
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 16 May 2022
Sec. Human and Medical Genomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.864228
This article is part of the Research Topic The genetics of inherited retinal diseases in understudied ethnic groups: novel associations, challenges, and perspectives View all 6 articles
 Lama Jaffal1,2†Hanane Akhdar1,2†Hawraa Joumaa1Mariam Ibrahim1Zahraa Chhouri1Alexandre Assi3Charles Helou3
Lama Jaffal1,2†Hanane Akhdar1,2†Hawraa Joumaa1Mariam Ibrahim1Zahraa Chhouri1Alexandre Assi3Charles Helou3 Hane Lee4Go Hun Seo4Wissam H. Joumaa1
Hane Lee4Go Hun Seo4Wissam H. Joumaa1 Said El Shamieh1,5*
Said El Shamieh1,5*The purpose of this study was to expand the mutation spectrum by searching the causative mutations in nine Lebanese families with Usher syndrome (USH) using whole-exome sequencing. The pathogenicity of candidate mutations was first evaluated according to their frequency, conservation, and in silico prediction tools. Then, it was confirmed via Sanger sequencing, followed by segregation analysis. Finally, a meta-analysis was conducted to calculate the prevalence of USH genes in the Lebanese population. Three missense mutations, two splice site mutations, and one insertion/deletion were detected in eight of the families. Four of these variants were novel: c.5535C > A; p.(Asn1845Lys) in exon 41 of CDH23, c.7130G > A; p.(Arg2377Gln) in exon 32 of ADGRV1, c.11390-1G > A in USH2A, and c.3999–6A > G in PCDH15. All the identified mutations were shown to be likely disease-causing through our bioinformatics analysis and co-segregated with the USH phenotype. The mutations were classified according to the ACMG standards. Finally, our meta-analysis showed that the mutations in ADGRV1, USH2A, and CLRN1 are the most prevalent and responsible for approximately 75% of USH cases in Lebanon. Of note, the frequency USH type 3 showed a relatively high incidence (23%) compared to the worldwide prevalence, which is around 2–4%. In conclusion, our study has broadened the mutational spectrum of USH and showed a high heterogeneity of this disease in the Lebanese population.
Usher syndrome (USH) is a genetic disorder combining various degrees of sensorineural hearing impairment with a gradual vision loss caused by retinitis pigmentosa (RP) (Boughman et al., 1983; Fuster-García et al., 2018; Castiglione and Möller, 2022). Hearing impairment is mainly due to changes in morphogenesis and stability of stereocilia projections on the cochlear hair cells that mediate sound transduction (Frolenkov et al., 2004; Millán et al., 2011). In addition, patients may suffer from balance defects due to disturbances in hair cells of the vestibular apparatus (Frolenkov et al., 2004; Millán et al., 2011). The visual loss driven by RP in USH patients starts with problems in night vision and reduced peripheral vision (Hartong et al., 2006). As the disease progresses, the central vision also begins to deteriorate, eventually leading to dysfunction and blindness (Edwards et al., 1998). The prevalence of USH ranges between 1 and 4 per 25,000 individuals among different populations, making it number 1 cause of deaf-blindness cases worldwide (Mathur and Yang, 2015) and in Lebanon (Saouda et al., 1998; Verpy et al., 2000; Audo et al., 2012; Reddy et al., 2014).
The clinical manifestation of most USH cases is divided into three distinct subtypes: type 1 (USH1), type 2 (USH2), and type 3 (USH3) (Petit, 2001; Mathur and Yang, 2015). The main differences between these clinical subtypes reside in the severity and progression of hearing impairment, the onset age of RP, and the implication of vestibular dysfunction or not (Petit, 2001; Mathur and Yang, 2015). USH3 is the rarest among these three subtypes as it accounts for only 2–4% of affected individuals (Yan and Liu, 2010). Moreover, some USH cases do not match the same clinical profile of any of these subtypes, and thus, they cannot be classified into any of them and are considered atypical USH forms (Liu et al., 1998; Mathur and Yang, 2015).
