
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet., 28 April 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.878274
 Ahmed Waqas1*
Ahmed Waqas1* Anam Nayab2Shabnam Shaheen3Safdar Abbas4
Anam Nayab2Shabnam Shaheen3Safdar Abbas4 Muhammad Latif1Misbahuddin M. Rafeeq5Ibtesam S. Al-Dhuayan6Amany I. Alqosaibi6Mashael M. Alnamshan6Ziaullah M. Sain7Alaa Hamed Habib8Qamre Alam9
Muhammad Latif1Misbahuddin M. Rafeeq5Ibtesam S. Al-Dhuayan6Amany I. Alqosaibi6Mashael M. Alnamshan6Ziaullah M. Sain7Alaa Hamed Habib8Qamre Alam9 Muhammad Umair9,10*Muhammad Arif Nadeem Saqib11
Muhammad Umair9,10*Muhammad Arif Nadeem Saqib11Intellectual disability (ID) has become very common and is an extremely heterogeneous disorder, where the patients face many challenges with deficits in intellectual functioning and adaptive behaviors. A single affected family revealed severe disease phenotypes such as ID, developmental delay, dysmorphic facial features, postaxial polydactyly type B, and speech impairment. DNA of a single affected individual was directly subjected to whole exome sequencing (WES), followed by Sanger sequencing. Data analysis revealed a novel biallelic missense variant (c.1511G>C; p.(Trp504Ser)) in the ALKBH8 gene, which plays a significant role in tRNA modifications. Our finding adds another variant to the growing list of ALKBH8-associated tRNA modifications causing ID and additional phenotypic manifestations. The present study depicts the key role of the genes associated with tRNA modifications, such as ALKBH8, in the development and pathophysiology of the human brain.
ALKBH8 (Alkylated DNA Repair Protein AlkB Homolog 8) is a nuclear and cytosolic protein essential for tRNA modifications, for normal survival after DNA damage, can inhibit apoptosis by helping boost cell survival and angiogenesis (Fu et al., 2010; Songe-Møller et al., 2010). There are eight different members of the AlkB family that have been identified in humans (ALKBH1–ALKBH8) and mice (Alkbh1–Alkbh8) (Aravind and Koonin, 2001; Kurowski et al., 2003; Gerken et al., 2007). Most ALKBH proteins have unknown functions, while ALKBH8 differs from the other versions by two annotated protein domains. The ALKBH8 protein contains three main domains, namely, an N-terminal RNA recognition motif (RRM), a C-terminal S-adenosyl-L-methionine (SAM)–dependent methyltransferase (MTase) motif, and a central conserved AlkB oxygenase domain. The MTase and AlkB-like domains have been reported to covalently catalyze hypermodifications of the wobble nucleotide base in specific tRNAs (Fu et al., 2010).
ALKBH8 performs the final methylation step during the formation of the wobble uridine modification mcm5U in mammals. The MTase domain catalyzes the methyl esterification of modified wobble uridine (U34) residues, resulting in the formation of mcm5U (5-methoxycarbonyl methyluridine) and mcm5s2U (5-methoxycarbonyl methyl-2-thiouridine) (Tsujikawa et al., 2007; van den Born et al., 2011). The mcm5U is a common modification intermediate for thiolation and ribose methylation (Kaneko et al., 2003).
Furthermore, ALKBH8 also interacts with TRM112 forming a heterodimeric complex, which aids in catalyzing the methyl esterification forming mcm5U and mcm5s2U. The mcm5Um is involved in the selenoprotein synthesis, and thus, a reduced level of Gpx1 (glutathione peroxidase 1) was observed in the Alkbh8−/− mice (Songe-Møller et al., 2010). Gpx1 is a selenium-containing enzyme that reduces hydrogen peroxide and alkyl hyperoxides, thus protecting against traumatic brain injury (TBI). Using transgenic knockout mice, overexpression of Gpx1 was observed in many organs, such as the brain. The Gpx1 deficient mice further revealed that this enzyme enhances neuroprotection in response to the oxidative challenge. The brains of GPx1−/− mice were reported to be more vulnerable to ischemia/reperfusion, cold-induced brain injury, and mitochondrial toxin treatment. The GPx1−/− mice also exhibited neurological deficits (Pitts et al., 2014).
This study illustrates that a novel homozygous missense variant in the ALKBH8 results in severe syndromic ID. The present investigation expands the mutational spectrum of ALKBH8-associated ID.
The present study was approved by the institutional review board of the University of Education (UOE), Lahore, Pakistan, and followed Helsinki protocols. Written informed consents for the publication of this case, photographs, and related data were obtained from the parents. In addition, a pedigree was constructed (Figure 1A), and the peripheral blood samples were obtained from all available normal and affected individuals of the family.
