 Hongguo Zhang1†
Hongguo Zhang1† Wei Li2†Yuting Jiang1
Wei Li2†Yuting Jiang1 Jia Li2Mucheng Chen2Ruixue Wang1Jing Zhao2Zhiyu Peng2Hui Huang2*Ruizhi Liu1*
Jia Li2Mucheng Chen2Ruixue Wang1Jing Zhao2Zhiyu Peng2Hui Huang2*Ruizhi Liu1*- 1Reproductive Medicine and Prenatal Diagnosis Center, The First Hospital, Jilin University, Changchun, China
- 2BGI Genomics, BGI-Shenzhen, Shenzhen, China
Background: Non-obstructive azoospermia (NOA) affects nearly 1% of men; however, the landscape of the causative genes is largely unknown.
Objective: To explore the genetic etiology which is the fundamental cause of NOA, a prospective case-control study and parental–proband trio linkage analysis were performed.
Materials: A total of 133 patients with clinicopathological NOA and 343 fertile controls were recruited from a single large academic fertility center located in Northeast China; in addition, eleven trio families were available and enrolled.
Results: Whole exome sequencing-based rare variant association study between the cases and controls was performed using the gene burden association testing. Linkage analysis on the trio families was also interrogated. In total, 648 genes were identified to be associated with NOA (three of which were previously reported), out of which six novel genes were found further associated based on the linkage analysis in the trio families, and involved in the meiosis-related network.
Discussion and Conclusion: The six currently identified genes potentially account for a fraction (3.76%, 5 out of 133 patients) of the heritability of unidentified NOA, and combining the six novel genes and the three previously reported genes together would potentially account for an overall 6.77% (9 out of 133 patients) heritability of unidentified NOA in this study.
Introduction
Approximately 7% of the male population worldwide suffer from infertility, and one of the main causes is azoospermia, which is a condition contributing to male infertility with the highest frequency of known genetic factors (about 25%) (Krausz and Riera-Escamilla 2018; Rodrigues, Polisseni et al., 2020). Given that nearly 50% of azoospermia cases are estimated to be associated with genetic defects, the etiology of azoospermia is still very elusive (Rodrigues, Polisseni et al., 2020). In terms of pathobiological mechanism, azoospermia usually is categorized into two groups: non-obstructive azoospermia (NOA) which is defined as no spermatozoa due to failure of spermatogenesis and obstructive azoospermia which occurs due to an obstruction of the seminal ducts and is characterized by the absence of spermatozoa in the ejaculate despite the normal spermatogenesis.
NOA is the most severe form of azoospermia; currently, no treatment can restore spermatogenesis in the majority of patients; however, some patients can benefit from treatment with assisted reproductive technologies. NOA is a heterogenous condition with variable histopathological phenotypes: the Sertoli cell-only syndrome (SCOS) is characterized by a complete absence of germ cells, while spermatogenic arrest is a condition originating from the physiological spermatogenesis defect and is known as the interruption of germinal cell formation of specific cellular type.
Approximately 20% of NOA cases were caused by detectable structural anomalies including chromosomal abnormalities such as the sex-chromosome aneuploidy, the trans-locational aberrations, and microdeletions of azoospermia factors (AZFs) (Krausz and Riera-Escamilla 2018). In addition, monogenic variations have been identified to be a causative for or associated with NOA; the attested genes include X chromosome linked genes TEX11 (Yatsenko, Georgiadis et al., 2015), FAM47C (Chertman, Arora et al., 2019), MAGEB4 (Okutman, Muller et al., 2017), and AR (Mou and Gui 2016); and autosomal genes include PIWIL1 (Gou, Kang et al., 2017), BRCA2 (Zhoucun, Zhang et al., 2006), BOLL and DAZL (Zhang, Xue et al., 2016), TEX15 (Okutman, Muller et al., 2015), SYCP3 (Miyamoto, Hasuike et al., 2003), NR5A1 (Safari, Zare-Abdollahi et al., 2014), SOHLH1 (Choi, Jeon et al., 2010), WT1 (Wang, Li et al., 2013; Xu, Jiang et al., 2017), TAF4B and ZMYND15 (Ayhan, Balkan et al., 2014), FANCM (Kasak, Punab et al., 2018), SPINK2 (Kherraf, Christou-Kent et al., 2017), TEX14 (Gershoni, Hauser et al., 2017), DNAH6 (Gershoni, Hauser et al., 2017), MEIOB (Gershoni, Hauser et al., 2017), RNF212 and STAG3 (Riera-Escamilla, Enguita-Marruedo et al., 2019), XRCC2 (Zhang, Li et al., 2019), TDRD7 and TDRD9 (Arafat, Har-Vardi et al., 2017; Tan, Tu et al., 2019), DMC1 (He, Tu et al., 2018), SYCE1 (Maor-Sagie, Cinnamon et al., 2015), NPAS2 (Ramasamy, Bakircioglu et al., 2015), MCM8 (Tenenbaum-Rakover, Weinberg-Shukron et al., 2015), MEI1 (Ben Khelifa, Ghieh et al., 2018), and STX2 (Nakamura, Kobori et al., 2018). Despite these efforts, the etiology remains unknown in an enormously large part of NOA patients (Krausz and Riera-Escamilla 2018). Considering that a surprisingly large proportion of all genes (roughly 1 in 25 of all mammalian genes) are specifically expressed in the testis, hundreds of genes may be involved in the spermatogenesis process (Schultz, Hamra et al., 2003; Bell, Mello et al., 2020). Therefore, the physiological pathway complexity may contribute to a large number of unresolved patients.
