 Yanyan Jia1
Yanyan Jia1 Li Guo
Li Guo- 1School of Automation Science and Engineering, Faculty of Electronic and Information Engineering, Xi’an Jiaotong University, Xi’an, China
- 2School of Big Data, Weifang Institute of Technology, Weifang, China
- 3Peking University Institute of Advanced Agricultural Sciences, Weifang, China
Introduction
Chinese balloon flower (Platycodon grandiflorus) is the sole species in genus Platycoldon within the Campanulaceae family. The typical blue purple or white flowers of P. grandiflorus are frequently used for ornamental purposes (Lv et al., 2021). As a traditional oriental medicine used to treat chronic inflammatory diseases, P. grandiflorus roots have rich pharmacological activities such as expectorant antitussive, anti-inflammatory, immune regulatory and anti-tumor effects (Choi et al., 2010; Nyakudya et al., 2014; Buchwald et al., 2020; Ke et al., 2020; Lee et al., 2020). The dried form of the Platycodi radix is officially listed as a traditional herbal medicine in the Chinese, Korean and Japanese Pharmacopoeia (Su et al., 2021). Platycodi radix is also being pickled in northeast China, and made into kimchi in the Korean Peninsula. The market demand of P. grandiflorus follows the development and application of medicine, food, health products, cosmetics, ornamental and other fields (Ji et al., 2020), and its market prospects are bright.
Over 100 secondary metabolites have been isolated from P. grandiflorus including triterpenoid saponins, flavonoids, polyphenols, polysaccharide and so on (Zhang et al., 2015; Qiu et al., 2019; Huang et al., 2021). So far, the pharmacological and metabolic pathways of the main active ingredient triterpenoid saponins have been studied (Kim et al., 2020; Kim et al., 2021; Yu et al., 2021). However, the molecular basis of biochemical pathways for P. grandiflorus secondary metabolites is overall poorly understood, hindering the progress of molecular breeding and metabolic engineering of P. grandiflorus towards increased production and utilization of its natural products. A high-quality genome assembly of the P. grandiflorus will significantly accelerate the genetic characterization of secondary metabolic pathways, their regulatory mechanisms and genome-assisted breeding.
Previously, a draft genome sequence of P. grandiflorus (2n = 2x = 18) was assembled using Illumina short reads by Kim et al. yielding a quite fragmented assembly with scaffold N50 of 277 kb (Kim et al. 2020). In this study, we assembled and annotated a chromosome-scale reference genome for P. grandiflorus cultivar XJD. This genome assembly has a total length of 622.86 Mb anchored to nine chromosomes with a high contiguity (contig N50 = 29.34Mb, scaffold N50 = 65.83 Mb), representing a significant improvement over the previously published draft genome of P. grandiflorus (Kim et al., 2020). The chromosome-scale genome assembly will advance our understanding of genome function and evolution of P. grandiflorus, and facilitate its molecular breeding and metabolic engineering.
Results and Discussion
Genome Assembly
To produce a chromosome-level genome assembly of P. grandiflorus cultivar XJD. We generated about 73 Gb Nanopore long reads with an average read length of 24 kb, 112 Gb Illumina paired-end short reads of 150 bp, and 311 Gb high-throughput chromatin conformation capture (Hi-C) sequencing data. The P. grandiflorus genome was estimated to be 642.38 Mb in length with a heterozygosity rate of 0.92% and a repeat content of 60% based on K-mer analysis of Illumina reads (Supplementary Table S1, Supplementary Figure S1). Nanopore long reads were first used to produce the draft assembly by NextDenovo, which was 622.86 Mb with a contig N50 of 29.34 Mb (Supplementary Table S2) after base correction by Pilon using Illumina reads. The quality of the genome assembly was evaluated by mapping Illumina short reads to the assembly with 99.3% of short reads mapped to 96.8% of the assembled genome. Furthermore, we performed BUSCO analysis, showing that the genome assembly captured 98.1% complete BUSCOs, including 95.5% single-copy and 2.6% duplicated (Supplementary Table S3) indicating that the genome assembly had high completeness.
