
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 11 April 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.872056
This article is part of the Research TopicGenetics of Familial Hypercholesterolemia: New Insight - Volume IIView all 17 articles
 Hayato Tada1*
Hayato Tada1* Nobuko Kojima1
Nobuko Kojima1 Kan Yamagami1
Kan Yamagami1 Akihiro Nomura1Atsushi Nohara2
Akihiro Nomura1Atsushi Nohara2 Soichiro Usui1Kenji Sakata1Noboru Fujino1Masayuki Takamura1Masa-Aki Kawashiri1
Soichiro Usui1Kenji Sakata1Noboru Fujino1Masayuki Takamura1Masa-Aki Kawashiri1Objective: It has been shown that pathogenic variants are associated with poor clinical outcomes in patients with familial hypercholesterolemia (FH). However, data on the effect of different types of pathogenic variants on FH phenotype is limited.
Methods: We retrospectively investigated the associations between genotypes and phenotypes, including low-density lipoprotein (LDL) cholesterol level and the occurrence of major adverse cardiac events (MACEs), defined as cardiovascular death, myocardial infarction, unstable angina, or coronary artery revascularization, in patients with FH (N = 1,050, male/female = 490/560). Based on genotype, the patients were divided into the following three groups: patients without pathogenic variants, patients with missense variants, and patients with protein-truncating variants (PTVs). Cox proportional hazard model was used to identify the factors associated with MACEs.
Results: The median follow-up duration was 12.6 years (interquartile range = 9.5–17.9 years). There were 665 patients with FH-mutation (277 patients with missense variants and 388 patients with PTVs) and 385 patients without FH-mutation. Over the follow-up duration, 175 MACEs were observed. We identified 89 different pathogenic variants in the 665 patients with FH. LDL cholesterol level was found to be significantly higher in patients with PTVs (256 mg/dl) than in patients with missense variants (236 mg/dl) and patients without pathogenic variants (216 mg/dl). It was also found that PTVs and missense variants are significantly associated with MACEs (hazard ratio [HR] = 1.58, 95% confidence interval [CI] = 1.08–2.08, p = 0.0033 and HR = 3.24, 95% CI = 2.12–4.40, p = 3.9 × 10−6, respectively), independent of classical risk factors.
Conclusion: Pathogenic variants, especially PTVs, are significantly associated with poor outcomes in patients with FH. Genetic testing is useful for the diagnosis and risk stratification of patients with FH.
• Pathogenic variants are associated with higher risk for CVD among patients with FH.
• LDL cholesterol level differs according to types of pathogenic variants among patients with FH.
• Prognosis differs according to types of pathogenic variants among patients with FH.
Familial hypercholesterolemia (FH) is one of the most common Mendelian disorders, and its incidence in the general population is reported to be one in 300 (Beheshti et al., 2020; Hu et al., 2020). FH is caused by genetic variants associated with low-density lipoprotein (LDL) metabolism, such as the LDL receptor (LDLR), apolipoprotein B (APOB), proprotein convertase subtilisin/kexin type 9 (PCSK9), and LDLR adaptor protein 1 (LDLRAP1) (Mabuchi, 2017). Typically, FH is diagnosed based on clinical criteria (Austin et al., 2004; Harada-Shiba et al., 2018; Tada et al., 2021a); however, it was reported that pathogenic variants may be associated with increased risk of coronary artery disease (Khera et al., 2016; Tada et al., 2017). Therefore, genetic testing is recommended for the diagnosis and risk stratification of patients with FH. More than 2000 pathogenic variants have been identified worldwide, and most of them are LDLR variants (Nohara et al., 2021). Based on genotype, pathogenic variants can be classified as missense variants (which can estimate residual LDLR activity) or protein-truncating variants (PTVs), which may have lost their LDLR function. Only a few studies have investigated the effects of pathogenic variants on the phenotype of patients with FH (Tada et al., 2020). The aim of this study was to investigate the associations between genotypes and phenotypes, including LDL cholesterol level and occurrence of major adverse cardiac events (MACEs) in patients with FH diagnosed using clinical diagnostic criteria.