However, current tools for understanding the unidentified genetic etiology of NOA are very limited due to the lack of large pedigrees with infertility; therefore, the discovery of novel genetic factors for unexplained NOA is still the main challenge. In this study we performed whole exome sequencing (WES), one of the most promising and powerful approach for exploring the pathogenic factors of inherent diseases, on 133 unrelated patients and 343 independent controls for rare variant association study (RVAS) (Lee, Abecasis et al., 2014; Artomov, Stratigos et al., 2017; Locke, Steinberg et al., 2019), which was a kind of analysis derived from genome-wide association study (GWAS); meanwhile, 11 trios were analyzed in a maternal inherited or a de novo pattern to delineate the causative genes. Protein-protein interaction (PPI) network functional enrichment analysis was also applied to provide potential evidence to further address the abovementioned questions.
Results
The workflow diagram of this study is shown in Figure 1, a total of 1739 patients were enrolled, amidst which 299 cases (about 17%) with abnormal karyotyping results (such as sex-autosome translocation, and Y chromosome polymorphisms (Yqh–or Yqh+)) were excluded, followed by 281 cases (about 16%) exclusion for harboring the AZF region (AZFa, AZFb, or AZFc) microdeletions; then 1,023 cases who did not consent to accept further sequencing test were not included. Furthermore, a custom panel for detecting the known genes of azoospermia (Supplementary Table S2) was developed, from which three cases with positive results (two with TEX11 mutation, one with AR mutation) were excluded. Eventually, 133 patients and 343 controls were subjected to WES; linkage analysis for 11 trios was also performed in this study.
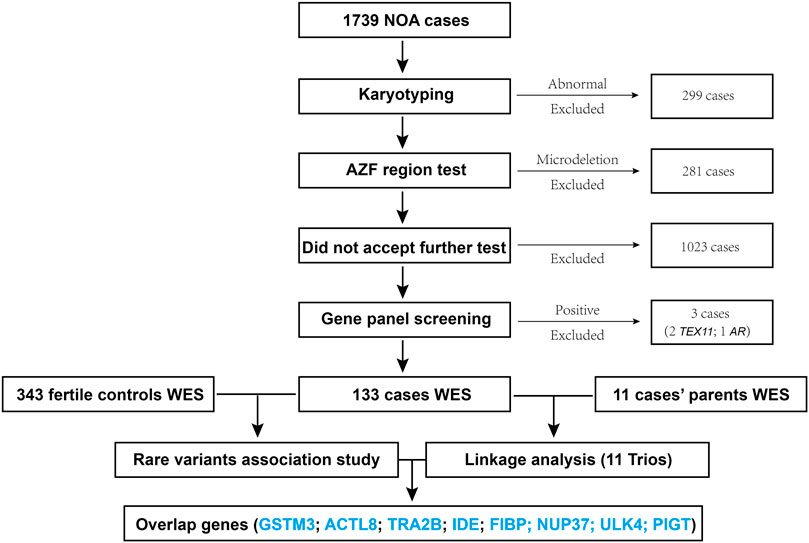
FIGURE 1. Enrollment of the patients and the workflow diagram for this study.
Among the 133 patients, WES identified 7 LOFs in six known genes (Table 1). Since FAM47C was identified to be highly associated with NOA previously, the X chromosome-linked (XL) FAM47C mutation (p.Val118Leufs*40, frameshift mutation similar with previous literature (Chertman, Arora et al., 2019)) from patient 58 in our study was also identified as the potential genetic etiology accordingly; the BRCA2 mutation in patient 58 (p.Ser 1955*) and patient 82 (p.Thr2197_Glu2198delinsLys) may be associated with NOA. The causative gene SYCP3 mutation in patient 88 (c.-13-2A > C) may be pathogenic (functional evidence needed) according to an autosomal dominant (AD) inheritance pattern. In autosomal recessive (AR) inheritance, the other genes are uncertainly causative concerning the individuals owing to another pathogenic allele not determined. Pathogenic CNVs were not identified among the 133 patients under WES.

TABLE 1. Attested causative genes mutations identified by WES in our study.
Whole exome-wide RVAS results were summarized in Supplementary Table S3, including the p-values and the OR values, the normalized expression (NE) of genes in testis tissue from the Human Protein Atlas database were also provided. NE was calculated according to the transcript profiling based on a combination of three transcriptomics datasets HPA, GTEx, and FANTOM5 (genes with no NE data were ruled out in this study). We quantified genes with the OR value >1 or with OR value not estimable due to the absence of the rare variations among the controls, meanwhile we employed a flexible cutoff of p < 0.05 due to the statistical power limitation of the relatively small sample size (133 patients) and the low proportion of rare variations among the entire samples (a lot of rare variations absent in controls). As a result, a total of 648 genes (65 genes with p < 0.01) were identified to be associated with NOA (three of which were previously reported). The most significant genes reported were DHRS4, WARS1, PICK1, RRBP1, and ENTPD2 (red), whereas the other 179 significant genes were shown in blue; meanwhile, three known genes BRCA2, SYCP3, and TDRD7 (yellow) were also identified by the RVAS test in our study (Figure 2).The 187 genes identified by RVAS (red, blue, and yellow) and 28 previously reported proteins (green) were analyzed for the PPI network as shown in Figure 3. Interestingly, the previously reported proteins, except for only MAGEB4 and FAM47C, tended to be involved in the meiosis-related network, which is centered around PIWIL1, TEX11, TEX15, DAZL, and TEX14, etc. The majority of RVAS identified proteins (117), such as WARS, RRBP1, DHRS4 (red), and PIWIL2, DDX39B, SLX4, HNRNPC (blue), were involved and enriched in the meiosis-related PPI network (FDR value for the male gamete generation pathway was 9.56 × 10–31), thereby betraying the functional potential to be strongly associated with NOA; whereas 70 proteins, like PICK1 and ENTPD2 (red), were not interacted with the PPI network, attenuating the possibility that these genes play major functional roles in the development of NOA.