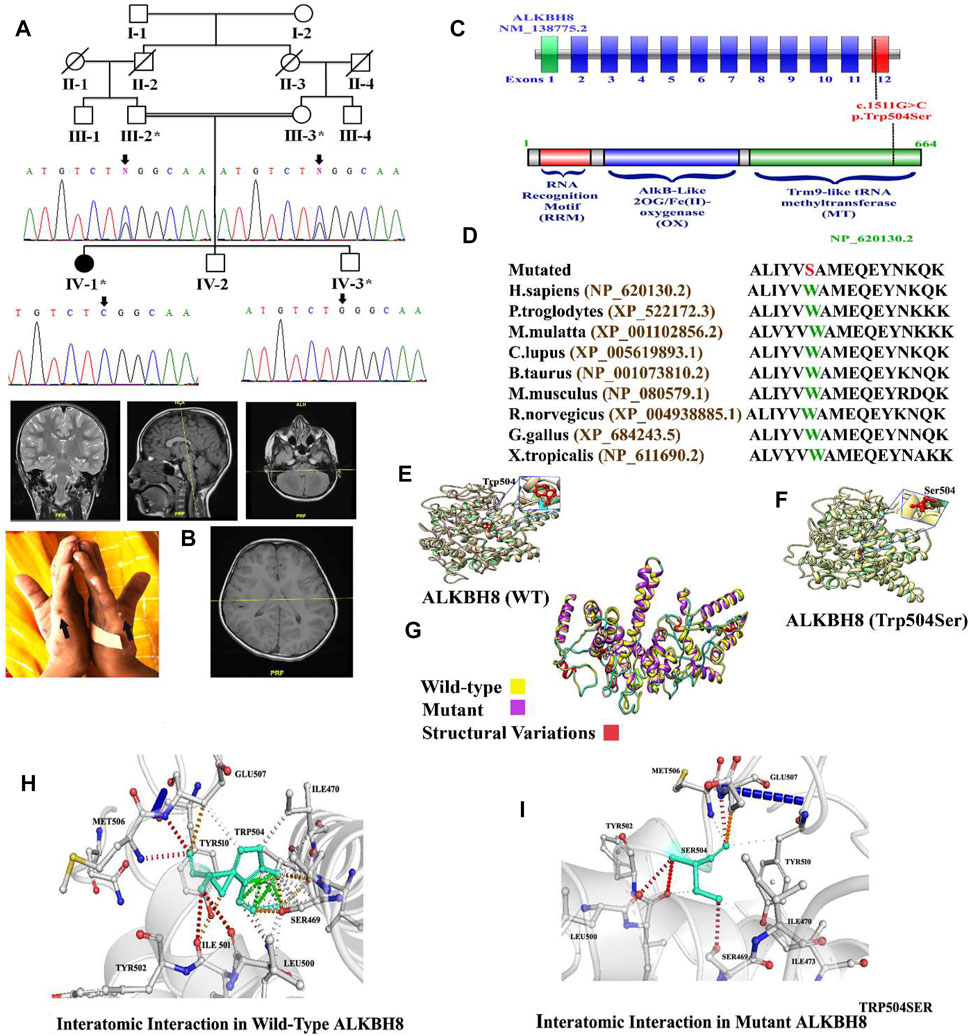
FIGURE 1. (A) Pedigree of the investigated family. Sanger sequencing electrograms shown below each member. (B) Brain MRI of the index (IV-1) and bilateral postaxial polydactyly type B. (C) Schematic representation of the ALKBH8 exons and protein domain. Dotted line represents the variant identified in the present study and its location in the exon and domain. (D) Partial amino acid sequence of ALKBH8 protein showing conservation of Trp504 amino acid across different species. (E–I) 3D protein modeling comparison of the ALKBH8 wild-type and mutated proteins.
The genomic DNA was extracted from the fresh blood of all the available members of the family using the QIAamp DNA Micro kit using standard procedures and was quantified using NanoDrop™ spectrophotometer using standard methods (Younus et al., 2019).
DNA of the single affected individual (IV-1) was subjected to WES using the Agilent SureSelect target enrichment workflow from the created DNA library. WES was performed using the Illumina work system with a minimum coverage of 20× of 98% of the total reads (Asiri et al., 2020; Umair et al., 2020; Umair et al.,2021). After sequencing, the sequence was aligned to the reference human genome build UCSC hg19 reference system [GRCh38/hg38]. An online Illumina data analysis tool (BaseSpace, Illumina; https://basespace.illumina.com) was used to analyze the patient VCF to identify the culprit gene/variant.
The identified candidate variant was Sanger sequenced bidirectionally in all the available members of the family. Sanger sequencing was performed as described earlier (Umair et al., 2019). Primer sequences were designed using the Primer3 online software (http://frodo.wi.mit.edu/primer3/) and will be provided upon request.
The pathogenicity variant was calculated via different online software: MutationTaster, VarSome, PolyPhen-2, PROVEAN, SIFT, and FATHMM-MKL. The frequency of the identified variants in the general population was checked using online databases such as EVS, 1000 Genomes, ExAC, gnomAD, and 2000+ in-house exomes. Finally, the amino acid conservation across different species was performed online using HomoloGene (NCBI; https://www.ncbi.nlm.nih.gov/homologene).