Today, mutations in sixteen genes have been associated with USH, all inherited in an autosomal recessive manner (RetNet: https://sph.uth.edu/retnet/). Noteworthy, some cases of digenic inheritance of USH were reported in the literature; for instance, PDZD7 variants combined with ADGRV1 (NG_007083) variants (Ebermann et al., 2010) and CDH23 (NG_008835) variants combined with PCDH15 (NG_009191) variants were also reported (Zheng et al., 2005). Herein, we aimed to expand the mutation spectrum by searching the causative genetic mutations in nine Lebanese USH families, using targeted and whole-exome sequencing (WES).
The Institutional Review Board of Beirut Arab University approved the study (2017H-0030-HS-R-0208). Written informed consent was obtained from all patients, and all procedures adhered to the tenets of the Declaration of Helsinki. As previously described, the affected individuals underwent clinical ophthalmic examination at Beirut Eye and ENT Specialist Hospital (Beirut, Lebanon) (Jaffal et al., 2019).
Genomic DNA was extracted from whole blood samples of the affected individuals and their family members using the QIAamp DNA Mini extraction kit (Qiagen, Hilden, Germany). Extracted DNA samples were quantified using a dsDNA HS Assay kit on a Qubit3.0 fluorometer (Thermo Fisher Scientific, ShahAlam, Malaysia). The DNA samples of all the index patients were investigated by whole-exome sequencing (WES) as previously described (Jaffal et al., 2019), except the sample of the index in family 13, which was analyzed using targeted NGS, as described elsewhere (Audo et al., 2012; Kikuchi et al., 2015).
To identify the top 10 candidate mutations in every index patient, we have searched the public genome databases, including Genome Aggregation Database (gnomAD) (Karczewski et al., 2019), Trans-Omics for Precision Medicine (TOPMed) program (Millán et al., 2011), Ensembl GRCh37 genome browser (Zerbino et al., 2018), and Single Nucleotide Polymorphism Database (dbSNP build 152) (https://www.ncbi.nlm.nih.gov/snp/). These databases were used to filter out common variants with a minor allele frequency (MAF) higher than 0.01. Among the remaining mutations, splice site, non-sense mutations, missense, and insertions/deletions (indels) were prioritized. For the pathogenicity assessment, the level of evolutionary conservation of the affected residues across different species was checked using the genome browser of the University of California at Santa Cruz (UCSC) (Supplementary Figures S1–S3) (Kent et al., 2002). The RetNet database (https://sph.uth.edu/retnet/) was used to check the association of the mutated genes with USH and confirm the clinical diagnosis. PolyPhen-2 (Adzhubei et al., 2010), scale-invariant feature transform (SIFT) (Kumar et al., 2009), and MutationTaster (Schwarz et al., 2014) computational prediction programs were used to predict the possible clinical significance of the detected mutations. Human Gene Mutation Database (Stenson et al., 2014), Leiden Open Variation Database (Fokkema et al., 2005), PubMed (https://www.ncbi.nlm.nih.gov/pubmed/), and Online Mendelian Inheritance in Man (https://omim.org/) were used to check if the identified mutations were novel or previously reported. Approximately half of the samples showed one homozygous candidate mutation after the filtering approach (Supplementary Table S1).
The candidate mutations detected by NGS were validated through conventional polymerase chain reaction (PCR) using a T100 thermal cycler (Biorad, Kaki Bukit, Singapore), followed by Sanger sequencing on a 3730xl DNA Sequencer (Applied Biosystems, Courtaboeuf, Les Ulis, France). For co-segregation analysis in available family members, DNA fragments harboring the mutations were amplified and then Sanger sequenced. The used primers are available upon request.
The available medical history and clinical findings of the patients are summarized in Table 1. Stringent filtration and analysis of NGS data revealed the presence of homozygous variants that are likely associated with USH in eight of the families (Table 2), while we were not able to find any likely causative variant in one family.