Hi-C data were then used to anchor the assembled contigs into individual chromosomes using ALLHiC (Zhang et al., 2019) and Juicerbox (Robinson et al., 2018), yielding nine pseudomolecules ranging from 47.09 to 104.37 Mb accounting for 95% of the assembly. Hi-C contact map showed that the nine pseudochromosomes could be distinguished clearly (Figure 1; Supplementary Table S4), consistent with the karyotype results (2n = 2x = 18) based on literature reports (Yang et al., 2016). The final genome assembly of P. grandiflorus was 622.86 Mb, with a contig N50 of 28.34 Mb, and a scaffold N50 of 65.83 Mb, the level of this genome assembly is much higher than a previous reported P. grandiflorus (Jangbaek-doraji cultivar) genome assembly (Kim et al., 2020) with a scaffold N50 of only 0.277 Mb (Supplementary Table S2). Whole genome sequence comparison showed that the two genome assemblies aligned well, where 4,815 scaffolds of Jangbaek-doraji assembly can be aligned to 99 scaffolds (95% anchored to nine chromosomes) of our XJD assembly (Supplementary Figure S2).
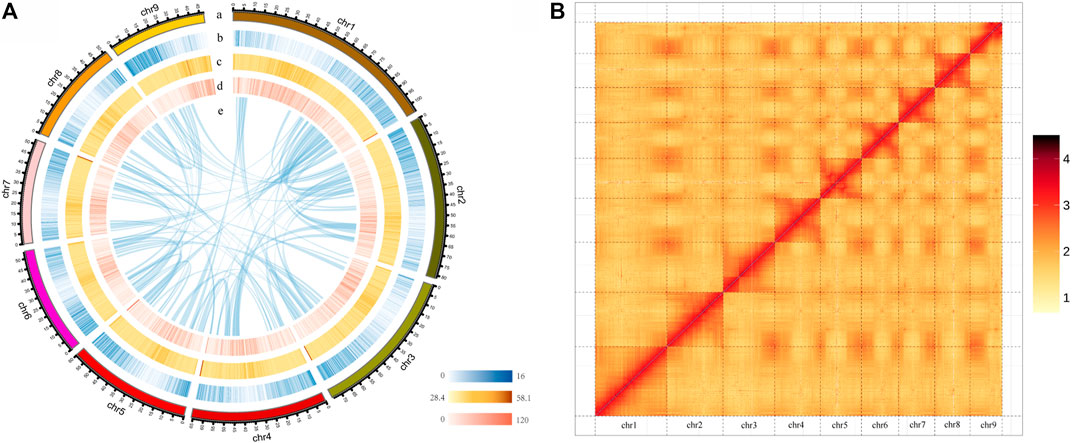
FIGURE 1. Overview of chromosome-level Platycodon grandiflorus genome assembly. (A) P. grandiflorus genomic features. Track a is the circular representation of nine pseudochromosomes. Track b-d represents the distribution of gene density, GC density, and repeat density, respectively, with densities calculated in 100 kb windows. Track e shows syntenic blocks identified within P. grandiflorus genome. (B) Hi-C interaction heatmap for the P. grandiflorus genome.
Genome Annotation
We then performed genome annotations combining ab initio prediction, protein homology and transcriptome data from leaves, roots and stems (Methods). The genome annotation identified 360.46 Mb repeat sequences in the P. grandiflorus genome, accounting for 57.87% of the genome. The top two categories of repetitive elements were long terminal repeats (LTRs: 51.2%) and DNA elements (2.64%). A total of 22,358 protein-coding genes were predicted in the genome, 96.91% of which can be predicted gene function, by aligning against a library of known proteins in related plant species (Supplementary Table S5). Furthermore, non-coding RNAs were predicted across the P. grandiflorus genome, detecting a total of 1,867 microRNAs (miRNAs), 989 transfer RNAs (tRNAs), 780 ribosomal RNAs (rRNAs), and 1,114 small nuclear RNAs (snRNAs).
Comparative Phylogenomics of P. grandiflorus
To determine the evolutionary relationships among P. grandiflorus and other species, we identified 1,436 single-copy orthologs from 10 representative plant species using OrthoMCL (Li et al., 2003) (Figure 2A). The protein sequence alignment of these orthologs were generated by MUSCLE (Edgar, 2004) and were used to generate a phylogenetic tree using Oryza sativa as outgroup (Figure 2B). Mikania micrantha, Helianthus annuus, Lactuca sativa were most closely related to P. grandiflorus with a divergence time around 73.8 million years ago (Mya) (Figure 2B). Gene family evolution analysis using CAFE on the 10 plant speices suggested that P. grandiflorus has 27 and 64 significantly expanded and contracted gene families (Figure 2C). Expansion gene families were enriched in 19 GO categories and 12 KEGG pathways, most of which were related to biosynthesis of secondary metabolites such as brassinosteroid, flavonoid, stilbenoid, and gingerol, and signaling pathway such as MAPK pathway (Supplementary Tables S6 and S7). Notably, P. grandiflorus contained 1,079 species-specific gene families consisting 1,914 genes relative to M. micrantha, H. annuus and L. sativa (Figure 2D). Then the GO enrichment analyses of these specific genes were performed (Supplementary Table S8). Positively selected genes in P. grandiflorus were identified by comparing with H. annuus and M. micrantha, the results of GO and KEGG analysis showed that the positively selected genes were significantly involved in DNA repair, cellular response to stress and stimulus, DNA metabolic process, nucleic acid metabolic process, DNA recombination, and so on (Supplementary Tables S9 and S10).