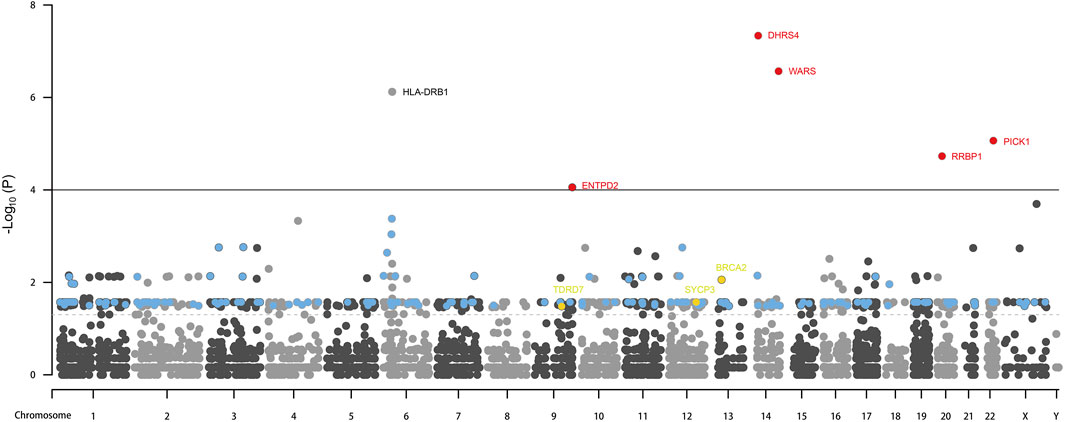
FIGURE 2. RVAS analysis results. Red, the most significant genes with p value lower than 0.0001 identified by RVAS test, and with OR values higher than 1. Blue: the significant genes with p value between 0.05 and 0.0001 identified by the RVAS test, with NE higher than 16 (taking the previously reported genes as a reference), and with OR values higher than 6 or with OR values not estimable. Yellow: the significant genes with p values between 0.05 and 0.0001 identified by the RVAS test while overlapping with known genes. The solid line denotes the p value equals 0.0001; the dashed line denotes the p value equals 0.05.
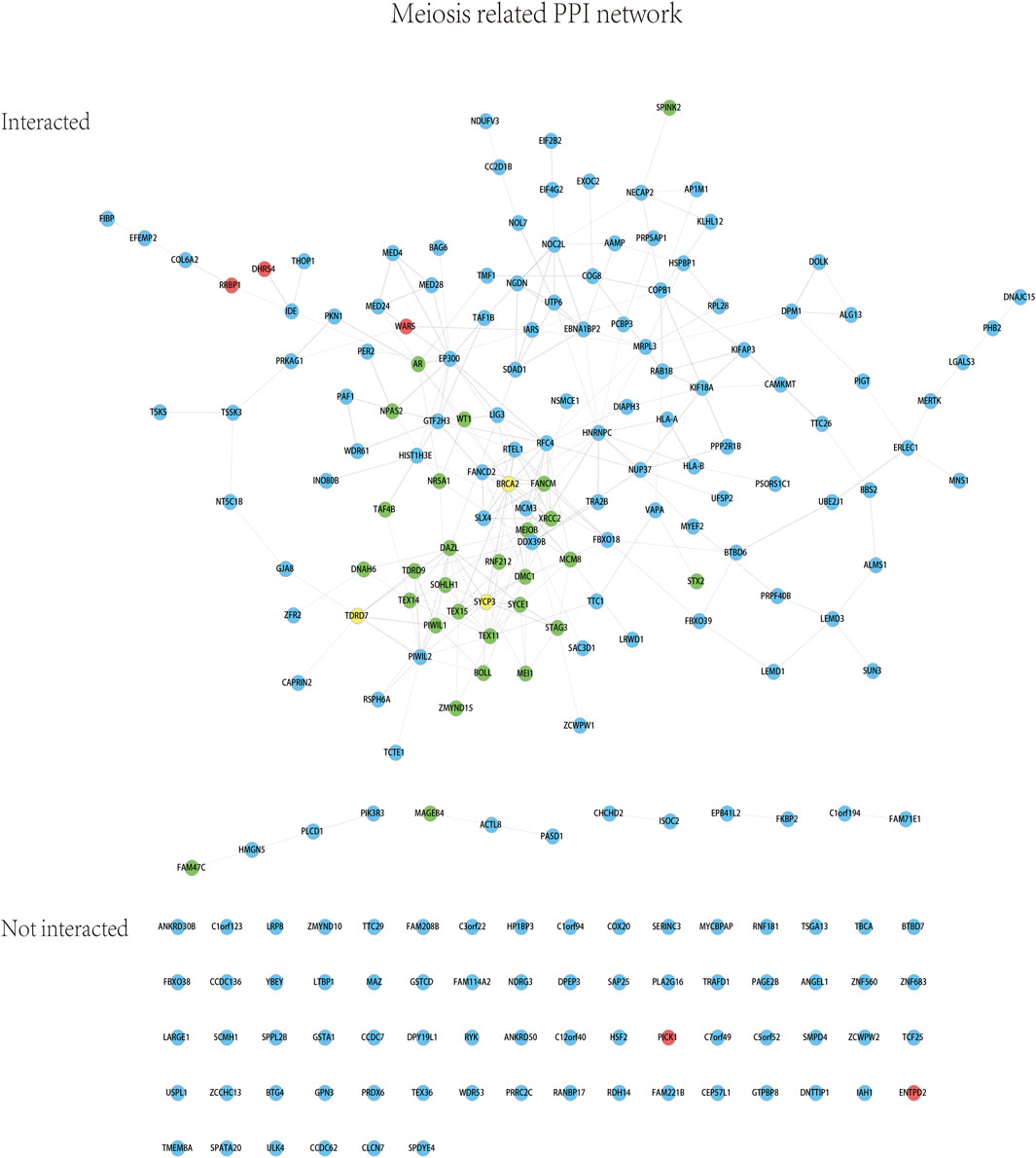
FIGURE 3. Meiosis-related PPI network based on the RVAS analysis. Red, blue, and yellow: the color code is identical with that in Figure 2. Green: 28 previously known proteins associated with NOA.