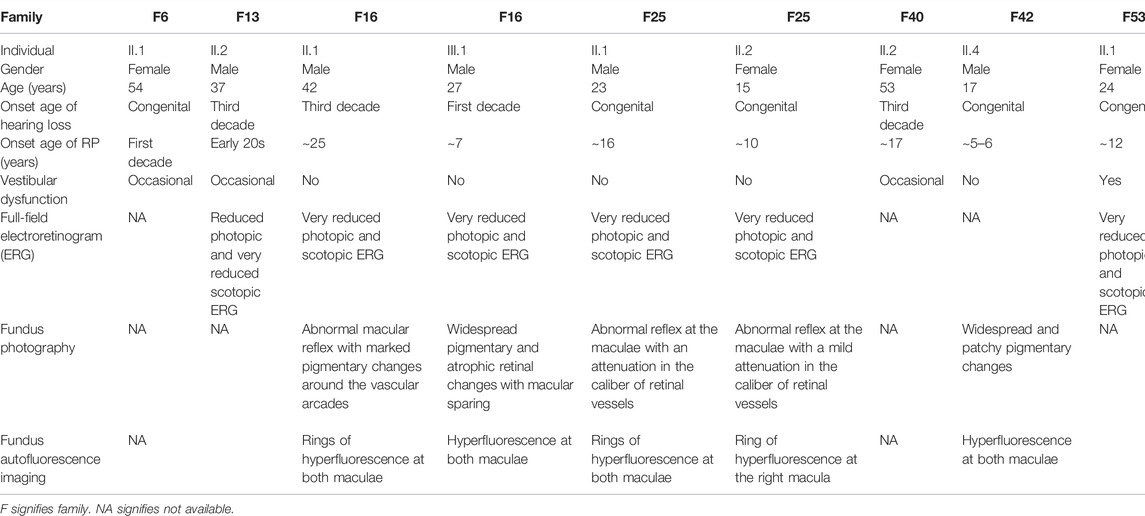
TABLE 1. Summary of medical history and clinical findings of Usher patients belonging to eight Lebanese families.
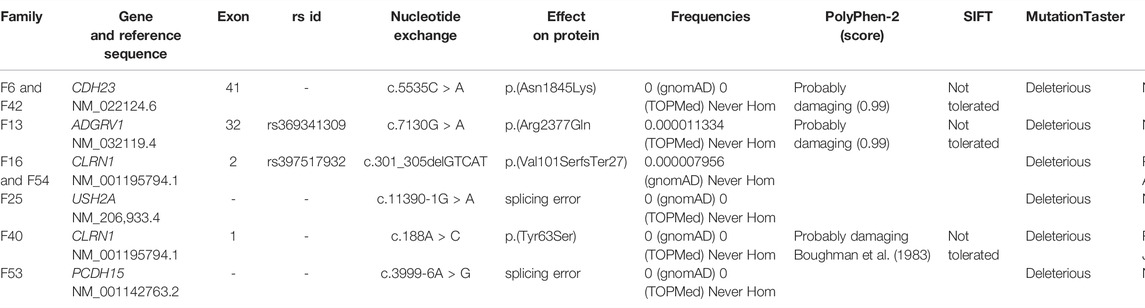
TABLE 2. Details of mutations identified in eight Lebanese USH families.
In family 6, proband II.1 (a 54-year-old woman) suffered from a congenital profound hearing loss, with no intelligible speech, in addition to occasional dizziness and balance problems. She started experiencing RP symptoms during her first decade (Table 1). NGS showed that she carried the novel homozygous missense variant: c.5535C > A; p.(Asn1845Lys) in exon 41 of CDH23 (Figure 1). According to UCSC, the Asn residue affected by this variant is highly conserved across species. The variant was absent in gnomAD and TOPMed populations (Table 2). It was also predicted to be probably damaging, not tolerated, and deleterious according to PolyPhen-2, SIFT, and MutationTaster, respectively (Table 2). Sanger sequencing showed that the affected sibling (II.2) also carried the variant in a homozygous state. At the same time, the unaffected mother (I.2) was a heterozygous carrier, indicating that the variant co-segregated well with the phenotype (Figure 1). According to the standards developed by the American College of Medical Genetics and Genomics (ACMG), this variant is classified as likely pathogenic (Supplementary Table S1).