We evaluated the data of 2011 patients with FH diagnosed clinically using the Japan Atherosclerosis Society (JAS) 2017 criteria at Kanazawa University Hospital between 1990 and 2020. All the patients in this study fulfilled at least two of the three essential clinical criteria stipulated by the JAS for FH diagnosis. The criteria are as follows: 1) LDL cholesterol level ≥180 mg/dl, 2) tendon xanthoma on the backs of the hands, elbows, knees, or other areas; Achilles tendon hypertrophy or Achilles tendon thickness on X-ray ≥ 9 mm; or xanthoma tuberosum, and 3) family history of FH or premature coronary artery disease diagnosed in a first- or second-degree relative. Nine hundred and sixty-one patients were excluded due to missing data (such as data on blood lipids and genetic analysis or data on homozygous and compound heterozygous FH. Finally, 1,050 patients were included in this study (Supplementaary Figure S1).
We defined hypertension as systolic blood pressure ≥140 mmHg, diastolic blood pressure ≥90 mmHg, or use of antihypertensive agents. Further, we used the definition of diabetes given by the Japan Diabetes Society (Araki et al., 2020). Smoking status was defined as a current smoking status. Cardiovascular disease (CVD) was defined as angina pectoris, myocardial infarction, or severe stenotic region(s) in the coronary artery (≥75% stenosis), identified on angiography or computed tomography. Serum levels of total cholesterol, triglycerides, and high-density lipoprotein cholesterol were determined enzymatically using automated instrumentation. LDL cholesterol level was calculated using the Friedewald formula if triglyceride level was <400 mg/dl; otherwise, it was determined enzymatically.
We assessed genotypes using a next-generation sequencer. In brief, the coding regions of LDLR, APOB, PCSK9, and LDLRAP1 were sequenced as described in a previous study (Tada et al., 2018). Further, copy number variations at the LDLR were assessed using the eXome Hidden Markov Model software as described in an earlier study (Yamamoto et al., 2016). We evaluated the pathogenicity of the genetic variants according to the standard American College of Medical Genetics and Genomics criteria (Richards et al., 2015). We classified pathogenic variants as missense variants or PTVs, which include frameshift variants, large deletion or duplication variants, nonsense variants, and splice site variants.
This study was approved by the Ethics Committee of Kanazawa University. All procedures were conducted in accordance with the ethical standards of the Human Research Committee (institutional and national) and the Helsinki Declaration (1975, revised in 2008). Informed consent for genetic analysis was obtained from all the study participants.
Categorical variables were reported as numbers and percentages, and they were compared using Fisher’s exact test or chi-square test. Normally distributed continuous variables were reported as means ± standard deviations. Not normally distributed continuous variables were reported as medians and interquartile ranges. Mean values of continuous variables were compared using Student’s t-test for independent variables, and median values were compared using non-parametric Wilcoxon–Mann–Whitney rank sum test. Chi-square or Fisher’s post-hoc test was used for categorical variables as indicated. Cox proportional hazard model was used to assess relationships between all the variables. Cumulative Kaplan–Meier survival curves starting at baseline were constructed to compare times to the first MACE. All statistical analyses were conducted using R statistics (https://www.r-project.org). p-values < 0.05 were considered statistically significant.
The clinical characteristics of the study participants are shown in Table 1. The mean age of patients was 49 years, and almost half of the patients were men. The median LDL cholesterol level at baseline was 244 mg/dl, and it decreased to 110 mg/dl at follow-up. A total of 776 patients (73.9%) had a family history of FH and/or premature CVD. Furthermore, 290 patients (27.6%) had a history of CVD. Upon division of patients into two groups based on the occurrence of MACEs, we observed significant differences in all variables (except family history of FH and/or premature CVD) between the two groups. In addition, we observed differences in the trend of characteristics; for example, we observed, following division of patients with pathogenic mutations into three groups based on genotype, that the proportion of patients with diabetes decreased (Supplementary Table S1). The medical treatments administered during follow-up are summarized in Table 2. Statin therapy, frequently followed by ezetimibe and colestimide therapy, was administered to most of the patients.