For association study, OR values were not estimable for most of the 187 genes (Supplementary Table S3) due to the absence of rare variations among the controls, we summarized a short list of genes with OR values to evaluate the genetic risk (Figure 4). All the genes with p values lower than 0.05, with the testis expression (NE > 16), and with the OR values higher than 1 were included. Consistent with the previous literature, BRCA2 (OR, 7.48; 95% CI, 1.37–40.89; p value, 8.79 × 10–3) was identified to be associated with NOA in our study. The following genes with high OR values were also involved in the meiosis-related PPI network (Figure 3), including ENTPD2 (OR, 23.34; 95% CI, 2.78–195.61; p value, 8.79 × 10–5) which had the highest OR; while RRBP1 (OR, 15.32; 95% CI, 3.23–72.57; p value, 1.86 × 10–5), WARS (OR, 8.83; 95% CI, 3.29–23.71; p value, 2.71 × 10–7), DHRS4 (OR, 7.54; 95% CI, 3.46–16.42; p value, 4.61 × 10–8), NOL7 (OR, 15.32; 95% CI, 1.70–138.23; p value, 2.28 × 10–3), HLA-A (OR, 15.03; 95% CI, 1.67–134.84; p value, 9.13 × 10–4), EBNA1BP2 (OR, 11.28; 95% CI, 1.17–108.90; p value, 1.06 × 10–2), CC2D1B (OR, 11.22; 95% CI, 1.17–108.12; p value, 1.07 × 10–2), DDX39B (OR, 7.64; 95% CI, 2.28–25.58; p value, 4.20 × 10–4), and COPB1 (OR, 7.64; 95% CI, 1.38–42.19; p value, 8.55 × 10–3) were shown.

FIGURE 4. Associated genes with odds ratio and p value for the RVAS analysis. 95% CI (whiskers) was shown as the solid line around the ln (odds ratio) value dot (circles). The black vertical line indicates an OR value of 1. The color description was identical with that in Figure 3.
In total, 11 trio families were recruited as shown in Figure 1, and their data were analyzed and summarized in Table 2. For linkage analysis, according to a maternally inherited pattern or the de novo mutation mechanism to which the gender-specific disease NOA condition was supposed to be genetically attributed, the paternal inherited variations were excluded; meanwhile, the probands’ LOFs were preserved only if the LOFs were not found in the 343 male controls. Sanger sequencing was performed for validating the variations. To facilitate the screening for the mutations, the p-value and OR value of RVAS for each gene were also rendered. Eventually, six genes (in bold and double-underlined in Table 2), ACTL8 and TRA2B for patient 62, IDE for patient 117, FIBP for patient 124, NUP37 for patient 130, and PIGT for patient 131 were regarded as the top candidates of associated genes (p < 0.05; OR values not estimable due to the absence of variations among the controls) based on the RVAS test (Figure 2) and their involvement in the meiosis-related PPI network (Figure 3).
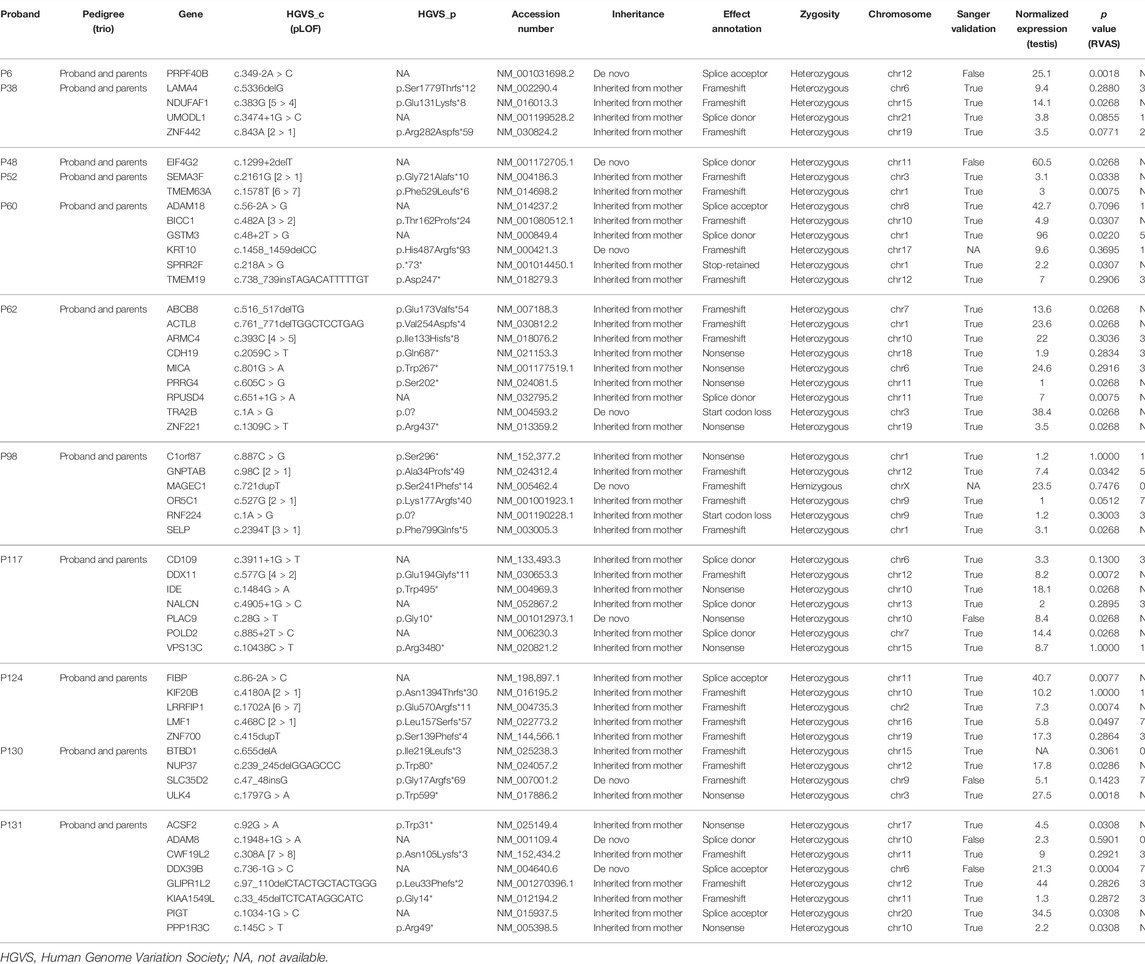
TABLE 2. Novel gene mutations identified in the trio families.