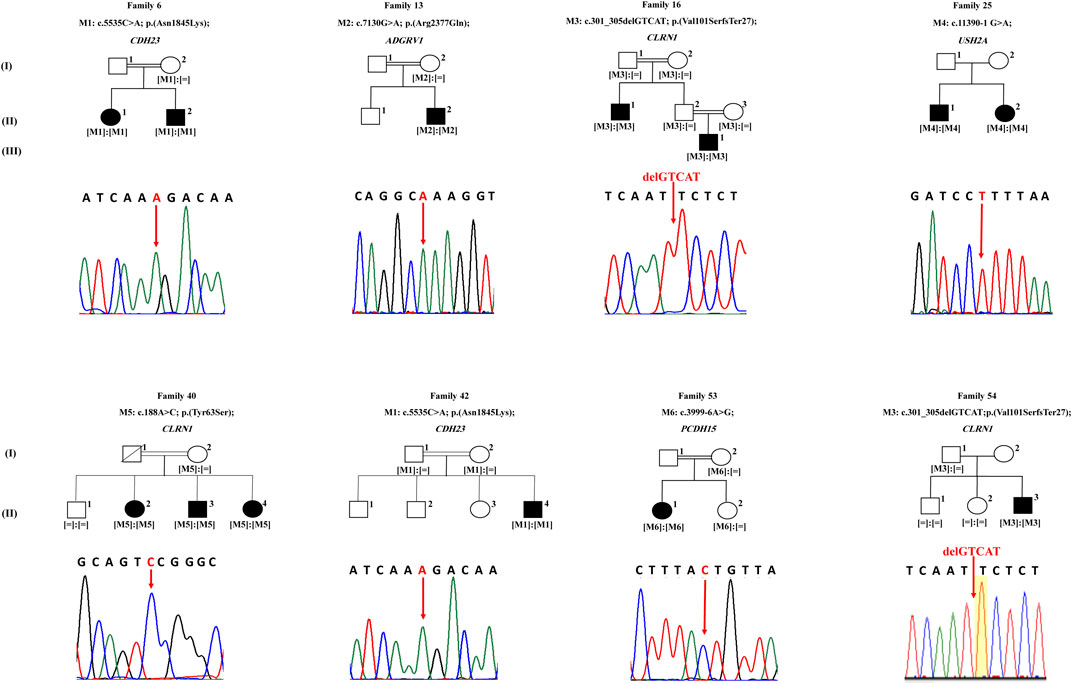
FIGURE 1. Pedigrees of eight families with Usher syndrome. Sequencing results of each proband are shown under the corresponding pedigree, noting that sequences of probands of families 25 and 53 are in reverse direction. White symbols indicate unaffected members. Black symbols indicate affected members. Square and round symbols represent male and female individuals, respectively. The slash indicates deceased individuals. Double horizontal lines represent consanguineous unions. [M] defines mutated alleles. [ = ] defines wild type alleles.
The patient of family 13 (a 37 year old male) was initially diagnosed with RP around the age of 25, but later on, he started experiencing a mild hearing impairment within his third decade (Table 1). NGS revealed that he harbored a novel homozygous missense variant: c.7130G > A; p.(Arg2377Gln); rs369341309; in exon 32 of ADGRV1 (Figure 1). According to the UCSC genome browser, the Arg residue affected by this variant is highly conserved. The variant was not detected in the gnomAD population, while it was shown to be very rare and never homozygous in the TOPMed population (0.000011334) (Table 2). Moreover, it was predicted to be probably damaging, not tolerated, and deleterious according to PolyPhen-2, SIFT, and MutationTaster, respectively (Table 2). The DNA sample of the unaffected mother (I.2) was screened and showed the candidate variant to be heterozygous in her, indicating that it co-segregated well with the phenotype (Figure 1). According to the standards of the ACMG, this variant is classified as having uncertain significance (Supplementary Table S1).