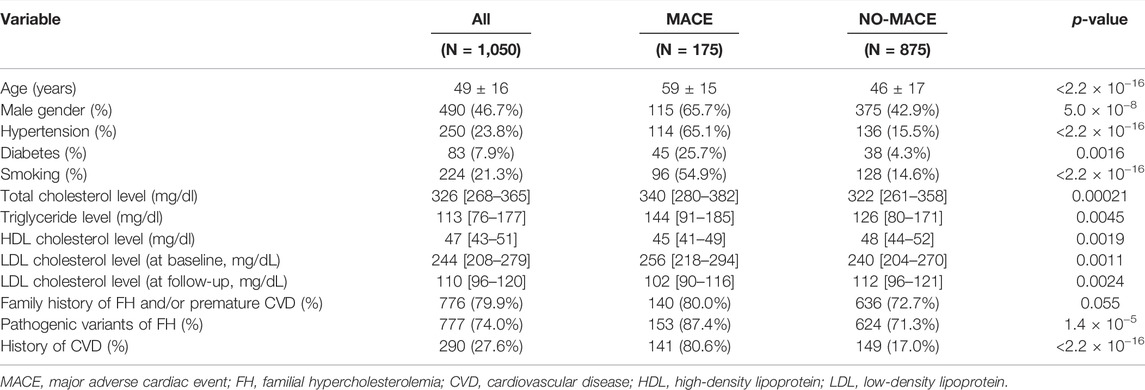
TABLE 1. Baseline characteristics.

TABLE 2. Medical therapies.
We identified 89 pathogenic variants in 665 patients. Of the pathogenic variants, 277 were classified as missense variants, and 388 were classified as PTVs (46 were frameshift variants, 69 were large deletion or duplication variants, 204 were nonsense variants, and 69 were splice site variants; Figure 1). The details are shown in Supplementary Table S2.
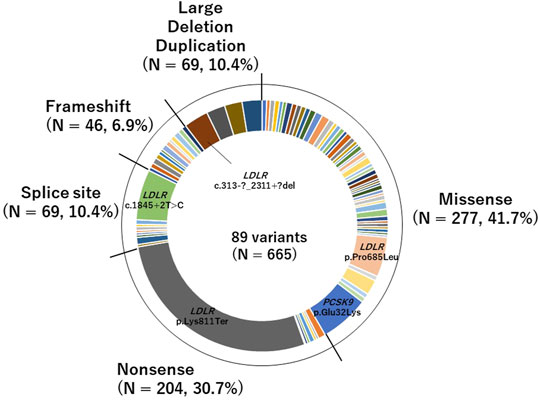
FIGURE 1. Pie chart of pathogenic variants identified in this study of the pathogenic variants, 277 were classified as missense variants, and 388 were classified as PTVs (46 were frameshift variants, 69 were large deletion or duplication variants, 204 were nonsense variants, and 69 were splice site variants).
Based on genotype, we divided the patients into the following three groups: patients without pathogenic variants, patients with missense variants, and patients with PTVs. We found that LDL cholesterol level was highest in patients with PTVs, and patients with missense variants were found to have higher LDL cholesterol levels than patients without pathogenic variants (Figure 2A). The median LDL cholesterol levels of patients without pathogenic variants, patients with missense variants, and patients with PTVs were 216 mg/dl, 236 mg/dl, and 256 mg/dl, respectively. The LDL cholesterol levels of the three groups showed deformed trimodal distributions (Figure 2B).