The partial amino acid (663aa) sequence of ALKBH8-encoding protein was retrieved from the UniProt Knowledgebase database with accession number Q96BT7-1 in the FASTA format. In the absence of an experimentally known structure, comparative modeling is one of the most precise computational approaches to predict a consistent three-dimensional (3D) design from sequence information (Källberg et al., 2012). Due to the absence of an experimentally known structure for ALKBH8, its protein sequence was submitted to the I-TASSER server (Yang et al., 2015) for structure prediction. From models generated by I-TASSER, the model was selected on the basis of the I-TASSER evaluation score. The selected structure was optimized through 1,000 steps of steepest descent (Wardi, 1988) and 1,000 steps of conjugate-gradient (Dai et al., 2000) minimization by UCSF Chimera version 1.11 (Meng et al., 2006), and through Amber ff14SB force field, the 3D structure of mutated protein was generated by MODELLER (9.19) (Webb and Sali, 2014). The MODELLER assists in 3D structure prediction of the proteins by satisfying spatial restraints (Eswar et al., 2008). The model was selected based on the MODELLER evaluation score from the models generated by MODELLER.
The recognition of errors in the experimental and theoretical models of protein structures is a significant problem in structural bioinformatics. Unfortunately, there is no single method that consistently and accurately predicts the errors in 3D structure (Melo et al., 1997). Therefore, different evaluation tools were used for the assessment of the protein structure. The model was further processed by RAMPAGE (Lovell et al., 2003) and ERRAT (Colovos and Yeates, 1993). RAMPAGE generates a Ramachandran plot for the assessment of models along with the distribution of residues in favored, allowed, and outlier regions. ERRAT generates a plot indicating the confidence and overall quality of the model.
In the present study, a consanguineous Pakistani family having a single affected individual was genetically and clinically evaluated. The affected individual (IV-1) was born at full term through vaginal delivery. At the age of 5–6 months, the parents observed developmental delay compared to other infants of the same age. At the age of 8–9 months, the index suffered from atonic seizures, diagnosed using EEG. The proband (IV-1) exhibited complex syndromic intellectual disability (ID) phenotypes, such as global developmental delay (GDD), hypotonia, speech impairment, seizure disorder, dysmorphic facial features (bulged eyes, prominent philtrum, high arched palate, large ears, thin upper lips, and lower nasal bridge), and bilateral postaxial polydactyly type B in the hands (Figure 1B). The parents were consanguineous, and the family pedigree depicts autosomal recessive inheritance. The parents of the affected individuals were healthy and did not reveal any phenotypic abnormality (Figure 1B; Table 1).
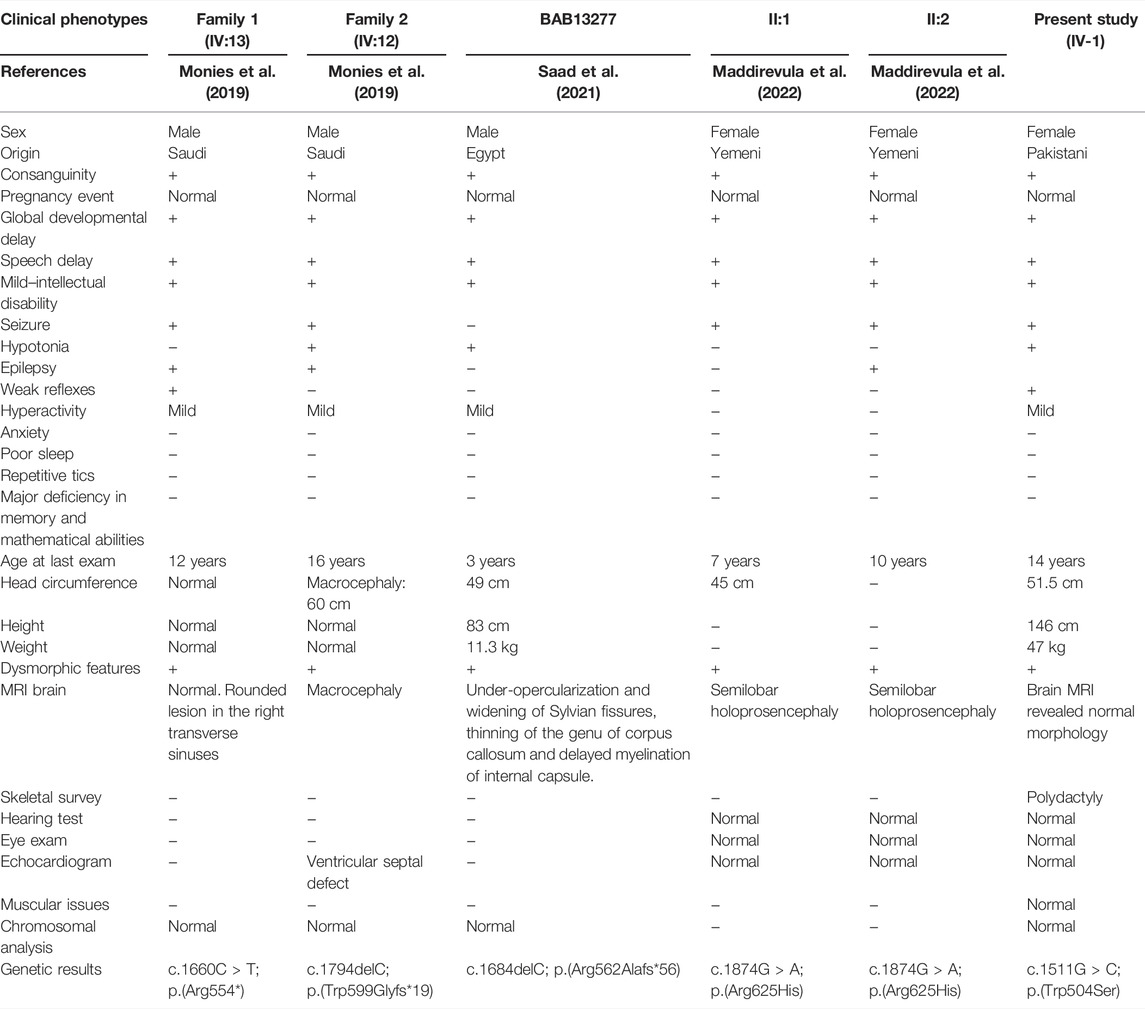
TABLE 1. Comparative clinical description of patients reported to date.