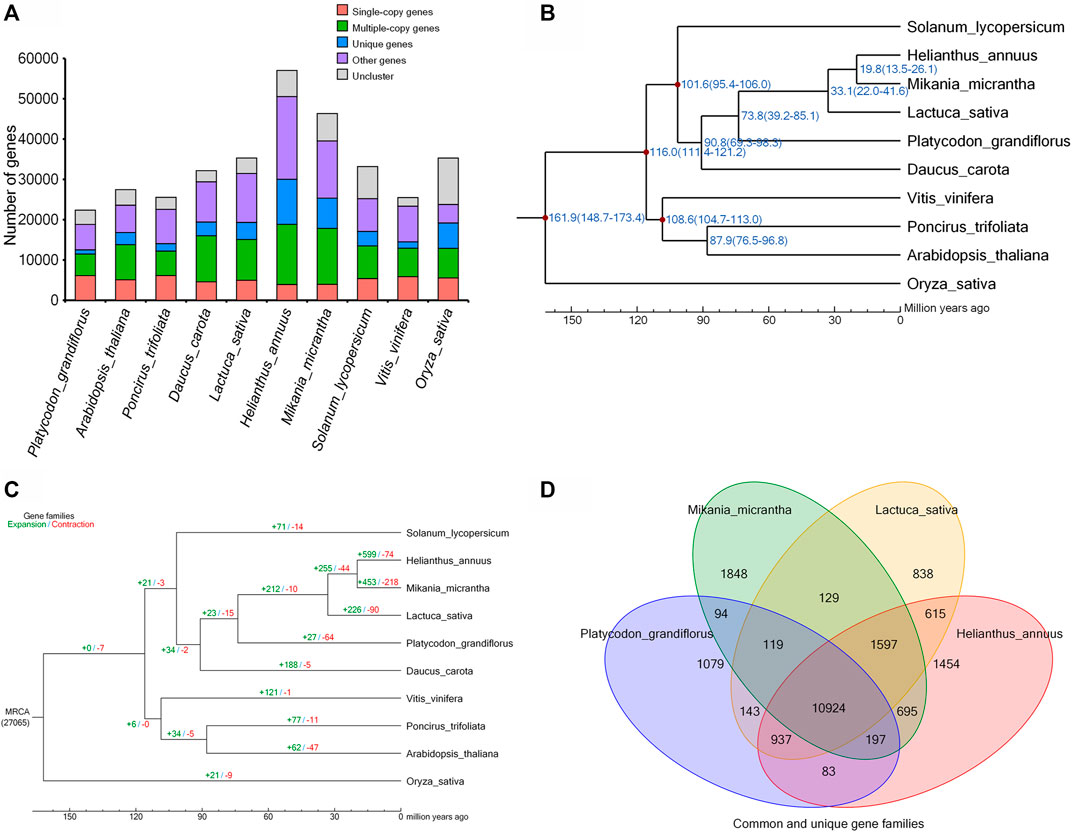
FIGURE 2. Platycodon grandiflorus phylogenomics. (A) The distribution of single-copy, multiple-copy, unique, and other genes in the 10 plant species. (B) Phylogenetic tree of the 10 plant species. The blue numbers denote divergence time of each node (MYA: million years ago). (C) Expansion and contraction in gene families of the 10 plant species. (D) Venn diagram represents the common and unique gene families among four closely related plants.
