raskett, C. M., Gilbert, C. A., Ross, A. H., and Altieri, D. C. (2010). Sorafenib Exerts Anti-glioma Activity In Vitro and In Vivo. Neurosci. Lett. 478 (3), 165–170. doi:10.1016/j.neulet.2010.05.009
Singh, A. K., Sidhu, G. S., Deepa, T., and Maheshwari, R. K. (1996). Curcumin Inhibits the Proliferation and Cell Cycle Progression of Human Umbilical Vein Endothelial Cell. Cancer Lett. 107 (1), 109–115. doi:10.1016/0304-3835(96)04357-1
Siveen, K. S., Nguyen, A. H., Lee, J. H., Li, F., Singh, S. S., Kumar, A. P., et al. (2014). Negative Regulation of Signal Transducer and Activator of Transcription-3 Signalling cascade by Lupeol Inhibits Growth and Induces Apoptosis in Hepatocellular Carcinoma Cells. Br. J. Cancer 111 (7), 1327–1337. doi:10.1038/bjc.2014.422
Siveen, K. S., Sikka, S., Surana, R., Dai, X., Zhang, J., Kumar, A. P., et al. (2014). Targeting the STAT3 Signaling Pathway in Cancer: Role of Synthetic and Natural Inhibitors. Biochim. Biophys. Acta (Bba) - Rev. Cancer 1845 (2), 136–154. doi:10.1016/j.bbcan.2013.12.005
Sivridis, E., Giatromanolaki, A., Karpathiou, G., Karpouzis, A., Kouskoukis, C., and Koukourakis, M. I. (2011). LC3A-Positive "Stone-like" Structures in Cutaneous Squamous Cell Carcinomas. The Am. J. dermatopathology 33 (3), 285–290. doi:10.1097/dad.0b013e3181f10de0
Souissi, I., Najjar, I., Ah-Koon, L., Schischmanoff, P. O., Lesage, D., Le Coquil, S., et al. (2011). A STAT3-Decoy Oligonucleotide Induces Cell Death in a Human Colorectal Carcinoma Cell Line by Blocking Nuclear Transfer of STAT3 and STAT3-Bound NF-Κb. BMC Cel Biol 12 (1), 14–19. doi:10.1186/1471-2121-12-14
Steelman, L. S., Pohnert, S. C., Shelton, J. G., Franklin, R. A., Bertrand, F. E., and McCubrey, J. A. (2004). JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in Cell Cycle Progression and Leukemogenesis. Leukemia 18 (2), 189–218. doi:10.1038/sj.leu.2403241
Su, J.-C., Tseng, P.-H., Hsu, C.-Y., Tai, W.-T., Huang, J.-W., Ko, C.-H., et al. (2014). RFX1-dependent Activation of SHP-1 Induces Autophagy by a Novel Obatoclax Derivative in Hepatocellular Carcinoma Cells. Oncotarget 5 (13), 4909–4919. doi:10.18632/oncotarget.2054
Subramaniam, A., Shanmugam, M. K., Ong, T. H., Li, F., Perumal, E., Chen, L., et al. (2013). Emodin Inhibits Growth and Induces Apoptosis in an Orthotopic Hepatocellular Carcinoma Model by Blocking Activation of STAT3. Br. J. Pharmacol. 170 (4), 807–821. doi:10.1111/bph.12302
Tai, W.-T., Shiau, C.-W., Chen, H.-L., Liu, C.-Y., Lin, C.-S., Cheng, A.-L., et al. (2013). Mcl-1-dependent Activation of Beclin 1 Mediates Autophagic Cell Death Induced by Sorafenib and SC-59 in Hepatocellular Carcinoma Cells. Cel Death Dis 4 (2), e485. doi:10.1038/cddis.2013.18
Talbot, J. J., Song, X., Wang, X., Rinschen, M. M., Doerr, N., LaRiviere, W. B., et al. (2014). The Cleaved Cytoplasmic Tail of Polycystin-1 Regulates Src-dependent STAT3 Activation. Jasn 25 (8), 1737–1748. doi:10.1681/asn.2013091026
Talukdar, S., Das, S. K., Pradhan, A. K., Emdad, L., Windle, J. J., Sarkar, D., et al. (2020). MDA-9/Syntenin (SDCBP) Is a Critical Regulator of Chemoresistance, Survival and Stemness in Prostate Cancer Stem Cells. Cancers 12 (1), 53. doi:10.3390/cancers12010053
Timofeeva, O. A., Tarasova, N. I., Zhang, X., Chasovskikh, S., Cheema, A. K., Wang, H., et al. (2013). STAT3 Suppresses Transcription of Proapoptotic Genes in Cancer Cells with the Involvement of its N-Terminal Domain. Proc. Natl. Acad. Sci. U.S.A. 110 (4), 1267–1272. doi:10.1073/pnas.1211805110