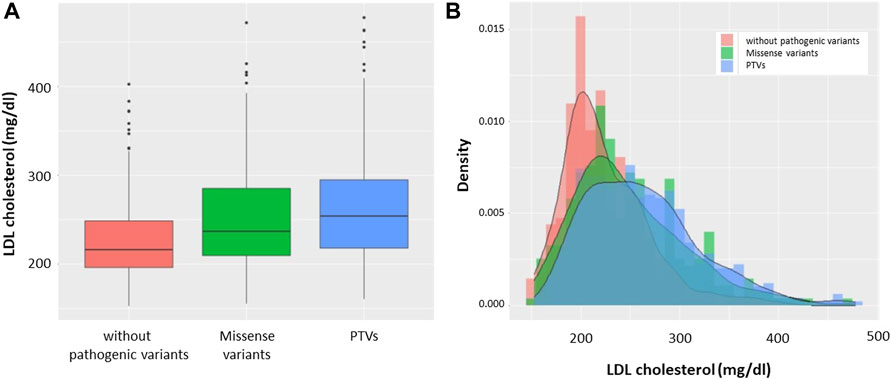
FIGURE 2. LDL cholesterol levels according to mutation status. (A) Boxplots: Red indicates patients without pathogenic variants. Green indicates patients with missense variants. Blue indicates patients with PTVs. (B) Histograms with density: Red indicates patients without pathogenic variants. Green indicates patients with missense variants. Blue indicates patients with PTVs.
Over the median follow-up duration of 12.6 years, 175 patients had MACEs, which include CVD-associated death, myocardial infarction, unstable angina, and staged revascularization (Table 3). Using Cox proportional hazard model, we assessed factors associated with MACEs and found that age (hazard ratio [HR] = 1.09, 95% confidence interval [CI] = 1.04–1.14, p = 2.2 × 10−14), male gender (HR = 1.58, 95% CI = 1.08–2.08, p = 0.0079), hypertension (HR = 3.11, 95% CI = 2.10–4.25, p = 6.9 × 10−6), diabetes (HR = 2.44, 95% CI = 1.46–3.52, p = 0.0021), smoking (HR = 2.56, 95% CI = 1.56–3.18, p = 0.00041), LDL cholesterol level (per 10 mg/dl; HR = 1.01, 95% CI = 1.00–1.02, p = 0.022), and prior CVD (HR = 3.45, 95% CI = 2.02–4.80, p < 2.2 × 10−16) are significantly associated with MACEs (Table 4). We also found that missense variants and PTVs are associated with MACEs (HR = 1.58, 95% CI = 1.08–2.08, p = 0.0033 and HR = 3.24, 95% CI = 2.12–4.40, p = 3.9 × 10−6, respectively).

TABLE 3. Types of MACEs.
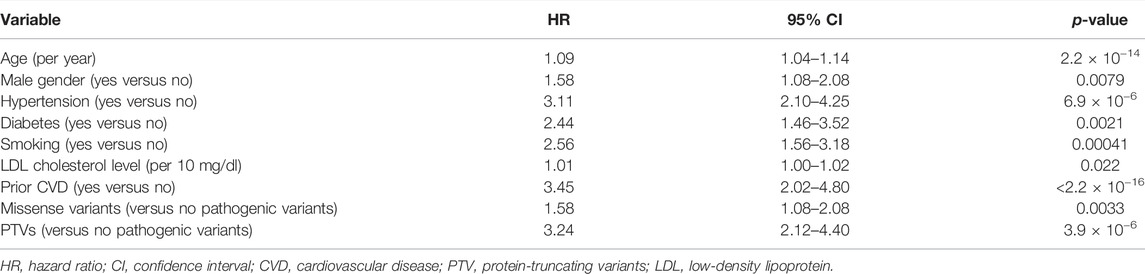
TABLE 4. Factors associated with MACEs.
We assessed the survival curve and found that patients with PTVs have the worst outcome of the three groups and that patients with missense variants have worse outcomes than patients without pathogenic variants (Figure 3).