Brain MRI revealed normal morphology and signal intensity of the supra- and infratentorial structures. No restriction diffusion was noted; however, a short corpus callosum was observed, with no intracranial susceptibility artifact. The ventricular system appeared normal in size and configuration, and no hydrocephalus was observed. Preserved flow voids of the major intracranial vessels and orbital cavities were within normal limits. The clinical description of all the reported cases has been summarized in Table 1.
As the pedigree depicted autosomal recessive inheritance pattern, the filtration of the variants was performed according to the autosomal recessive pattern and preference was given to the homozygous variants, yet compound heterozygous variants were not ignored. Initial screening included all the reported ID genes. We focused only on pathogenic disease–causing non-synonymous (NS) variants causing nonsense, missense, splice site variants (SS), and frameshift coding insertions or deletions (indel).
WES identified a biallelic missense variant (c.1511G>C; p.(Trp504Ser)) in the exon 12 of the ALKBH8 gene (NM_138775.3) located on chromosome 11q22.3, which segregated with the disease phenotype as validated using Sanger sequencing (Figure 1A). The identified homozygous variant (c.1511G>C; p.(Trp504Ser)) was not reported in the gnomAD/ExAC database containing 15,708 whole-genome sequences and 125,748 human exome sequences. A list of other filtered variants obtained after WES is given in Supplementary Material S1. The variant Trp504 was highly conserved across the different species (Figure 1D). The pathogenicity index of the identified mutation was calculated using various online available tools (Table 2).
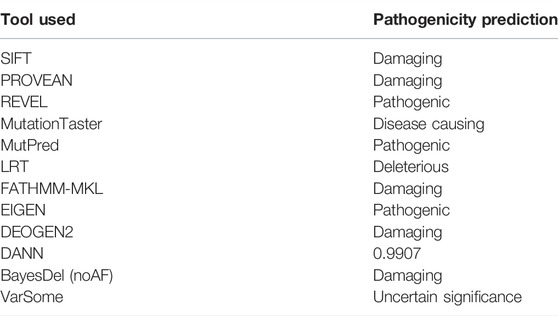
TABLE 2. Pathogenicity of the identified ALKBH8 variant (c.1511G>C; p.(Trp504Ser)).
In this study, in silico methodology such as homology modeling for wild-type and mutant was carried out. The 3D structure of ALKBH8 was modeled by the I-TASSER server (Nayab et al., 2021). The predicted structure of ALKBH8 has a good degree of accuracy. Different evaluation programs assessed the final refined model. Using homology modeling, 3D models of wild-type and mutated ALKBH8 protein (p.(Trp504Ser)) were predicted and evaluated by online structure analysis tools. The Ramachandran plot indicated that approximately 89 and 93% of the residues in the wild-type and mutant structure lay in allowed regions of the torsion angles, respectively. The 3D structure of both wild-type and mutant of ALKBH8 was subjected into the ERRAT protein structure verification server (Figures 1E–G).
The missense mutations at Trp504 might affect the secondary structure of the protein. Our analysis revealed that Trp504 interacts with Ser469, Ile470, Leu500, Ile501, Met506, Glu507, and Tyr510 (Figures 1H,I). Although both valine and leucine contain nonpolar neutral side chains, the substitution of a smaller valine to the bigger leucine disturbed interaction with the surrounding amino acid residues differently. These new interactions, in turn, might potentially disrupt both protein secondary structure and function. Using DUET, SDM, ENCoM, and mCSM, we predicted that Trp504Ser mutation would cause a −2.929, −3.470, −1.169, and −3.115 kcal/mole change in ΔΔG, respectively, indicating that the variant would significantly destabilize the protein structure and hence disrupt its function (Figures 1E–I).