These six novel genes potentially accounted for a fraction (3.76%, 5 out of 133 patients) of the heritability of NOA in question. Combined with the three known genes (yellow), the six novel genes potentially accounted for an overall 6.77% (9 out of 133 patients) heritability of NOA in this study. In addition, the 19 following genes (p < 0.05; OR > 1 or values not estimable; NE values not less than 1) were also regarded as expansive candidate genes for each proband, including NDUFAF1 for patient 38, TMEM63A and SEMA3F for patient 52, BICC1, SPRR2F, and GSTM3 for patient 60, ABCB8, PRRG4, RPUSD4, and ZNF221 for patient 62, SELP and GNPTAB for patient 98, DDX11 and POLD2 for patient 117, LRRFIP1 and LMF1 for patient 124, ULK4 for patient 130, ACSF2 and PPP1R3C for patient 131.
Discussion
Whole exome-based RVAS analysis identified a series of genes associated with NOA in this study. Especially, the most significant proteins, WARS1 (tryptophanyl-tRNA synthetase), DHRS4 (dehydrogenase/reductase 4), and RRBP1 (ribosome binding protein 1), were also involved in the meiosis related pathway, indicating they are associated with NOA. Meanwhile, three previously reported genes, TDRD7, SYCP3, and BRCA2, were also identified by RVAS and involved in the meiosis-related pathway, which partially supported the efficacy of the RVAS test flow in this study.
Combining RVAS analysis and trios linkage analysis showed the overlapped genes, ACTL8 (encodes actin-like 8 protein, restricted expression toward testis), TRA2B (encodes transformer 2 beta homolog protein), IDE (encodes insulin-degrading enzyme), FIBP (encodes FGF1 intracellular binding protein), NUP37 (encodes nucleoporin 37, a constituent of the nuclear pore complexes required for mitosis and probably for meiosis) (Loiodice, Alves et al., 2004), and PIGT (encodes phosphatidylinositol glycan anchor biosynthesis class T protein), were associated with NOA, as well as involved in the meiosis-related PPI network. Especially, the gene NUP37 was previously identified as indispensable for mitosis, we speculate that NUP37 (p.Trp80*, a frameshift mutation found in one out of 133 patients while absent in controls) may also play important roles during meiosis and spermatogenesis. In addition, the expression of ACTL8 gene is restricted in testis, the actin-like 8 protein ACTL8 (p.Val254Aspfs*4, a frameshift mutation found in one out of 133 patients, while being absent in controls) is assumed to execute the functions during spermatogenesis and play a role in NOA.
The other genes identified by RVAS or parental-proband trios analysis, especially those involved in the meiosis-related PPI network like PIWIL2, HNRNPC, and DDX39B may also play a role in NOA. It has not escaped our notice that PIWIL1 (encodes Human P-element-induced wimpy testis 1 protein, a paralog of human PIWIL2) was a previously identified gene causative for NOA; Human P-element-induced wimpy testis (PIWI) proteins act as protectors of germline, and are expressed mainly in the germline cells (Gou, Kang et al., 2017), the phenotype of Piwil2-deficient (knockout) male mice exhibited azoospermia with complete spermatogenic arrest according to the Mouse Genome Database, hence PIWIL2 (p.Gln11Serfs*78, a frameshift mutation found in one, P129, out of 133 patients while absent in controls) may play a similar role during spermatogenesis and in NOA.
Despite significant advances have been achieved in understanding the etiology of NOA, this study has certain limitations. First, prospectively more functional evidence like the cellular or physiological experiments should be investigated to address this question better. Second, the limitation lies in that non-LOFs like the missense mutations and small insertions or deletions which may be pathogenic were not considered. Finally, the false-positive rate of the de novo mutations (Table 2) is relatively high, which is in part attributable to sequencing errors; therefore, we suggest that de novo mutations should be treated with caution in clinical practice.
We demonstrate that RVAS identified a pool of genes, especially the most significant novel genes WARS, DHRS4, and RRBP1 which were also involved in the meiosis-related PPI network, indicating they are associated with NOA. In addition, six novel genes, ACTL8, TRA2B, IDE, FIBP, NUP37, and PIGT were revealed to be associated with NOA in both the RVAS and trios linkage analysis, as well as involved in the meiosis-related pathway; we conclude that these six novel genetic risk factors potentially account for a fraction (3.76%, 5 out of 133 patients) of the heritability of NOA in question, together with the three previously reported genes would potentially account for 6.77% (9 out of 133 patients) heritability of NOA in question in this study.
This study not only sheds light on the underlying pathological mechanism of NOA, but also offers valuable insight into the genomic landscape of NOA to constitute a potential basis for more efficient diagnosis yield in the future clinical application, and might have broad implications on men’s health.
Materials and Methods
Study Design and Patients
From this center, 133 patients with idiopathic NOA and large sets of controls (343 external male and 22 family-based controls) were recruited. Of 133 patients, 11 cases together with their parents were enrolled as trios. All patients underwent semen analysis at least on three different occasions, whereas no sperm were observed in the ejaculate even after centrifugation, and those with a history of orchitis, obstruction of vas deferens, or endocrine disorders were excluded. All the controls had fathered at least one child.
The clinical characteristics of the patients were summarized in Supplementary Table S1. These patients were selected according to the following criteria: 1) azoospermia due to either spermatogenic arrest (at spermatogonial or spermatocyte level) or SCOS (Supplementary Figure S1); 2) normal karyotype; 3) absence of Y-chromosome microdeletions and TEX11 mutations (including the copy number variation (CNV) of TEX11); and 4) absence of a list (Supplementary Table S2) of known causes for azoospermia.