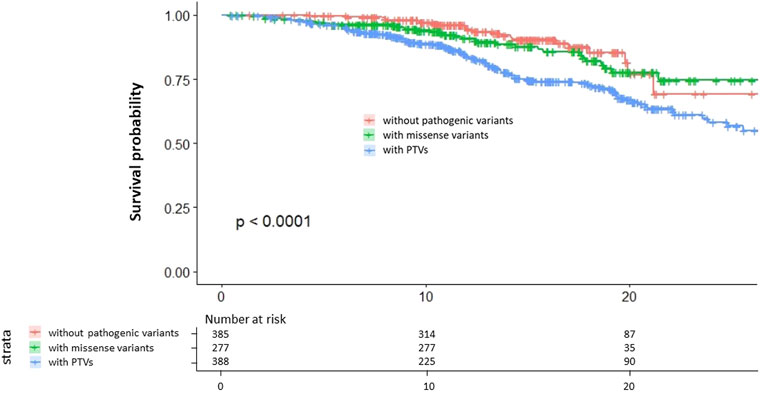
FIGURE 3. Kaplan–Meier survival curves. Red indicates patients without pathogenic variants. Green indicates patients with missense variants. Blue indicates patients with PTVs.
In this study, we evaluated the effects of pathogenic variants on the clinical phenotypes of FH and found that 1) LDL cholesterol level is highest in patients with PTVs and higher in patients with missense variants than in patients without pathogenic variants, 2) prognosis is worst in patients with PTVs and worse in patients with missense variants than in patients without pathogenic variants.
There are many factors, including traditional risk factors and genetic factors that have been associated with CVD in patients with FH. Indeed, a clinical risk score named Montreal-FH-score comprising traditional risk factors has been one of the useful tools predicting CVD risk in patients with FH (Paquette et al., 2017). In addition, we and others have shown that rare and common genetic variations contributed to increase or decrease risks for CVD among patients with FH on top of FH-mutation (Tada et al., 2018; Fahed et al., 2020). Based on different standpoints, including diagnosis, cascade screening, and risk stratification, genetic testing is currently recommended for FH (Sturm et al., 2018). Patients with FH are associated with significantly high risk of CVD due to cumulative exposure to elevated LDL cholesterol level (Tada et al., 2021b). Thus, early diagnosis and intervention are considered vital. Previous studies reveal that FH diagnosis is improved by early treatment of patients (Luirink et al., 2019; Tada et al., 2021c). Since patients with FH typically do not have tendon xanthomas (one of the most important clinical diagnostic criteria) in childhood, genetic testing is essential for early diagnosis of FH (Tada et al., 2021d; Matsunaga et al., 2021; Nagahara et al., 2021). Furthermore, given the criteria of pathogenicity of genetic variants and the catalog of pathogenic variants of FH, there is a growing demand for further risk stratification of patients with FH based on genotype (Chora et al., 2022). The growing demand is natural in this personalized medicine era, when phenotypes can be estimated using genotypes and the best treatments for many inherited diseases can be determined based on genotype (Sukrithan et al., 2019; Pujol et al., 2021; Tada, 2021). We expect that patients with PTVs will receive intensive treatment and that cascade screening will be recommended for patients with pathogenic variants, especially PTVs, to identify patients with high CVD risk.