ID is a major developmental disorder defined by substantial limitations in intellectual presentation and reduced social, conceptual, and practical skills. It is likely to affect almost 1% of the overall population (Maulik et al., 2011). Although characterized as syndromic and non-syndromic forms, the non-syndromic forms of ID seem more common. At the same time, more patients with similar gene defects are identified, the syndromic features become ostensible. Although environmental causes have been reported, severe forms of ID are often linked with genetic defects in single genes (Najmabadi et al., 2011; Gilissen et al., 2014). Autosomal recessive ID (monogenic) has been excessively studied with the expansion of next-generation sequencing techniques, expanding genetic heterogeneity and phenotypic variability. According to Kochinke et al. (2016), 650 genes for ID have been characterized including ∼62% (autosomal recessive), ∼16% (X-linked), ∼3% (autosomal dominant), and 19% (de novo). The mutated genes in ID patients are involved in coding for highly diverse groups of proteins, suggesting crucial and nonredundant functions of these genes in multiple biological processes.
In the present study, using WES, we identified a homozygous missense variant (c.1511G>C; p.(Trp504Ser)) in exon 12 of the ALKBH8 gene (NM_138775.3) that segregated with the disease phenotype. The identified variant is located in the highly conserved tRNA methyltransferase domain (MT) of the ALKBH8 protein located at the C-terminus (Figure 1C). The affected individual showed ID, facial dysmorphism, speech delay, and learning disability, which were the common features observed in the patients reported by Monies et al. (2019). Homozygous loss-of-function variants in the ALKBH8 gene were first associated with syndromic ID (Monies et al., 2019). Saad et al. (2021) reported a novel ALKBH8 variant in an Egyptian family associated with a neurodevelopmental disorder. We report, to the best of our knowledge, on the fourth novel variant in ALKBH8 associated with syndromic ID. Our index’s phenotypes mirror the features reported previously (Monies et al., 2019; Saad et al., 2021). Previously, all three patients revealed a loss-of-function variant; however, more recently, a single family having two affected individuals revealed a biallelic missense variant (c.1874G>A; (p.Arg625His)) in the ALKBH8 gene associated with syndromic ID (Maddirevula et al., 2022). Thus supporting our claim that missense variants in ALKBH8 also cause syndromic ID in humans.
Novel gene discoveries have introduced a new emerging ID group associated with tRNA modifications, namely, the ADAT3 gene, which edits adenosine to inosine at the wobble position 34 of mature tRNA (Alazami et al., 2013). Other genes include NSUN2 (Abbasi-Moheb et al., 2012), WDR4 (Shaheen et al., 2015), TRMT10A (Igoillo-Esteve et al., 2013), TRIT1 (Yarham et al., 2014; Kernohan et al., 2017), TRMT1 (Najmabadi et al., 2011), ELP2 (Najmabadi et al., 2011), PUS3 (Nøstvik et al., 2021), and FTSJ1 (Freude et al., 2004).
Transfer RNAs (tRNAs) are vital molecules that participate in the final protein synthesis. Defects in tRNA modifications have been associated with different types of disorders such as myopathies, respiratory defects, metabolic (diabetes type II), mitochondrial disorders such as encephalopathy, myopathy, stroke-like episodes (MELAS), lactic acidosis, familial dysautonomia, and ID (Bednářová et al., 2017). The human brain is susceptible, and defects in tRNA modifications and mutations in specific genes involved in posttranscriptional modifications can be attributed to causing severe neurological disorders (Bednářová et al., 2017). tRNA modifications have been reported to cause ID phenotypes in humans, such as PUS3 (OMIM 616283), which isomerizes uracil to pseudouridine in human tRNA homozygous pathogenic mutations in PUS3 causing autosomal recessive ID (OMIM 617051; Nøstvik et al., 2021). In addition, methylation of tRNA at specific residues is performed by the FTSJ1 (OMIM 300499), and mutations in these genes lead to non-syndromic X-linked mental retardation and ID (Guy et al., 2015).
Pathogenic disease–causing mutations in the tRNA modification genes exhibited severe clinical phenotypes. Understanding the proper function of these tRNA-modifying genes/enzymes will help us understand the precise biological roles, which might develop therapeutic strategies for this new class of disorders either by controlling tRNA gene expression or altering the low or highly expressed tRNA using CRISPR technologies (Khan et al., 2020).
In conclusion, we reveal that disease-causing biallelic missense variants in the ALKBH8 are associated with an autosomal recessive form of syndromic neurological disorder. Our finding further supports the evidence that variants in ALKHB8 cause syndromic neurological diseases and add to the growing class of tRNA modification-related disorders.