Materials and Methods
Plant Materials, Library Construction, and Sequencing
Fresh leaf, stem and root samples were collected from four-week-old seedlings of P. grandiflorus cultivar XJD grown in a plant growth chamber with a 16-h light photoperiod. The tissues were flash-frozen in liquid nitrogen and used for total genomic DNA or RNA extraction. Total genomic DNA of P. grandiflorus leaves were extracted using a DNeasy Plant Mini Kit (Qiagen), followed by PCR-free library construction using Illumina TruSeq DNA PCR-Free Library Preparation Kit following the manufacturer’s instructions. The libraries were sequenced on Illumina HiseqX Ten platform to generate 150 bp paired-end reads used to perform genome survey, polish the genome assembly, and evaluate the quality of assemblies.
For ONT and Hi-C sequencing, fresh young leaves were used for DNA isolation and library construction. For ONT sequencing, total genomic DNA was extracted from leaf samples using the CTAB method. ONT libraries were constructed and used for sequencing in the following steps: fragment repair, connecting reactions, quantitative detection, and library construction. Finally, single-molecule real-time sequencing was carried out on the Nanopore PromethION sequencer to obtain the raw data prior to error correction to obtain high fidelity sequence data. The Hi-C sequencing libraries were generated following a standard procedure described previously (Rao et al., 2014) involving crosslink DNA, restriction enzyme digestion, filling ends and biotin labeling, ligation, DNA purification and capture using antibody. The Hi-C libraries were subjected to quality control before being sequenced on Illumina HiseqX Ten platform. For transcriptome sequencing, total RNA was extracted from leaves, stems and roots of P. grandiflorus using the Plant RNA Purification Reagent (Qiagen) according to the manufacturer’s instructions. RNA-seq transcriptome libraries were prepared using the TruSeq RNA sample preparation Kit (Illumina), and sequencing was performed on an Illumina HiseqX Ten platform.
De Novo Genome Assembly
K-mer frequency analysis was performed using Jellyfish V2.0 (Marçais and Kingsford, 2011) to estimate the P. grandiflorus genome size, heterozygosity and repeat content. The NextDenovo (https://github.com/Nextomics/NextDenovo) was used to assemble the P. grandiflorus genome with ONT long reads, and then the Nanopore-assembled genome was polished using the Illumina DNA short reads by NextPolish V1.3.1 (Hu et al., 2020) to improve base accuracy using default parameters. Next, the ALLHiC V0.9.8 (Zhang et al., 2019) was used to reorder and anchor preliminarily assembled contigs into chromosomes based on Hi-C data using default parameters. Finally, we use the Juicerbox V1.1 (Robinson et al., 2018) to adjust the heatmap and assemble it into a chromosome version of the genome. To assess the accuracy and completeness of the assemblies, Illumina clean reads were mapped to our assembly using BWA (Li and Durbin, 2009). In addition, BUSCO (Simão et al., 2015) was used to access the completeness of the genome assembly.
Genome Annotation
Genome annotation mainly includes repetitive sequence annotation, gene annotation and non-coding RNA annotation. Firstly, transcriptome read assemblies were generated with Trinity (Grabherr et al., 2013) for the genome annotation. To optimize the genome annotation, the RNA-Seq reads from different tissues were aligned to draft genome using Hisat2 (Kim et al., 2015) with default parameters to identify exons region and splice positions. The alignment results were then used as input for Stringtie (Pertea et al., 2015) with default parameters for genome-based transcript assembly.
Repeat sequences were annotated based on homology and ab initio. Tandem Repeat was extracted using Tandem Repeats Finder (Benson, 1999) by ab initio prediction. RepeatModeler (Flynn et al., 2020), RepeatScout (Price et al., 2005), and LTR-Finder (Xu and Wang, 2007), were applied to ab initio repeat element library construction with default parameters, and RepeatMasker (Tarailo-Graovac and Chen, 2009) were used to annotate repetitive elements with the database. RepeatMasker and RepeatproteinMask were used to search the genome sequence for known repetitive elements, with the genome sequences used as queries against the repbase database (Jurka, 2000).