Family 16 comprised two affected members: proband II.1 (a 42-year-old man) and his nephew III.1 (a 27-year-old man) who started experiencing hearing impairment within their third and first decades, respectively. Both of them did not report balance problems. Patient II.1 has fully intelligible speech. Patient III.1 also has intelligible speech, although he presented speech difficulties that improved with speech therapy at a younger age. RP onset ages were around 27 years for patient II.2 and around 7 years for his nephew III.1 (Table 1). WES was carried in index patient II.1, who was found to have a homozygous frameshift deletion in exon 2 of CLRN1 (NG_009168): c.301_305delGTCAT; p.(Val101SerfsTer27); rs397517932; resulting in the introduction of a premature termination codon, 27 amino acids downstream (Figure 1). Sanger sequencing showed that patient III.1 also carried this variant in the homozygous state. The variant was shown to be rare heterozygous in gnomAD (0.000007956), while it was not detected in TOPMed populations (Table 2). It co-segregated well with the phenotype as parents of both patients (I.1, I.2, II.2, and II.3) were shown to be heterozygous carriers (Figure 1). Interestingly, this 5-bp deletion was previously reported in another Lebanese USH family in 2011 (Akoury et al., 2011). According to the standards of the ACMG, this variant is classified as pathogenic (Supplementary Table S1).
Patients of family 25 (a 23-year-old man and a 15-year-old woman) suffered from a congenital hearing loss, with no balance problems. They started experiencing RP symptoms around 16 and 10, respectively (Table 1). WES was carried in the proband II.1 and showed her to harbor the homozygous acceptor splice site variant: c.11390-1G > A in USH2A (NG_009497) (Figure 1). Sanger sequencing revealed that his affected sister (II.2) also carried the variant in a homozygous state (Figure 1). The variant was not found in gnomAD nor TOPMed populations (Table 2). According to MutationTaster, it was predicted to be deleterious (Table 2). No DNA samples of additional family members were available for co-segregation analysis. According to the standards of the ACMG, this variant is classified as pathogenic (Supplementary Table S1).
Proband II.2 of family 40 (a 53-year-old woman) started experiencing RP symptoms around 17 years, while hearing impairment started in her third decade. She also reported experiencing occasional balance problems (Table 1). WES showed her to carry a missense homozygous variant c.188A > C: p.(Tyr63Ser) in exon 1 of CLRN1 (Figure 1). This variant affects a highly conserved amino acid according to UCSC. It is absent in gnomAD and TOPMed populations and predicted to be probably damaging, not tolerated, and deleterious according to PolyPhen-2, SIFT, and MutationTaster, respectively (Table 2). Sanger sequencing showed that her affected siblings (II.3 and II.4) also carried the variant in a homozygous state, while the unaffected mother (I.2) was a heterozygous carrier. At the same time, the unaffected sibling (II.1) did not carry it, indicating that it co-segregated well with the phenotype (Figure 1). Of note that we previously detected this same CLRN1 variant in another Lebanese USH patient in 2019 (Jaffal et al., 2019). According to the standards of the ACMG, this variant is classified as likely pathogenic (Supplementary Table S1).
Patient II.1 of family 42 (a 17-year-old boy) suffered from a congenital profound hearing loss, with no intelligible speech at all. No balance problems were reported. He started experiencing RP symptoms around the age of 5. NGS showed him to carry the same CDH23 homozygous variant that is detected in patients of family 6: the c.5535C > A; p(Asn1845Lys) in exon 41 (Figure 1; Table 2). It co-segregated well with the phenotype as both parents (I.1 and I.2) were shown to be heterozygous carriers (Figure 1). According to the standards of the ACMG, this variant is classified as likely pathogenic (Supplementary Table S1).
The patient of family 53 (a 24-year-old woman) suffered from a congenital profound hearing loss, with no intelligible speech, in addition to frequent dizziness and disequilibrium. She started experiencing RP symptoms around the age of 12. WES showed her to carry the homozygous splice region variant c.3999--6A > G in PCDH15 (Figure 1). The variant was not found in gnomAD nor TOPMed populations (Table 2). According to MutationTaster, it was predicted to be deleterious (Table 2). We have tested this variation by splice AI, and it showed a probability of 62 and 99% to affect the −6 and −1 positions of the pre-mRNA. It co-segregated well with the phenotype as the unaffected mother (I.2) and sister (II.2) were shown to be heterozygous carriers (Figure 1). According to the standards of the ACMG, this variant is classified as having uncertain significance (Supplementary Table S1).