The data sets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The studies involving human participants were reviewed and approved by IRB UOE, Lahore. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
AW performed the experiments and drafted the manuscript, AN and MU analyzed the genetic data and reviewed the manuscript. SS analyzed the genomic data. ML and SA recruited the family and performed protein modeling. ISA-D, AA, MA, ZS,MANS,QA and AH performed clinical evaluation and data analysis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are grateful to the patient and his family reported in this article for their genuine support.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.878274/full#supplementary-material
Abbasi-Moheb, L., Mertel, S., Gonsior, M., Nouri-Vahid, L., Kahrizi, K., Cirak, S., et al. (2012). Mutations in NSUN2 Cause Autosomal- Recessive Intellectual Disability. Am. J. Hum. Genet. 90, 847–855. doi:10.1016/j.ajhg.2012.03.021
Alazami, A. M., Hijazi, H., Al-Dosari, M. S., Shaheen, R., Hashem, A., Aldahmesh, M. A., et al. (2013). Mutation inADAT3, Encoding Adenosine Deaminase Acting on Transfer RNA, Causes Intellectual Disability and Strabismus. J. Med. Genet. 50 (7), 425–430. doi:10.1136/jmedgenet-2012-101378
Aravind, L., and Koonin, E. V. (2001). The DNA-Repair Protein AlkB, EGL-9, and Leprecan Define New Families of 2-oxoglutarate- and Iron-dependent Dioxygenases. Genome Biol. 2 (3), RESEARCH0007. doi:10.1186/gb-2001-2-3-research0007
Asiri, A., Aloyouni, E., Umair, M., Alyafee, Y., Al Tuwaijri, A., Alhamoudi, K. M., et al. (2020). MutatedRAP1GDS1causes a New Syndrome of Dysmorphic Feature, Intellectual Disability & Speech Delay. Ann. Clin. Transl Neurol. 7 (6), 956–964. doi:10.1002/acn3.51059
Bednářová, A., Hanna, M., Durham, I., VanCleave, T., England, A., Chaudhuri, A., et al. (2017). Lost in Translation: Defects in Transfer RNA Modifications and Neurological Disorders. Front. Mol. Neurosci. 10, 135. doi:10.3389/fnmol.2017.00135
Colovos, C., and Yeates, T. O. (1993). Verification of Protein Structures: Patterns of Nonbonded Atomic Interactions. Protein Sci. 2, 1511–1519. doi:10.1002/pro.5560020916
Dai, Y., Han, J., Liu, G., Sun, D., Yin, H., and Yuan, Y.-X. (2000). Convergence Properties of Nonlinear Conjugate Gradient Methods. SIAM J. Optim. 10 (2), 345–358. doi:10.1137/s1052623494268443
Eswar, N., Eramian, D., Webb, B., Shen, M.-Y., and Sali, A. (2008). Protein Structure Modeling with MODELLER. Methods Mol. Biol. 426, 145–159. doi:10.1007/978-1-60327-058-8_8
Freude, K., Hoffmann, K., Jensen, L.-R., Delatycki, M. B., des Portes, V., Moser, B., et al. (2004). Mutations in the FTSJ1 Gene Coding for a Novel S-Adenosylmethionine-Binding Protein Cause Nonsyndromic X-Linked Mental Retardation. Am. J. Hum. Genet. 75 (2), 305–309. doi:10.1086/422507
Fu, D., Brophy, J. A. N., Chan, C. T. Y., Atmore, K. A., Begley, U., Paules, R. S., et al. (2010). Human AlkB Homolog ABH8 Is a tRNA Methyltransferase Required for Wobble Uridine Modification and DNA Damage Survival. Mol. Cel Biol 30 (10), 2449–2459. doi:10.1128/MCB.01604-09
Gerken, T., Girard, C. A., Tung, Y. C., Webby, C. J., Saudek, V., Hewitson, K. S., et al. (2007). The Obesity-Associated FTO Gene Encodes a 2-oxoglutarate-dependent Nucleic Acid Demethylase. Science 318 (5855), 1469–1472. doi:10.1126/science.1151710
Gilissen, C., Hehir-Kwa, J. Y., Thung, D. T., van de Vorst, M., van Bon, B. W. M., Willemsen, M. H., et al. (2014). Genome Sequencing Identifies Major Causes of Severe Intellectual Disability. Nature 511 (7509), 344–347. doi:10.1038/nature13394
Guy, M. P., Shaw, M., Weiner, C. L., Hobson, L., Stark, Z., Rose, K., et al. (2015). Defects in tRNA Anticodon Loop 2′-O-Methylation Are Implicated in Nonsyndromic X-Linked Intellectual Disability Due to Mutations inFTSJ1. Hum. Mutat. 36 (12), 1176–1187. doi:10.1002/humu.22897