For gene structure prediction, Augustus (Stanke et al., 2008), GlimmerHMM (Majoros et al., 2004) and SNAP (Korf, 2004) were used in our de novo prediction study. Blast (Kent, 2002) and Genewise software (Birney et al., 2004) were used for homologous annotation performation. Based on homology prediction and de novo prediction results, combined with the transcriptome-based prediction data, the EvidenceModeler (Haas et al., 2008) was applied to integrate the prediction results for obtaining a non-redundant, more complete gene set. Finally, we used PASA (Haas et al., 2003), combined with the transcriptome assembly results, to correct the EVM annotation results, add UTR and variable shear and other information to get the final gene set. This final gene set was compared to public databases, including SwissProt (Bairoch and Apweiler, 2000), NR (Marchler-Bauer et al., 2011), Pfam (Griffiths-Jones et al., 2005), KEGG (Kanehisa et al., 2013), GO (Ashburner et al., 2000) and InterPro (Zdobnov and Apweiler, 2001) for function annotation of protein-coding genes. In addition, we also predicted different non-coding RNAs. The tRNAs were predicted using the program tRNAscan-SE (Chan and Lowe, 2019). For rRNAs are highly conserved, we predict rRNA sequences using BLAST. Other ncRNAs were identified by searching against the Rfam database with default parameters using the infernal software (Griffiths-Jones et al., 2005).
Phylogenomic Analysis
Synteny analysis was conducted using MCScanX (Wang et al., 2012) applied to BLASTp results of P. grandiflorus protein sequences. For the phylogeny analysis, OrthoMCL (Li et al., 2003) was firstly used for detecting multi-copy gene families and single-copy gene families between P. grandiflorus and other representative species, and then all the single-copy gene families were performed for multiple sequence alignment using MUSCLE (Edgar, 2004), all the comparison results were combined together to form a super alignment matrix, RAxML (Stamatakis, 2014) was used to construct phylogenetic tree species. the Oryza sativa as an outgroup, and the bootstrap value was set to 100. The MCMCTREE of PAML (Yang, 1997) was implemented to estimate the differentiation time. Time correction points are: Solanum lycopersicum - Helianthus annuus (95–106 Mya), Vitis Vinifera - Arabidopsis thaliana (105–115 Mya), P. grandiflorus - Vitis vinifera (111–131 Mya), P. grandiflorus–Oryza sativa (148–173 Mya). The time correction points are taken from the TimeTree website (Sudhir et al., 2017).
Gene Family Analysis
The CAFE software (Han et al., 2013) was used to analyze gene family expansion and contraction, based on the results of divergence times and phylogenetic relationships. In order to avoid false positive results, CAFE results were filtered, and the screening conditions for significant enrichment results were family-wide p-value < 0.05 and Viterbi p-value < 0.05. The enrichment analyses based on GO and KEGG annotations were performed to identify functional implications of the expanded and contracted genes.
Positive Selection Analysis
The protein sequences of single-copy gene families were extracted and aligned by MUSCLE (Edgar, 2004). The Codeml program of PAML software was applied for positive selection analysis using the branch-site model with H. annuus and M. micrantha as the background branch. The likelihood ratio test was used to detect candidates that underwent positive selection with a cutoff p value of 0.05. Fisher’s test and FDR correction (q-value < 0.05) were used for functional enrichment analysis of these positively selected genes.
Data Availability Statement
The whole genome sequence data reported in this paper have been deposited in the Genome Warehouse in National Genomics Data Center (BioProject: PRJCA003843), Beijing Institute of Genomics, Chinese Academy of Sciences/China National Center for Bioinformation (GWH: GWHARYT00000000.1) publicly accessible at https://ngdc.cncb.ac.cn/gwh. The raw sequencing data for the ONT long reads, Illumina short reads, Hi-C Illumina and RNA-seq reads have been deposited in the Genome Sequence Archive at the National Genomics Data Center (GSA: CRA003503) publicly accessible at http://bigd.big.ac.cn/gsa. The genome annotation has been deposited in https://doi.org/10.6084/m9.figshare.19093331.v1.
Author Contributions
Project design and oversight: LG; Sample collection and curation: YJ; Conducting experiment and data analysis: YJ, SC, LZ, MX, ZS; Figure and table preparation: YJ, WC, SC; Result interpretation and discussion: YJ, PZ, LG; Manuscript writing and revision: YJ, WC, PZ, LG; Funding acquisition: LG, YJ. All authors read and approve the final version of this manuscript.