Unno, J., Satoh, K., Hirota, M., Kanno, A., Hamada, S., Ito, H., et al. (2009). LIV-1 Enhances the Aggressive Phenotype through the Induction of Epithelial to Mesenchymal Transition in Human Pancreatic Carcinoma Cells. Int. J. Oncol. 35 (4), 813–821. doi:10.3892/ijo_00000394
Vogl, D. T., Stadtmauer, E. A., Tan, K.-S., Heitjan, D. F., Davis, L. E., Pontiggia, L., et al. (2014). Combined Autophagy and Proteasome Inhibition. Autophagy 10 (8), 1380–1390. doi:10.4161/auto.29264
Wakahara, R., Kunimoto, H., Tanino, K., Kojima, H., Inoue, A., Shintaku, H., et al. (2012). Phospho-Ser727 of STAT3 Regulates STAT3 Activity by Enhancing Dephosphorylation of Phospho-Tyr705 Largely through TC45. Genes to Cells 17 (2), 132–145. doi:10.1111/j.1365-2443.2011.01575.x
Wang, J., Zhang, L., Chen, G., Zhang, J., Li, Z., Lu, W., et al. (2014). Small Molecule 1′-acetoxychavicol Acetate Suppresses Breast Tumor Metastasis by Regulating the SHP-1/STAT3/MMPs Signaling Pathway. Breast Cancer Res. Treat. 148 (2), 279–289. doi:10.1007/s10549-014-3165-6
Wang, Y., Wu, J., Xu, J., and Lin, S. (2019). Clinical Significance of High Expression of Stanniocalcin-2 in Hepatocellular Carcinoma. Biosci. Rep. 39 (4). doi:10.1042/BSR20182057
Wu, J., Chen, Z.-P., Shang, A.-Q., Wang, W.-W., Chen, Z.-N., Tao, Y.-J., et al. (2017). Systemic Bioinformatics Analysis of Recurrent Aphthous Stomatitis Gene Expression Profiles. Oncotarget 8 (67), 111064–111072. doi:10.18632/oncotarget.22347
Wu, J., Cui, L.-L., Yuan, J., Wang, Y., and Song, S. (2017). Clinical Significance of the Phosphorylation of MAPK and Protein Expression of Cyclin D1 in Human Osteosarcoma Tissues. Mol. Med. Rep. 15 (4), 2303–2307. doi:10.3892/mmr.2017.6224
Wu, J., Lu, W.-Y., and Cui, L.-L. (2015). Clinical Significance of STAT3 and MAPK Phosphorylation, and the Protein Expression of Cyclin D1 in Skin Squamous Cell Carcinoma Tissues. Mol. Med. Rep. 12 (6), 8129–8134. doi:10.3892/mmr.2015.4460
Xia, T. H. H., Zhu, Q. A., and Xiong, X. (2018). Experimental Study of miRNA-26b Targeting IL-6/STAT3 Signaling Pathway Induces Autophagy in Hepatocellular Carcinoma Cells. Chin. Clin. Oncol. 23, 593–597. doi:10.3969/j.issn.1009-0460.2018.07.004
Xia, Z., Xiao, J., Dai, Z., and Chen, Q. (2022). Membrane Progesterone Receptor α (mPRα) Enhances Hypoxia-Induced Vascular Endothelial Growth Factor Secretion and Angiogenesis in Lung Adenocarcinoma through STAT3 Signaling. J. Transl Med. 20 (1), 72–14. doi:10.1186/s12967-022-03270-5
Xiong, A., Yang, Z., Shen, Y., Zhou, J., and Shen, Q. (2014). Transcription Factor STAT3 as a Novel Molecular Target for Cancer Prevention. Cancers 6 (2), 926–957. doi:10.3390/cancers6020926
Xu, S., and Neamati, N. (2013). gp130: a Promising Drug Target for Cancer Therapy. Expert Opin. Ther. Targets 17 (11), 1303–1328. doi:10.1517/14728222.2013.830105
Yamada, E., Bastie, C. C., Koga, H., Wang, Y., Cuervo, A. M., and Pessin, J. E. (2012). Mouse Skeletal Muscle Fiber-type-specific Macroautophagy and Muscle Wasting Are Regulated by a Fyn/STAT3/Vps34 Signaling Pathway. Cel Rep. 1 (5), 557–569. doi:10.1016/j.celrep.2012.03.014
Yang, B., Li, Y., Zhang, R., Liu, L., Miao, H., Li, Y., et al. (2020). MOB1A Regulates Glucose Deprivation-Induced Autophagy via IL6-STAT3 Pathway in Gallbladder Carcinoma. Am. J. Cancer Res. 10 (11), 3896–3910.