Igoillo-Esteve, M., Genin, A., Lambert, N., Désir, J., Pirson, I., Abdulkarim, B., et al. (2013). tRNA Methyltransferase Homolog Gene TRMT10A Mutation in Young Onset Diabetes and Primary Microcephaly in Humans. Plos Genet. 9 (10), e1003888. doi:10.1371/journal.pgen.1003888
Källberg, M., Wang, H., Wang, S., Peng, J., Wang, Z., Lu, H., et al. (2012). Template-based Protein Structure Modeling Using the RaptorX Web Server. Nat. Protoc. 7 (8), 1511–1522. doi:10.1038/nprot.2012.085
Kaneko, T., Suzuki, T., Kapushoc, S. T., Rubio, M. A., Ghazvini, J., Watanabe, K., et al. (2003). Wobble Modification Differences and Subcellular Localization of tRNAs in Leishmania Tarentolae: Implication for tRNA Sorting Mechanism. EMBO J. 22 (3), 657–667. doi:10.1093/emboj/cdg066
Kernohan, K. D., Dyment, D. A., Pupavac, M., Cramer, Z., McBride, A., Bernard, G., et al. (2017). Matchmaking Facilitates the Diagnosis of an Autosomal-Recessive Mitochondrial Disease Caused by Biallelic Mutation of the tRNA Isopentenyltransferase (TRIT1 ) Gene. Hum. Mutat. 38 (5), 511–516. doi:10.1002/humu.23196
Khan, A., Miao, Z., Umair, M., Ullah, A., and Alshabeeb, M. A. (2020). Two Cases of Recessive Intellectual Disability Caused by NDST1 and METTL23 variants. Genes 11 (9), 1021. doi:10.3390/genes11091021
Kochinke, K., Zweier, C., Nijhof, B., Fenckova, M., Cizek, P., Honti, F., et al. (2016). Systematic Phenomics Analysis Deconvolutes Genes Mutated in Intellectual Disability into Biologically Coherent Modules. Am. J. Hum. Genet. 98 (1), 149–164. doi:10.1016/j.ajhg.2015.11.024
Kurowski, M. A., Bhagwat, A. S., Papaj, G., and Bujnicki, J. M. (2003). Phylogenomic Identification of Five New Human Homologs of the DNA Repair Enzyme AlkB. BMC Genomics 4, 48. doi:10.1186/1471-2164-4-48
Lovell, S. C., Davis, I. W., Arendall, W. B., de Bakker, P. I., Word, J. M., Prisant, M. G., et al. (2003). Structure Validation by Cα Geometry: ϕ,ψ and Cβ Deviation. Proteins 50 (3), 437–450. doi:10.1002/prot.10286
Maddirevula, S., Alameer, S., Ewida, N., de Sousa, M. M. L., Bjørås, M., Vågbø, C. B., et al. (2022). Insight into ALKBH8-Related Intellectual Developmental Disability Based on the First Pathogenic Missense Variant. Hum. Genet. 141 (2), 209–215. doi:10.1007/s00439-021-02391-z
Maulik, P. K., Mascarenhas, M. N., Mathers, C. D., Dua, T., and Saxena, S. (2011). Prevalence of Intellectual Disability: a Meta-Analysis of Population-Based Studies. Res. Dev. Disabilities 32 (2), 419–436. doi:10.1016/j.ridd.2010.12.018
Melo, F., Devos, D., Depiereux, E., and Feytmans, E. (1997). ANOLEA: a Www Server to Assess Protein Structures. Proc. Int. Conf. Intell. Syst. Mol. Biol. 5, 187–190.
Meng, E. C., Pettersen, E. F., Couch, G. S., Huang, C. C., and Ferrin, T. E. (2006). Tools for Integrated Sequence-Structure Analysis with UCSF Chimera. BMC Bioinformatics 7, 339. doi:10.1186/1471-2105-7-339
Monies, D., Vågbø, C. B., Al-Owain, M., Alhomaidi, S., and Alkuraya, F. S. (2019). Recessive Truncating Mutations in ALKBH8 Cause Intellectual Disability and Severe Impairment of Wobble Uridine Modification. Am. J. Hum. Genet. 104 (6), 1202–1209. doi:10.1016/j.ajhg.2019.03.026
Najmabadi, H., Hu, H., Garshasbi, M., Zemojtel, T., Abedini, S. S., Chen, W., et al. (2011). Deep Sequencing Reveals 50 Novel Genes for Recessive Cognitive Disorders. Nature 21478 (7367), 57–63. doi:10.1038/nature10423
Nøstvik, M., Kateta, S. M., Schönewolf‐Greulich, B., Afenjar, A., Barth, M., Boschann, F., et al. (2021). Clinical and Molecular Delineation of PUS3 ‐associated Neurodevelopmental Disorders. Clin. Genet. 100 (5), 628–633. doi:10.1111/cge.14051
Nayab, A., Alam, Q., Alzahrani, O. R., Khan, R., Sarfaraz, S., Albaz, A. A., et al. (2021). Targeted exome sequencing identified a novel frameshift variant in the PGAM2 gene causing glycogen storage disease type X. Eur. J. Med. Gent. 64 (9), 103954. doi:10.1016/j.ejmg.2021.104283
Pitts, M. W., Byrns, C. N., Ogawa-Wong, A. N., Kremer, P., and Berry, M. J. (2014). Selenoproteins in Nervous System Development and Function. Biol. Trace Elem. Res. 161 (3), 231–245. doi:10.1007/s12011-014-0060-2