This study has several limitations. First, this is a retrospective study conducted in a single center. Therefore, the study findings may not be applicable to other patients. However, our institute has a long history of treating patients with FH and has one of the largest databases in Japan. Second, we could not account for treatments administered during follow-up, and this may affect the study results. Third, many patients were excluded from analysis due to missing data or loss to follow-up. This exclusion may also affect study results. When we compared the baseline characteristics (only the key variables, including age, gender, and LDL cholesterol) between study subjects and those excluded. We found that the mean age of the patients excluded from this study was significantly younger than that of patients included in this study, although there were no significant differences between these 2 groups in gender and LDL cholesterol (Supplementary Table S2). Fourth, in this study, functional analysis was not performed to validate the pathogenicity of genetic variants. Fifth, polygenic factors were not considered in this study. Sixth, we did not account for functional analyses of the variants, especially in missense variants. In fact, some of the missense variants, such as p.Ser177Leu, p.Glu228Gln, p.Asp266Asn, and p.Val429Leu have been considered as “null alleles” based on functional assays (Hobbs et al., 1992). However, the number of individuals with these variants were so small that it is unlikely to affect our results. In addition, it is difficult to classify all of the variants of this study clearly because of lack of such data. Furthermore, a simple classification of PTVs appears to have great impact on phenotypes in our data. Accordingly, we believe that it is beneficial for us to divide PTVs and missense variants in case of FH. Seventh, we did not fully observe other atherosclerotic disease, such as cerebrovascular disease, peripheral artery disease and aortic valve stenosis unless they exhibited any symptoms suggesting these conditions. Further studies will full assessments of these conditions will be useful to estimate their comprehensive risk assessments.
In conclusion, pathogenic variants, especially PTVs, are significantly associated with poor outcomes in patients with FH. Genetic testing is useful for the diagnosis and further risk stratification of patients with FH.
The datasets presented in this article are not readily available because the IRB of Kanazawa University did not give us an approval to deposit genomic data even in a public repository. Requests to access the datasets should be directed to the corresponding author, HT.
The studies involving human participants were reviewed and approved by the IRB of Kanazawa University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
HT and M-AK conceived of the presented idea. HT, NK, KY, ATN, AKN, SU, KS, NF, MT, and M-AK collected clinical data. HT, NK, KY, and AKN performed genetic analyses. HT performed statistical analyses. MT and M-AK supervised the findings of this work. All authors contributed to write the manuscript. All authors discussed the results and contributed to the final manuscript.
This work has been supported by scientific research grants from the Ministry of Education, Science and Culture of Japan (19K08575); a grant from Ministry of Health, Labour and Welfare (MHLW) of Japan (Sciences Research Grant for Research on Rare and Intractable Diseases) and Japanese Circulation Society (project for genome analysis in cardiovascular diseases); and the Japan Agency for Medical Research and Development (19188592, JP22ek0109487, and 16ek0210075h0001) to HT.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are thankful to Kazuko Honda and Sachio Yamamoto for their outstanding technical assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.872056/full#supplementary-material