The patient of family 54 (a 12-year-old boy) started experiencing both hearing impairment and RP symptoms in his first decade. He also shares occasional balance problems (Table 1). WES showed him to carry the same CLRN1 homozygous variant that was detected in patients of family 16: c.301_305delGTCAT; p.(Val101SerfsTer27) (Figure 1; Table 2). Sanger sequencing showed that the unaffected mother (I.2) was a heterozygous carrier, while both unaffected siblings (II.1 and II.2) were wild type, indicating that the variant co-segregated well with the phenotype (Figure 1). According to the standards of the ACMG, this variant is classified as pathogenic (Supplementary Table S1).
In our nine solved families, each of ADGRV1 and PCDH15 were responsible for USH in one patient, USH2A was responsible for USH in two patients, CDH23 was responsible for USH in three patients, and CLRN1 was responsible for USH in seven patients. In addition, ten other Lebanese families were genetically screened in previous studies (Saouda et al., 1998; Verpy et al., 2000; Akoury et al., 2011; Reddy et al., 2014). In those ten families, MYO7A was responsible for USH in one patient, each of CDH23 and CLRN1 were responsible for USH in two patients, USH1C was found mutated in three patients, USH2A was found mutated in seven patients, and ADGRV1 was found mutated in ten patients. Overall, seven USH genes have been responsible for USH in the 39 Lebanese affected individuals published so far (Figure 2). Mutations in ADGRV1 (28%), USH2A (23%), and CLRN1 (23%) were the most prevalent. CDH23 followed with 13%, while PCDH15, USH1C, and MYO7A were the least prevalent, with a frequency ranging from ∼8 to 3% (Figure 2).
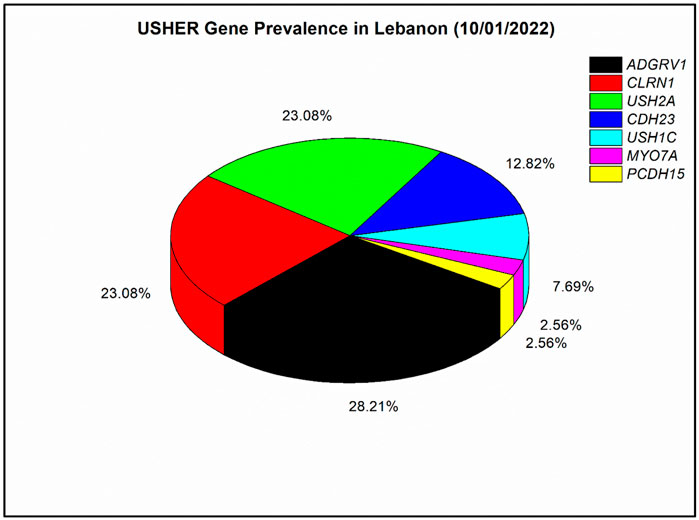
FIGURE 2. Prevalence of mutated Usher genes detected in the Lebanese population to date (till 10/01/2022). A meta-analysis with four previously published articles was done (Verpy et al., 2000; Akoury et al., 2011; Reddy et al., 2014; Jaffal et al., 2019); in total, 39 affected individuals with Usher syndrome were included. The prevalence analysis is based on the number of affected individuals and not families as in Reddy et al. (2014).
This study used mainly WES to determine the likely causative mutations in nine Lebanese families with a presumed diagnosis with USH. Extensive bioinformatics analysis of NGS data revealed the presence of six homozygous mutations in eight families, while the case of one family remained unsolved. Of note, consanguineous marriages were present in six out of the eight solved families (Figure 1). Of the detected variants, four were novel (Table 2). Those variants were shown to be heterozygous or absent in the available unaffected family members, indicating that they co-segregate adequately with the USH phenotype (Figure 1).