Saad, A. K., Marafi, D., Mitani, T., Du, H., Rafat, K., Fatih, J. M., et al. (2021). Neurodevelopmental Disorder in an Egyptian Family with a Biallelic ALKBH8 Variant. Am. J. Med. Genet. 185 (4), 1288–1293. doi:10.1002/ajmg.a.62100
Shaheen, R., Abdel-Salam, G. M. H., Guy, M. P., Alomar, R., Abdel-Hamid, M. S., Afifi, H. H., et al. (2015). Mutation in WDR4 Impairs tRNA m7G46 Methylation and Causes a Distinct Form of Microcephalic Primordial Dwarfism. Genome Biol. 2816, 210. doi:10.1186/s13059-015-0779-x
Songe-Møller, L., van den Born, E., Leihne, V., Vågbø, C. B., Kristoffersen, T., Krokan, H. E., et al. (2010). Mammalian ALKBH8 Possesses tRNA Methyltransferase Activity Required for the Biogenesis of Multiple Wobble Uridine Modifications Implicated in Translational Decoding. Mol. Cel Biol 30 (7), 1814–1827. doi:10.1128/MCB.01602-09
Tsujikawa, K., Koike, K., Kitae, K., Shinkawa, A., Arima, H., Suzuki, T., et al. (2007). Expression and Sub-cellular Localization of Human ABH Family Molecules. J. Cell. Mol. Med. 11 (5), 1105–1116. doi:10.1111/j.1582-4934.2007.00094.x
Umair, M., Ballow, M., Asiri, A., Alyafee, Y., Tuwaijri, A., Alhamoudi, K. M., et al. (2020). EMC10 Homozygous Variant Identified in a Family with Global Developmental Delay, Mild Intellectual Disability, and Speech Delay. Clin. Genet. 98 (6), 555–561. doi:10.1111/cge.13842
Umair, M., Khan, A., Amin, W., Abbas, S., Younus, M., Alfadhel, M., et al. (2019). Biallelic Missense Biallelic Missense Mutation in the ECEL1 Underlies Distal Arthrogryposis Type 5 (DA5D). Front. Pediatr. 7, 343. doi:10.3389/fped.2019.00343
Umair, M., Palander, O., Bilal, M., Almuzzaini, B., Alam, Q., Ahmad, F., et al. (2021). Biallelic Variant in DACH1, Encoding Dachshund Homolog 1, Defines a Novel Candidate Locus for Recessive Postaxial Polydactyly Type A. Genomics 113, 2495–2502. doi:10.1016/j.ygeno.2021.05.015
van den Born, E., Vågbø, C. B., Songe-Møller, L., Leihne, V., Lien, G. F., Leszczynska, G., et al. (2011). ALKBH8-mediated Formation of a Novel Diastereomeric Pair of Wobble Nucleosides in Mammalian tRNA. Nat. Commun. 2, 172. doi:10.1038/ncomms1173
Wardi, Y. (1988). A Stochastic Steepest-Descent Algorithm. J. Optim. Theor. Appl. 59 (2), 307–323. doi:10.1007/bf00938315
Webb, B., and Sali, A. (2014). Protein Structure Modeling with MODELLER. Methods Mol. Biol. 1137, 1–15. doi:10.1007/978-1-4939-0366-5_1
Yang, J., Yan, R., Roy, A., Xu, D., Poisson, J., and Zhang, Y. (2015). The I-TASSER Suite: Protein Structure and Function Prediction. Nat. Methods 12 (1), 7–8. doi:10.1038/nmeth.3213
Yarham, J. W., Lamichhane, T. N., Pyle, A., Mattijssen, S., Baruffini, E., Bruni, F., et al. (2014). Defective i6A37 Modification of Mitochondrial and Cytosolic tRNAs Results from Pathogenic Mutations in TRIT1 and its Substrate tRNA. Plos Genet. 10 (6), e1004424. doi:10.1371/journal.pgen.1004424
Keywords: missense variant, biallelic variant, whole exome sequencing, tRNA methyl transferase, intellectual disability, posttranscriptional modification
Citation: Waqas A, Nayab A, Shaheen S, Abbas S, Latif M, Rafeeq MM, Al-Dhuayan IS, Alqosaibi AI, Alnamshan MM, Sain ZM, Habib AH, Alam Q, Umair M and Saqib MAN (2022) Case Report: Biallelic Variant in the tRNA Methyltransferase Domain of the AlkB Homolog 8 Causes Syndromic Intellectual Disability. Front. Genet. 13:878274. doi: 10.3389/fgene.2022.878274
Received: 17 February 2022; Accepted: 18 March 2022;
Published: 28 April 2022.
Edited by:
Lawrence Todd Reiter, University of Tennessee Health Science Center (UTHSC), United StatesReviewed by:
Asmat Ullah, University of Copenhagen, DenmarkCopyright © 2022 Waqas, Nayab, Shaheen, Abbas, Latif, Rafeeq, Al-Dhuayan, Alqosaibi, Alnamshan, Sain, Habib, Alam, Umair and Saqib. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ahmed Waqas, cmF2aWFuNjQzQGdtYWlsLmNvbQ==; Muhammad Umair, a2h1Z29vNHVAeWFob28uY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.