Funding
This project is supported by the National Natural Science Foundation of China (Grant No. 31970317), Chinese Postdoctoral Research Foundation (Grant No. 2020M683514). LG is also supported by a faculty startup package from Peking University Institute of Advanced Agricultural Sciences.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors also would like to thank Dr. Bo Wang at Xi’an Jiaotong University for technical assistance, and anonymous reviewers for their comments and suggestions to improve this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.869784/full#supplementary-material
References
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 25 (1), 25–29. doi:10.1038/75556
Bairoch, A., and Apweiler, R. (2000). The SWISS-PROT Protein Sequence Database and its Supplement TrEMBL in 2000. Nucleic Acids Res. 28 (1), 45–48. doi:10.1093/nar/28.1.45
Benson, G. (1999). Tandem Repeats Finder: a Program to Analyze DNA Sequences. Nucleic Acids Res. 27 (2), 573–580. doi:10.1093/nar/27.2.573
Birney, E., Clamp, M., and Durbin, R. (2004). GeneWise and Genomewise. Genome Res. 14 (5), 988–995. doi:10.1101/gr.1865504
Buchwald, W., Szulc, M., Baraniak, J., Derebecka, N., Kania-Dobrowolska, M., Piasecka, A., et al. (2020). The Effect of Different Water Extracts from Platycodon Grandiflorum on Selected Factors Associated with Pathogenesis of Chronic Bronchitis in Rats. Molecules 25 (21), 5020. doi:10.3390/molecules25215020
Chan, P. P., and Lowe, T. M. (2019). tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. Methods Mol. Biol., 1962. 1–14. doi:10.1007/978-1-4939-9173-0_1
Choi, Y. H., Yoo, D. S., Cha, M.-R., Choi, C. W., Kim, Y. S., Choi, S.-U., et al. (2010). Antiproliferative Effects of Saponins from the Roots of Platycodon Grandiflorum on Cultured Human Tumor Cells. J. Nat. Prod. 73 (11), 1863–1867. doi:10.1021/np100496p
Edgar, R. C. (2004). MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 32 (5), 1792–1797. doi:10.1093/nar/gkh340
Flynn, J. M., Hubley, R., Goubert, C., Rosen, J., Clark, A. G., Feschotte, C., et al. (2020). RepeatModeler2 for Automated Genomic Discovery of Transposable Element Families. Proc. Natl. Acad. Sci. USA 117 (17), 9451–9457. doi:10.1073/pnas.1921046117
Grabherr, M., Haas, B., Yassour, M., Levin, J., Thompson, D., Amit, I., et al. (2013). Trinity: Reconstructing a Full-Length Transcriptome without a Genome from RNA-Seq Data. Nat. Biotechnol. 29 (7), 644–652.
Griffiths-Jones, S., Moxon, S., Marshall, M., Khanna, A., Eddy, S. R., and Bateman, A. (2005). Rfam: Annotating Non-coding RNAs in Complete Genomes. Nucleic Acids Res. 33, D121–D124. doi:10.1093/nar/gki081
Haas, B. J., Delcher, A., Mount, S., Wortman, J., Smith, R., Hannick, L., et al. (2003). Improving the Arabidopsis Genome Annotation Using Maximal Transcript Alignment Assemblies. Nucleic Acids Res. 31 (19), 5654–5666. doi:10.1093/nar/gkg770
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated Eukaryotic Gene Structure Annotation Using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9 (1), R7. doi:10.1186/gb-2008-9-1-r7
Han, M. V., Thomas, G. W. C., Lugo-Martinez, J., and Hahn, M. W. (2013). Estimating Gene Gain and Loss Rates in the Presence of Error in Genome Assembly and Annotation Using CAFE 3. Mol. Biol. Evol. 30 (8), 1987–1997. doi:10.1093/molbev/mst100
Hu, J., Fan, J., Sun, Z., and Liu, S. (2020). NextPolish: a Fast and Efficient Genome Polishing Tool for Long-Read Assembly. Bioinformatics 36 (7), 2253–2255. doi:10.1093/bioinformatics/btz891
Huang, W., Zhou, H., Yuan, M., Lan, L., Hou, A., and Ji, S. (2021). Comprehensive Characterization of the Chemical Constituents in Platycodon Grandiflorum by an Integrated Liquid Chromatography-Mass Spectrometry Strategy. J. Chromatogr. A 1654, 462477. doi:10.1016/j.chroma.2021.462477