Genomic Analysis
The genomic DNA was extracted from the peripheral blood. All the WES data underwent the same quality control filtering and pruning procedures to maximize parity between cases and controls. The average sequencing depth of whole-exome target regions for each sample was higher than 100, and ×20 coverage for more than 95% targeted bases was achieved for each sample. The process of bioinformatics analysis includes data filtering, alignment, mutation detection, and result annotation. The raw data were first evaluated for quality to remove low-quality and adapter contaminated reads. The clean data containing pair-end reads were mapped to the human genome (NCBI37/hg19 genome assembly) using BWA software (Burrows-Wheeler Aligner http://sourceforge.net/projects/bio-bwa/, version 0.7.15). The PCR-induced duplication was eliminated using Picard software. SNVs and Indels were tested using the genomic analysis toolkit (GATK). The GATK pipeline was used to identify the variations. For subsequent analyses, variants with minor allele frequencies (MAF) lower than one percent or uncatalogued in the Genome Aggregation Database (gnomAD) and the 1,000 Genomes were annotated with ANNOVAR. Loss of function (LOF) variations were defined as frameshift mutations, initiation mutations, premature or stopless mutations, or disruption of canonical splicing sites (± 2 bp). CNVs were analyzed using ExomeDepth and CNVkit. PPI network was built by the search tool for the retrieval of interacting genes/proteins (STRING) and further analyzed by Cytoscape_v4.0.2 software.
For linkage analysis, according to a maternally inherited pattern or the de novo mutation mechanism to which the gender-specific disease NOA condition was supposed to be genetically attributed, the paternally inherited variations were excluded; meanwhile, the probands’ LOFs were preserved only if the LOFs were not found in the 343 male controls. Sanger sequencing was performed for validating the variations.
Statistical Analysis
A total of 282 LOFs were removed due to the exact test for Hardy-Weinberg equilibrium (p < 0.05). The remaining LOFs were analyzed in the gene burden association test using the sequence kernel association test (SKAT) package (Ionita-Laza, Lee et al., 2013). The two-tailed p values were calculated, p < 0.05 was considered as statistically significant. False discovery rate (FDR) was calculated for PPI network enrichment analysis. The resulting p values were adjusted using Benjamini and Hochberg’s approach for controlling the false discovery rate. The odds ratio (OR) and 95% confidence interval (CI) were calculated based on the genotype matrix. The quartile–quartile plot (Q–Q plot) for the RVAS analysis was drawn (Supplementary Figure S2). All analyses were performed using R v4.0.2.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The name of the repository and accession number can be found below: CNSA (https://db.cngb.org/cnsa/); CNP0001080. The Human Protein Atlas database, http://www.proteinatlas.org/ 1,000 Genomes, http://www.internationalgenome.org/ Genome Aggregation Database, https://gnomad.broadinstitute.org/ Mouse Genome Database, http://www.informatics.jax.org/.
Ethics Statement
The studies involving human participants were reviewed and approved by the Medical Ethics Review Board (18K039-002) of The First Hospital affiliated to Jilin University (China), and the study was carried out in compliance with the Helsinki Declaration. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
Conceptualization: RL, HZ, WL and HH; collection and processing of clinical samples: HZ, YJ, WL, XZ, HH, LL, JH and RW; formal analysis and investigation: WL, HH, RL, HZ, JL, JZ, MC and JW; statistical and bioinformatics analyses: WL, HZ, HH, RL and YJ; writing—original draft preparation: WL; funding acquisition: RL; supervision: RL, ZP, WL and HH; and review and approval of text: all authors.
Funding
This study was supported by the funding from the Department of Science and Technology of Jilin Province, China (20210101302JC).
Conflict of Interest
WL, JL, JZ, MC, JW, ZP and HH were employed by the company BGI Genomics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank all the participants in this study for support. Thank Xinyue Zhang, Leilei Li, Jing He, Jing Wu for advice, assistant and help.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.872179/full#supplementary-material
Supplementary Figure S1 | The histopathology examination for patients with NOA (spermatogenic arrest or Sertoli cell-only syndrome). The hematoxylin-eosin (HE) staining was performed to facilitate the diagnosis for patients. The two upper images showed a patient diagnosed as Sertoli cell-only syndrome; and the two lower photos showed another patient diagnosed as spermatogenic arrest type I. Only Sertoli cells, or primary spermatocytes, or spermatogonium could be detected, whereas no mature spermatozoa could be seen in either case. The red arrow indicated the corresponding cell types.
Supplementary Figure S2 | The quartile–quartile plot (Q–Q plot) for the RVAS analysis. 95% CI was shown as the blue solid line around the uniform distribution line (red dashed line).
Supplementary Figure S3 | The histopathology examination for 5 independent patients with NOA. The hematoxylin–eosin (HE) staining was performed to facilitate the diagnosis for patients P62, P117, P124, P130, and P131, respectively. The red arrow indicated the corresponding cell types.