Araki, E., Goto, A., Kondo, T., Noda, M., Noto, H., Origasa, H., et al. (2020). Japanese Clinical Practice Guideline for Diabetes 2019. J. Diabetes Investig. 11, 1020–1076. doi:10.1111/jdi.13306
Austin, M. A., Hutter, C. M., Zimmern, R. L., and Humphries, S. E. (2004). Genetic Causes of Monogenic Heterozygous Familial Hypercholesterolemia: a HuGE Prevalence Review. Am. J. Epidemiol. 160, 407–420. doi:10.1093/aje/kwh236
Beheshti, S. O., Madsen, C. M., Varbo, A., and Nordestgaard, B. G. (2020). Worldwide Prevalence of Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 75, 2553–2566. doi:10.1016/j.jacc.2020.03.057
Chora, J. R., Iacocca, M. A., Tichý, L., Wand, H., Kurtz, C. L., Zimmermann, H., et al. (2022). The Clinical Genome Resource (ClinGen) Familial Hypercholesterolemia Variant Curation Expert Panel Consensus Guidelines for LDLR Variant Classification. Genet. Med. 24, 293–306. doi:10.1016/j.gim.2021.09.012
Fahed, A. C., Wang, M., Homburger, J. R., Patel, A. P., Bick, A. G., Neben, C. L., et al. (2020). Polygenic Background Modifies Penetrance of Monogenic Variants for Tier 1 Genomic Conditions. Nat. Commun. 11, 3635. doi:10.1038/s41467-020-17374-3
Harada-Shiba, M., Arai, H., Ishigaki, Y., Ishibashi, S., Okamura, T., Ogura, M., et al. (2018). Guidelines for Diagnosis and Treatment of Familial Hypercholesterolemia 2017. J. Atheroscler. Thromb. 25, 751–770. doi:10.5551/jat.cr003
Hobbs, H. H., Brown, M. S., and Goldstein, J. L. (1992). Molecular Genetics of the LDL Receptor Gene in Familial Hypercholesterolemia. Hum. Mutat. 1, 445–466. doi:10.1002/humu.1380010602
Hu, P., Dharmayat, K. I., Stevens, C. A. T., Sharabiani, M. T. A., Jones, R. S., Watts, G. F., et al. (2020). Prevalence of Familial Hypercholesterolemia Among the General Population and Patients with Atherosclerotic Cardiovascular Disease. Circulation 141, 1742–1759. doi:10.1161/circulationaha.119.044795
Khera, A. V., Won, H.-H., Peloso, G. M., Lawson, K. S., Bartz, T. M., Deng, X., et al. (2016). Diagnostic Yield and Clinical Utility of Sequencing Familial Hypercholesterolemia Genes in Patients with Severe Hypercholesterolemia. J. Am. Coll. Cardiol. 67, 2578–2589. doi:10.1016/j.jacc.2016.03.520
Luirink, I. K., Wiegman, A., Kusters, D. M., Hof, M. H., Groothoff, J. W., de Groot, E., et al. (2019). 20-year Follow-Up of Statins in Children with Familial Hypercholesterolemia. N. Engl. J. Med. 381, 1547–1556. doi:10.1056/nejmoa1816454
Mabuchi, H. (2017). Half a century Tales of Familial Hypercholesterolemia (FH) in Japan. Jat 24, 189–207. doi:10.5551/jat.rv16008
Matsunaga, K., Mizobuchi, A., Fu, H. Y., Ishikawa, S., Tada, H., Kawashiri, M.-a., et al. (2021). Universal Screening for Familial Hypercholesterolemia in Children in Kagawa, Japan. Jat, 62780. doi:10.5551/jat.62780
Nagahara, K., Nishibukuro, T., Ogiwara, Y., Ikegawa, K., Tada, H., Yamagishi, M., et al. (2021). Genetic Analysis of Japanese Children Clinically Diagnosed with Familial Hypercholesterolemia. Jat, 62807. doi:10.5551/jat.62807
Nohara, A., Tada, H., Ogura, M., Okazaki, S., Ono, K., Shimano, H., et al. (2021). Homozygous Familial Hypercholesterolemia. J. Atheroscler. Thromb. 28, 665–678. doi:10.5551/jat.RV17050
Paquette, M., Dufour, R., and Baass, A. (2017). The Montreal-FH-SCORE: A New Score to Predict Cardiovascular Events in Familial Hypercholesterolemia. J. Clin. Lipidol. 11, 80–86. doi:10.1016/j.jacl.2016.10.004
Pujol, P., Barberis, M., Beer, P., Friedman, E., Piulats, J. M., Capoluongo, E. D., et al. (2021). Clinical Practice Guidelines for BRCA1 and BRCA2 Genetic Testing. Eur. J. Cancer 146, 30–47. doi:10.1016/j.ejca.2020.12.023