Ji, M.-Y., Bo, A., Yang, M., Xu, J.-F., Jiang, L.-L., Zhou, B.-C., et al. (2020). The Pharmacological Effects and Health Benefits of Platycodon grandiflorus-A Medicine Food Homology Species. Foods 9 (2), 142. doi:10.3390/foods9020142
Jurka, J. (2000). Repbase Update: a Database and an Electronic Journal of Repetitive Elements. Trends Genet. 16 (9), 418–420. doi:10.1016/s0168-9525(00)02093-x
Kanehisa, M., Goto, S., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2013). Data, Information, Knowledge and Principle: Back to Metabolism in KEGG. Nucl. Acids Res. 42, D199–D205. doi:10.1093/nar/gkt1076
Ke, W., Bonilla-Rosso, G., Engel, P., Wang, P., Chen, F., and Hu, X. (2020). Suppression of High-Fat Diet-Induced Obesity by Platycodon grandiflorus in Mice Is Linked to Changes in the Gut Microbiota. J. Nutr. 150 (9), 2364–2374. doi:10.1093/jn/nxaa159
Kent, W. J. (2002). Blat-the BLAST-like Alignment Tool. Genome Res. 12 (4), 656–664. doi:10.1101/gr.229202
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 12 (4), 357–360. doi:10.1038/nmeth.3317
Kim, J., Kang, S.-H., Park, S.-G., Yang, T.-J., Lee, Y., Kim, O. T., et al. (2020). Whole-genome, Transcriptome, and Methylome Analyses Provide Insights into the Evolution of Platycoside Biosynthesis in Platycodon grandiflorus, a Medicinal Plant. Hortic. Res. 7, 112. doi:10.1038/s41438-020-0329-x
Kim, Y.-K., Sathasivam, R., Kim, Y. B., Kim, J. K., and Park, S. U. (2021). Transcriptomic Analysis, Cloning, Characterization, and Expression Analysis of Triterpene Biosynthetic Genes and Triterpene Accumulation in the Hairy Roots of Platycodon Grandiflorum Exposed to Methyl Jasmonate. ACS Omega 6 (19), 12820–12830. doi:10.1021/acsomega.1c01202
Korf, I. (2004). Gene Finding in Novel Genomes. BMC Bioinformatics 5, 59. doi:10.1186/1471-2105-5-59
Lee, S., Han, E. H., Lim, M.-K., Lee, S.-H., Yu, H. J., Lim, Y. H., et al. (2020). Fermented Platycodon Grandiflorum Extracts Relieve Airway Inflammation and Cough Reflex Sensitivity In Vivo. J. Med. Food 23 (10), 1060–1069. doi:10.1089/jmf.2019.4595
Li, H., and Durbin, R. (2009). Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 13 (9), 2178–2189. doi:10.1101/gr.1224503
Lv, Y., Tong, X., Zhang, P., Yu, N., Gui, S., Han, R., et al. (2021). Comparative Transcriptomic Analysis on white and Blue Flowers of Platycodon Grandiflorus to Elucidate Genes Involved in the Biosynthesis of Anthocyanins. Iran J. Biotechnol. 19 (3), e2811.
Majoros, W. H., Pertea, M., and Salzberg, S. L. (2004). TigrScan and GlimmerHMM: Two Open Source Ab Initio Eukaryotic Gene-Finders. Bioinformatics 20 (16), 2878–2879. doi:10.1093/bioinformatics/bth315
Marchler-Bauer, A., Lu, S., Anderson, J. B., Chitsaz, F., Derbyshire, M. K., DeWeese-Scott, C., et al. (2011). CDD: a Conserved Domain Database for the Functional Annotation of Proteins. Nucleic Acids Res. 39, D225–D229. doi:10.1093/nar/gkq1189
Marçais, G., and Kingsford, C. (2011). A Fast, Lock-free Approach for Efficient Parallel Counting of Occurrences of K-Mers. Bioinformatics 27 (6), 764–770. doi:10.1093/bioinformatics/btr011
Nyakudya, E., Jeong, J. H., Lee, N. K., and Jeong, Y.-S. (2014). Platycosides from the Roots of Platycodon Grandiflorum and Their Health Benefits. Jfn 19 (2), 59–68. doi:10.3746/pnf.2014.19.2.059
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T.-C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie Enables Improved Reconstruction of a Transcriptome from RNA-Seq Reads. Nat. Biotechnol. 33 (3), 290–295. doi:10.1038/nbt.3122
Price, A. L., Jones, N. C., and Pevzner, P. A. (2005). De Novo identification of Repeat Families in Large Genomes. Bioinformatics 21, i351–i358. doi:10.1093/bioinformatics/bti1018