Yeo, S. K., Wen, J., Chen, S., and Guan, J.-L. (2016). Autophagy Differentially Regulates Distinct Breast Cancer Stem-like Cells in Murine Models via EGFR/Stat3 and Tgfβ/Smad Signaling. Cancer Res. 76 (11), 3397–3410. doi:10.1158/0008-5472.can-15-2946
Yokoyama, T., Kondo, Y., and Kondo, S. (2007). Roles of mTOR and STAT3 in Autophagy Induced by Telomere 3' Overhang-specific DNA Oligonucleotides. Autophagy 3 (5), 496–498. doi:10.4161/auto.4602
Yoshihara, N., Takagi, A., Ueno, T., and Ikeda, S. (2014). Inverse Correlation between Microtubule-Associated Protein 1A/1B-Light Chain 3 and P62/sequestosome-1 Expression in the Progression of Cutaneous Squamous Cell Carcinoma. J. Dermatol. 41 (4), 311–315. doi:10.1111/1346-8138.12439
You, L., Shou, J., Deng, D., Jiang, L., Jing, Z., Yao, J., et al. (2015). Crizotinib Induces Autophagy through Inhibition of the STAT3 Pathway in Multiple Lung Cancer Cell Lines. Oncotarget 6 (37), 40268–40282. doi:10.18632/oncotarget.5592
You, L., Wang, Z., Li, H., Shou, J., Jing, Z., Xie, J., et al. (2015). The Role of STAT3 in Autophagy. Autophagy 11 (5), 729–739. doi:10.1080/15548627.2015.1017192
Yu, H., Pardoll, D., and Jove, R. (2009). STATs in Cancer Inflammation and Immunity: a Leading Role for STAT3. Nat. Rev. Cancer 9 (11), 798–809. doi:10.1038/nrc2734
Zamame Ramirez, J. A., Romagnoli, G. G., Falasco, B. F., Gorgulho, C. M., Sanzochi Fogolin, C., Dos Santos, D. C., et al. (2020). Blocking Drug-Induced Autophagy with Chloroquine in HCT-116 colon Cancer Cells Enhances DC Maturation and T Cell Responses Induced by Tumor Cell Lysate. Int. immunopharmacology 84, 106495. doi:10.1016/j.intimp.2020.106495
Zhang, J., Chu, D., Kawamura, T., Tanaka, K., and He, S. (2019). GRIM‐19 Repressed Hypoxia‐induced Invasion and EMT of Colorectal Cancer by Repressing Autophagy through Inactivation of STAT3/HIF‐1α Signaling axis. J. Cel Physiol 234 (8), 12800–12808. doi:10.1002/jcp.27914
Keywords: autophagy, JAK/STAT3, interaction, regulation, tumor
Citation: Xu J, Zhang J, Mao Q-F, Wu J and Wang Y (2022) The Interaction Between Autophagy and JAK/STAT3 Signaling Pathway in Tumors. Front. Genet. 13:880359. doi: 10.3389/fgene.2022.880359
Received: 21 February 2022; Accepted: 21 March 2022;
Published: 26 April 2022.
Edited by:
Zhenjian Zhuo, Guangzhou Medical University, ChinaReviewed by:
Borhane Annabi, Université du Québec à Montréal, CanadaCopyright © 2022 Xu, Zhang, Mao, Wu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuan Wang, d3kwMDM3QGhtYy5lZHUuY24=; Jian Wu, d3VqaWFuZ2xpbnhpbmdAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.