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Sturm, A. C., Knowles, J. W., Gidding, S. S., Ahmad, Z. S., Ahmed, C. D., Ballantyne, C. M., et al. (2018). Clinical Genetic Testing for Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 72, 662–680. doi:10.1016/j.jacc.2018.05.044
Sukrithan, V., Deng, L., Barbaro, A., and Cheng, H. (2019). Emerging Drugs for EGFR-Mutated Non-small Cell Lung Cancer. Expert Opin. Emerging Drugs 24, 5–16. doi:10.1080/14728214.2018.1558203
Tada, H., Hori, M., Matsuki, K., Ogura, M., Nohara, A., Kawashiri, M.-a., et al. (2021). Achilles Tendon Thickness Assessed by X-ray Predicting a Pathogenic Mutation in Familial Hypercholesterolemia Gene. J. Atheroscler. Thromb., 62869. doi:10.5551/jat.62869
Tada, H., Hori, M., Nomura, A., Hosomichi, K., Nohara, A., Kawashiri, M.-a., et al. (2020). A Catalog of the Pathogenic Mutations of LDL Receptor Gene in Japanese Familial Hypercholesterolemia. J. Clin. Lipidol. 14, 346–351. doi:10.1016/j.jacl.2020.03.002
Tada, H., Kawashiri, M.-a., Nohara, A., Inazu, A., Mabuchi, H., and Yamagishi, M. (2017). Impact of Clinical Signs and Genetic Diagnosis of Familial Hypercholesterolaemia on the Prevalence of Coronary Artery Disease in Patients with Severe Hypercholesterolaemia. Eur. Heart J. 38, 1573–1579. doi:10.1093/eurheartj/ehx004
Tada, H., Kawashiri, M.-a., Nomura, A., Teramoto, R., Hosomichi, K., Nohara, A., et al. (2018). Oligogenic Familial Hypercholesterolemia, LDL Cholesterol, and Coronary Artery Disease. J. Clin. Lipidol. 12, 1436–1444. doi:10.1016/j.jacl.2018.08.006
Tada, H., Okada, H., Nohara, A., Yamagishi, M., Takamura, M., and Kawashiri, M.-a. (2021). Effect of Cumulative Exposure to Low-Density Lipoprotein-Cholesterol on Cardiovascular Events in Patients with Familial Hypercholesterolemia. Circ. J. 85, 2073–2078. doi:10.1253/circj.cj-21-0193
Tada, H., Okada, H., Nomura, A., Nohara, A., Yamagishi, M., Takamura, M., et al. (2021). Prognostic Impact of cascade Screening for Familial Hypercholesterolemia on Cardiovascular Events. J. Clin. Lipidol. 15, 358–365. doi:10.1016/j.jacl.2020.12.012
Tada, H., Okada, H., Nomura, A., Usui, S., Sakata, K., Nohara, A., et al. (2021). Clinical Diagnostic Criteria of Familial Hypercholesterolemia ― A Comparison of the Japan Atherosclerosis Society and Dutch Lipid Clinic Network Criteria ―. Circ. J. 85, 891–897. doi:10.1253/circj.cj-20-0901
Tada, H. (2021). Personalized Medicine beyond Low-Density Lipoprotein Cholesterol to Combat Residual Risk for Coronary Artery Disease. J. Atheroscler. Thromb. 28, 1130–1132. doi:10.5551/jat.ed162
Yamamoto, T., Shimojima, K., Ondo, Y., Imai, K., Chong, P. F., Kira, R., et al. (2016). Challenges in Detecting Genomic Copy Number Aberrations Using Next-Generation Sequencing Data and the eXome Hidden Markov Model: a Clinical Exome-First Diagnostic Approach. Hum. Genome 3, 16025. doi:10.1038/hgv.2016.25
Keywords: familial hypercholesterolemia, LDL cholesterol, genetics, LDL receptor (LDLR), protein-truncating variants
Citation: Tada H, Kojima N, Yamagami K, Nomura A, Nohara A, Usui S, Sakata K, Fujino N, Takamura M and Kawashiri M-A (2022) Effects of Different Types of Pathogenic Variants on Phenotypes of Familial Hypercholesterolemia. Front. Genet. 13:872056. doi: 10.3389/fgene.2022.872056
Received: 09 February 2022; Accepted: 18 March 2022;
Published: 11 April 2022.
Edited by:
Alpo Juhani Vuorio, University of Helsinki, FinlandReviewed by:
Estibaliz Jarauta, University of Zaragoza, SpainCopyright © 2022 Tada, Kojima, Yamagami, Nomura, Nohara, Usui, Sakata, Fujino, Takamura and Kawashiri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hayato Tada, aHQyNDB6QHNhMy5zby1uZXQubmUuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.