Qiu, L., Xiao, Y., Liu, Y.-Q., Peng, L.-x., Liao, W., and Fu, Q. (2019). Platycosides P and Q, Two New Triterpene Saponins from Platycodon Grandiflorum. J. Asian Nat. Prod. Res. 21 (5), 419–425. doi:10.1080/10286020.2018.1488835
Rao, S. S. P., Huntley, M. H., Durand, N. C., Stamenova, E. K., Bochkov, I. D., Robinson, J. T., et al. (2014). A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 159 (7), 1665–1680. doi:10.1016/j.cell.2014.11.021
Robinson, J. T., Turner, D., Durand, N. C., Thorvaldsdóttir, H., Mesirov, J. P., and Aiden, E. L. (2018). Juicebox.js Provides a Cloud-Based Visualization System for Hi-C Data. Cel Syst. 6 (2), 256–258. doi:10.1016/j.cels.2018.01.001
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 31 (19), 3210–3212. doi:10.1093/bioinformatics/btv351
Stamatakis, A. (2014). RAxML Version 8: a Tool for Phylogenetic Analysis and post-analysis of Large Phylogenies. Bioinformatics 30 (9), 1312–1313. doi:10.1093/bioinformatics/btu033
Stanke, M., Diekhans, M., Baertsch, R., and Haussler, D. (2008). Using Native and Syntenically Mapped cDNA Alignments to Improve De Novo Gene Finding. Bioinformatics 24 (5), 637–644. doi:10.1093/bioinformatics/btn013
Su, X., Liu, Y., Han, L., Wang, Z., Cao, M., Wu, L., et al. (2021). A Candidate Gene Identified in Converting Platycoside E to Platycodin D from Platycodon grandiflorus by Transcriptome and Main Metabolites Analysis. Sci. Rep. 11 (1), 9810. doi:10.1038/s41598-021-89294-1
Sudhir, K., Glen, S., Michael, S., and Blair, H. (2017). TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 34 (7), 1812–1819.
Tarailo-Graovac, M., and Chen, N. (2009). Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinformatics. Chapter 4: Unit 4.10. doi:10.1002/0471250953.bi0410s25
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a Toolkit for Detection and Evolutionary Analysis of Gene Synteny and Collinearity. Nucleic Acids Res. 40 (7), e49. doi:10.1093/nar/gkr1293
Xu, Z., and Wang, H. (2007). LTR_FINDER: an Efficient Tool for the Prediction of Full-Length LTR Retrotransposons. Nucleic Acids Res. 35, W265–W268. doi:10.1093/nar/gkm286
Yang, F., Bao, G., Zhou, H., and Zhang, Y. (2016). Observation of Chromosome Number and Cytology Observation on Meiosis of Platycodon Grandiflorum. Gansu Agric. Sci. Technology 10, 14–16.
Yang, Z. (1997). PAML: a Program Package for Phylogenetic Analysis by Maximum Likelihood. Bioinformatics 13 (5), 555–556. doi:10.1093/bioinformatics/13.5.555
Yu, H., Liu, M., Yin, M., Shan, T., Peng, H., Wang, J., et al. (2021). Transcriptome Analysis Identifies Putative Genes Involved in Triterpenoid Biosynthesis in Platycodon grandiflorus. Planta 254 (2), 34. doi:10.1007/s00425-021-03677-2
Zdobnov, E. M., and Apweiler, R. (2001). InterProScan - an Integration Platform for the Signature-Recognition Methods in InterPro. Bioinformatics 17 (9), 847–848. doi:10.1093/bioinformatics/17.9.847
Zhang, L., Wang, Y., Yang, D., Zhang, C., Zhang, N., Li, M., et al. (2015). Platycodon grandiflorus - an Ethnopharmacological, Phytochemical and Pharmacological Review. J. Ethnopharmacology 164, 147–161. doi:10.1016/j.jep.2015.01.052
Keywords: platycodon grandiflorus, genome assembly, Oxford nanopore, phylogenomics, Hi-C
Citation: Jia Y, Chen S, Chen W, Zhang P, Su Z, Zhang L, Xu M and Guo L (2022) A Chromosome-Level Reference Genome of Chinese Balloon Flower (Platycodon grandiflorus). Front. Genet. 13:869784. doi: 10.3389/fgene.2022.869784
Received: 05 February 2022; Accepted: 03 March 2022;
Published: 08 April 2022.
Edited by:
Sunil Kumar Sahu, Beijing Genomics Institute (BGI), ChinaReviewed by:
Lei Zhang, Jiangsu Normal University, ChinaXiaojun Nie, Northwest A&F University, China
Copyright © 2022 Jia, Chen, Chen, Zhang, Su, Zhang, Xu and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Guo, bGkuZ3VvQHBrdS1pYWFzLmVkdS5jbg==