References
Arafat, M., Har-Vardi, I., Harlev, A., Levitas, E., Zeadna, A., Abofoul-Azab, M., et al. (2017). Mutation in TDRD9 Causes Non-obstructive Azoospermia in Infertile Men. J. Med. Genet. 54 (9), 633–639. doi:10.1136/jmedgenet-2017-104514
Artomov, M., Stratigos, A. J., Kim, I., Kumar, R., Lauss, M., Reddy, B. Y., et al. (2017). Rare Variant, Gene-Based Association Study of Hereditary Melanoma Using Whole-Exome Sequencing. J. Natl. Cancer Inst. 109 (12). doi:10.1093/jnci/djx083
Ayhan, Ö., Balkan, M., Guven, A., Hazan, R., Atar, M., Tok, A., et al. (2014). Truncating Mutations inTAF4BandZMYND15causing Recessive Azoospermia. J. Med. Genet. 51 (4), 239–244. doi:10.1136/jmedgenet-2013-102102
Bell, A. D., Mello, C. J., Nemesh, J., Brumbaugh, S. A., Wysoker, A., and McCarroll, S. A. (2020). Insights into Variation in Meiosis from 31,228 Human Sperm Genomes. Nature. doi:10.1038/s41586-020-2347-0
Ben Khelifa, M., Ghieh, F., Boudjenah, R., Hue, C., Fauvert, D., Dard, R., et al. (2018). A MEI1 Homozygous Missense Mutation Associated with Meiotic Arrest in a Consanguineous Family. Hum. Reprod. 33 (6), 1034–1037. doi:10.1093/humrep/dey073
Chertman, W., Arora, H., Griswold, A. J., Lopategui, D. M., and Ramasamy, R. (2019). Whole Exome Sequencing Identifies a Rare Nonsense Mutation in FAM47C as a Possible Cause of Severe Oligospermia in Brothers with Varicocele. Urology 129, 71–73. doi:10.1016/j.urology.2019.03.016
Choi, Y., Jeon, S., Choi, M., Lee, M.-h., Park, M., Lee, D. R., et al. (2010). Mutations in SOHLH1 Gene Associate with Nonobstructive Azoospermia. Hum. Mutat. 31 (7), 788–793. doi:10.1002/humu.21264
Gershoni, M., Hauser, R., Yogev, L., Lehavi, O., Azem, F., Yavetz, H., et al. (2017). A Familial Study of Azoospermic Men Identifies Three Novel Causative Mutations in Three New Human Azoospermia Genes. Genet. Med. 19 (9), 998–1006. doi:10.1038/gim.2016.225
Gou, L.-T., Kang, J.-Y., Dai, P., Wang, X., Li, F., Zhao, S., et al. (2017). Ubiquitination-Deficient Mutations in Human Piwi Cause Male Infertility by Impairing Histone-To-Protamine Exchange during Spermiogenesis. Cell 169 (6), 1090–1104. doi:10.1016/j.cell.2017.04.034
He, W.-B., Tu, C.-F., Liu, Q., Meng, L.-L., Yuan, S.-M., Luo, A.-X., et al. (2018). DMC1 Mutation that Causes Human Non-obstructive Azoospermia and Premature Ovarian Insufficiency Identified by Whole-Exome Sequencing. J. Med. Genet. 55 (3), 198–204. doi:10.1136/jmedgenet-2017-104992
Ionita-Laza, I., Lee, S., Makarov, V., Buxbaum, J. D., and Lin, X. (2013). Sequence Kernel Association Tests for the Combined Effect of Rare and Common Variants. Am. J. Hum. Genet. 92 (6), 841–853. doi:10.1016/j.ajhg.2013.04.015
Kasak, L., Punab, M., Nagirnaja, L., Grigorova, M., Minajeva, A., Lopes, A. M., et al. (2018). Bi-allelic Recessive Loss-Of-Function Variants in FANCM Cause Non-obstructive Azoospermia. Am. J. Hum. Genet. 103 (2), 200–212. doi:10.1016/j.ajhg.2018.07.005
Kherraf, Z. E., Christou‐Kent, M., Karaouzene, T., Amiri‐Yekta, A., Martinez, G., Vargas, A. S., et al. (2017). SPINK 2 Deficiency Causes Infertility by Inducing Sperm Defects in Heterozygotes and Azoospermia in Homozygotes. EMBO Mol. Med. 9 (8), 1132–1149. doi:10.15252/emmm.201607461
Krausz, C., and Riera-Escamilla, A. (2018). Genetics of Male Infertility. Nat. Rev. Urol. 15 (6), 369–384. doi:10.1038/s41585-018-0003-3
Lee, S., Abecasis, G. R., Boehnke, M., and Lin, X. (2014). Rare-variant Association Analysis: Study Designs and Statistical Tests. Am. J. Hum. Genet. 95 (1), 5–23. doi:10.1016/j.ajhg.2014.06.009
Locke, A. E., Steinberg, K. M., Steinberg, K. M., Chiang, C. W. K., Service, S. K., Havulinna, A. S., et al. (2019). Exome Sequencing of Finnish Isolates Enhances Rare-Variant Association Power. Nature 572 (7769), 323–328. doi:10.1038/s41586-019-1457-z
Loïodice, I., Alves, A., Rabut, G., Van Overbeek, M., Ellenberg, J., Sibarita, J.-B., et al. (2004). The Entire Nup107-160 Complex, Including Three New Members, Is Targeted as One Entity to Kinetochores in Mitosis. MBoC 15 (7), 3333–3344. doi:10.1091/mbc.e03-12-0878
Maor-Sagie, E., Cinnamon, Y., Yaacov, B., Shaag, A., Goldsmidt, H., Zenvirt, S., et al. (2015). Deleterious Mutation in SYCE1 Is Associated with Non-obstructive Azoospermia. J. Assist. Reprod. Genet. 32 (6), 887–891. doi:10.1007/s10815-015-0445-y
Miyamoto, T., Hasuike, S., Yogev, L., Maduro, M. R., Ishikawa, M., Westphal, H., et al. (2003). Azoospermia in Patients Heterozygous for a Mutation in SYCP3. The Lancet 362 (9397), 1714–1719. doi:10.1016/s0140-6736(03)14845-3
Mou, L., and Gui, Y. (2016). A Novel Variant of Androgen Receptor Is Associated with Idiopathic Azoospermia. Mol. Med. Rep. 14 (4), 2915–2920. doi:10.3892/mmr.2016.5587