In two of the families (Hartong et al., 2006; Jouret et al., 2019), the patients were shown to carry the same homozygous variant in CDH23 that encodes for cadherin 23, a protein needed to establish and/or maintain the proper growth and excellent cohesion of the hair bundle in the vestibular organ and the cochlea (El-Amraoui and Petit, 2005; Pan and Zhang, 2012). CDH23 is known to cause USH1 characterized by a congenital severe-to-profound hearing loss, vestibular dysfunction, and early onset of RP within the first decade, making it the most severe subtype (Petit, 2001; Mathur and Yang, 2015). The clinical profiles of the patients in these two families are in favor of USH1, since they suffer from a severe congenital hearing impairment with no intelligible speech at all (Table 1). The patients of family 6 also have balance problems. However, the patient of family 42 did not experience balance problems to date, keeping in mind that this patient is still young (17 years) and might develop vestibular dysfunction at an older age.
Patient II.2 of family 13 was initially diagnosed with RP in his early twenties. Still, genetic testing showed that he harbored a homozygous variant in ADGRV1, a gene related to USH and previously known as GPR98. Consequently, we re-investigated his clinical history and queried if he suffered from additional symptoms other than the visual impairment. Indeed, this revealed that he was experiencing a mild, stable hearing impairment and occasional dizziness that developed within his third decade, which caused him to use hearing aids. Thus, this led to a revised re-diagnosis with USH. This observation highlights the importance of genetic testing in getting an accurate clinical diagnosis, especially in syndromic inherited retinal diseases that can be misdiagnosed because the systemic symptoms may only appear late in life, or because ophthalmologists may only focus on the visual impairment without considering other symptoms and relating them to the case.
ADGRV1 encodes the adhesion of G-protein-coupled receptor V1 that is needed for the normal development and maintenance of auditory hair bundles of the cochlear sensory hair cells (McGee et al., 2006). It is usually known to cause USH2 characterized by congenital moderate-to-severe hearing loss, absence of vestibular dysfunction, and RP manifestation from the second decade onward (Petit, 2001; Weston et al., 2004). However, the clinical profile of this patient was compatible with neither USH2 features nor any of the other classical subtypes, since he reported a late mild hearing impairment that only appeared in adulthood and had an intelligible speech. This confirms that the hearing impairment occurrence was post-lingual and not congenital, unlike the cases of USH1 and USH2. The patient also stated that his hearing impairment is stable and not progressing over the years, in contrast to the hearing impairment in USH3 that is characterized by its progressiveness. Accordingly, this case may be considered an atypical USH form.
The affected members in families 16, 40, and 54 were shown to carry homozygous CLRN1 mutations. The clarin-1 protein encoded by this gene is involved in the morphogenesis of the hair bundle and the synaptic transmission between light-sensitive photoreceptor cells and their associated neurons, in addition to being potentially engaged in the maintenance and development of stereocilia (Zallocchi et al., 2009; Dulon et al., 2018). CLRN1 variants are usually associated with USH3, which is characterized by post-lingual hearing impairment with a progressive nature, variable vestibular dysfunction, and variable onset of RP (Audo et al., 2012; Jaffal et al., 2019). Patients of families 16 and 40 had intelligible speech, confirming the post-lingual hearing loss. They also reported that their hearing impairment is progressive and worsens over the years, which is in accordance with USH3. On the contrary, the patient of family 54 had a prelingual hearing impairment diagnosed as early as the age of 1, which is not in accordance with USH3.
We previously reported another Lebanese USH3 family having the same missense mutation in CLRN1; p(Tyr63Ser) (Jaffal et al., 2019). In addition, a Lebanese family comprising two USH patients, with one of them presenting symptoms of USH3, was reported by Akoury et al. (2011). Interestingly, the latter two USH patients carried the same CLRN1 5-bp deletion that is identified in families 16 and 54 in this study. This suggests that these two mutations might have a founder effect in the Lebanese population. Of note, most of CLRN1-affected individuals (from the current and previous studies) come from the south region of Lebanon.