Nakamura, S., Kobori, Y., Ueda, Y., Tanaka, Y., Ishikawa, H., Yoshida, A., et al. (2018). STX2 Is a Causative Gene for Nonobstructive Azoospermia. Hum. Mutat. 39 (6), 830–833. doi:10.1002/humu.23423
Okutman, O., Muller, J., Baert, Y., Serdarogullari, M., Gultomruk, M., Piton, A., et al. (2015). Exome Sequencing Reveals a Nonsense Mutation inTEX15causing Spermatogenic Failure in a Turkish Family. Hum. Mol. Genet. 24 (19), 5581–5588. doi:10.1093/hmg/ddv290
Okutman, O., Muller, J., Skory, V., Garnier, J. M., Gaucherot, A., Baert, Y., et al. (2017). A No-Stop Mutation in MAGEB4 Is a Possible Cause of Rare X-Linked Azoospermia and Oligozoospermia in a Consanguineous Turkish Family. J. Assist. Reprod. Genet. 34 (5), 683–694. doi:10.1007/s10815-017-0900-z
Ramasamy, R., Bakırcıoğlu, M. E., Cengiz, C., Karaca, E., Scovell, J., Jhangiani, S. N., et al. (2015). Whole-exome Sequencing Identifies Novel Homozygous Mutation in NPAS2 in Family with Nonobstructive Azoospermia. Fertil. Sterility 104 (2), 286–291. doi:10.1016/j.fertnstert.2015.04.001
Riera-Escamilla, A., Enguita-Marruedo, A., Moreno-Mendoza, D., Chianese, C., Sleddens-Linkels, E., Contini, E., et al. (2019). Sequencing of a 'mouse Azoospermia' Gene Panel in Azoospermic Men: Identification of RNF212 and STAG3 Mutations as Novel Genetic Causes of Meiotic Arrest. Hum. Reprod. 34 (6), 978–988. doi:10.1093/humrep/dez042
Rodrigues, V. O., Polisseni, F., Pannain, G. D., and Carvalho, M. A. G. (2020). “Genetics in Human Reproduction,” in JBRA Assist Reprod. doi:10.5935/1518-0557.20200007
Safari, S., Zare-Abdollahi, D., Mirfakhraie, R., Ghafouri-Fard, S., Pouresmaeili, F., Movafagh, A., et al. (2014). An Iranian Family with Azoospermia and Premature Ovarian Insufficiency Segregating NR5A1 Mutation. Climacteric 17 (3), 301–303. doi:10.3109/13697137.2013.847079
Schultz, N., Hamra, F. K., and Garbers, D. L. (2003). A Multitude of Genes Expressed Solely in Meiotic or Postmeiotic Spermatogenic Cells Offers a Myriad of Contraceptive Targets. Proc. Natl. Acad. Sci. U.S.A. 100 (21), 12201–12206. doi:10.1073/pnas.1635054100
Tan, Y.-Q., Tu, C., Meng, L., Yuan, S., Sjaarda, C., Luo, A., et al. (2019). Loss-of-function Mutations in TDRD7 lead to a Rare Novel Syndrome Combining Congenital Cataract and Nonobstructive Azoospermia in Humans. Genet. Med. 21 (5), 1209–1217. doi:10.1038/gim.2017.130
Tenenbaum-Rakover, Y., Weinberg-Shukron, A., Renbaum, P., Lobel, O., Eideh, H., Gulsuner, S., et al. (2015). Minichromosome Maintenance Complex Component 8 (MCM8) Gene Mutations Result in Primary Gonadal Failure. J. Med. Genet. 52 (6), 391–399. doi:10.1136/jmedgenet-2014-102921
Wang, X. N., Li, Z. S., Ren, Y., Jiang, T., Wang, Y. Q., Chen, M., et al. (2013). The Wilms Tumor Gene, Wt1, Is Critical for Mouse Spermatogenesis via Regulation of Sertoli Cell Polarity and Is Associated with Non-obstructive Azoospermia in Humans. Plos Genet. 9 (8), e1003645. doi:10.1371/journal.pgen.1003645
Xu, J., Jiang, L., Yu, W., Guo, H., Zhang, H., Wei, D., et al. (2017). A Novel Functional Variant in Wilms' Tumor 1 (WT1 ) Is Associated with Idiopathic Non-obstructive Azoospermia. Mol. Reprod. Dev. 84 (3), 222–228. doi:10.1002/mrd.22768
Yatsenko, A. N., Georgiadis, A. P., Röpke, A., Berman, A. J., Jaffe, T., Olszewska, M., et al. (2015). X-linked TEX11 Mutations, Meiotic Arrest, and Azoospermia in Infertile Men. N. Engl. J. Med. 372 (22), 2097–2107. doi:10.1056/nejmoa1406192
Zhang, C., Xue, P., Gao, L., Chen, X., Lin, K., Yang, X., et al. (2016). Highly Conserved Epigenetic Regulation of BOULE and DAZL Is Associated with Human Fertility. FASEB j. 30 (10), 3424–3440. doi:10.1096/fj.201500167r
Zhang, Y. X., Li, H. Y., He, W. B., Tu, C., Du, J., Li, W., et al. (2019). XRCC2 Mutation Causes Premature Ovarian Insufficiency as Well as Non‐obstructive Azoospermia in Humans. Clin. Genet. 95 (3), 442–443. doi:10.1111/cge.13475
Keywords: causative genes, linkage analysis, non-obstructive azoospermia, whole exome sequencing, rare variant association study
Citation: Zhang H, Li W, Jiang Y, Li J, Chen M, Wang R, Zhao J, Peng Z, Huang H and Liu R (2022) Whole Exome Sequencing Identifies Genes Associated With Non-Obstructive Azoospermia. Front. Genet. 13:872179. doi: 10.3389/fgene.2022.872179
Received: 09 February 2022; Accepted: 15 March 2022;
Published: 13 April 2022.
Edited by:
Fan Jin, Zhejiang University, ChinaReviewed by:
Zhao Liangyu, The Fifth Affiliated Hospital of Sun Yat-sen University, ChinaZhiqiang Li, Qingdao University, China
Copyright © 2022 Zhang, Li, Jiang, Li, Chen, Wang, Zhao, Peng, Huang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Huang, aHVhbmdodWlAZ2Vub21pY3MuY24=; Ruizhi Liu, bGl1cnpAamx1LmVkdS5jbg==
†These authors have contributed equally to this work