Patients of family 25 were shown to carry a homozygous acceptor splice site variant in USH2A encoding for Usherin, which is needed for retinal photoreceptor retention and normal cochlear hair cell growth (Liu et al., 2007). Disruption of this same splice site has been observed in a Caucasian USH family (but c.11390-1G > C instead of c.11390-1G > A) (Le Quesne Stabej et al., 2012). Acceptor and donor splice site variants usually result in a loss of protein function (Baralle and Baralle, 2005). USH2A mutations are the major cause of USH2 cases (Toualbi et al., 2020). USH2 description is in accordance with the clinical profile in the patients of this family who suffer from a congenital hearing impairment and RP manifestation from the second decade, with no vestibular dysfunction (Table 1).
The patient of family 53 carries the homozygous splice region variant c.3999-6A > G in PCDH15. PCDH15 is expressed in the neurosensory epithelium of the eye and ear to maintain the function of stereocilia and retinal photoreceptor cells (Ahmed et al., 2003). PCDH15 mutations are known to cause USH1 (Ahmed et al., 2003), which is in accordance with the clinical profile showing a severe congenital prelingual hearing impairment and vestibular dysfunction (Table 1).
To date, we were able to genetically screen 10 Lebanese USH families: one in a previous study (Jaffal et al., 2019) and nine in the current study; nine of those families were genetically solved, while one family remained unsolved. Therefore, until today, our success rate with USH families is 90%. This rate is similar to the diagnosis rates of USH in larger populations (77–93%) (Bonnet et al., 2016; Sun et al., 2018), which were also achieved using WES, proving this technique to be highly efficient in USH molecular diagnosis. Nevertheless, the use of whole-genome sequencing (WGS) might be efficient in improving those rates by identifying non-coding variants that WES and other standard sequencing techniques usually miss. For instance, a deep-intronic variant that escaped detection by panel-NGS, genome-wide linkage analysis, and WES was identified in CLRN1 (c.254–649T > G) using WGS (Bonnet et al., 2016). However, the high cost, the challenging filtration of extensive data, and the difficulty of proving the pathogenicity of non-coding variants are all factors that hinder WGS from being used as a routine diagnostic approach (Bonnet et al., 2016).
Our prevalence analysis shows heterogeneity in the genetics of USH among different populations. For instance, ADGRV1, which ranked first in Lebanese patients (Figure 2), is not usually among the most prevalent genes in other populations. A meta-analysis from 2019 showed it accounts for 5% of USH cases (Jouret et al., 2019); similarly, a recent study of a large Italian cohort also showed it accounts for 5% (Colombo et al., 2021). On the other hand, USH2A mutations usually show the highest prevalence in most other populations, such as previously reported Chinese, French, Italian, and Spanish cohorts (Fuster-García et al., 2018; Sun et al., 2018; Colombo et al., 2021; Mansard et al., 2021). Moreover, the relatively high prevalence of CLRN1 variants (23%) suggests that USH3 may have a high prevalence in the Lebanese population, while this subtype is the rarest among the three USH subtypes as it accounts for only 2–4% of all cases in most populations (Audo et al., 2012). Exceptionally, the frequency of USH3 is significant in the Finnish population (40%) (Ness et al., 2003), the Ashkenazi Jews (40%) (Pakarinen et al., 1995), the population of Birmingham, United Kingdom (20%) (Edwards et al., 1998), and now in the Lebanese population (23%), specifically in the south region, from where our CLRN1 patients originated. The prevalence of CDH23 was around 13% in this analysis (Hope et al., 1997), while it ranged between 2 and 8% in other countries (Fuster-García et al., 2018; Jouret et al., 2019; Colombo et al., 2021; Wafa et al., 2021). Regarding PCDH15 and USH1C, there was no significant difference with other cohorts. In contrast, MYO7A, which is among the least frequent genes in this analysis (around 3%), is detected in much higher frequencies in most other populations and considered as a major USH gene (Fuster-García et al., 2018; Sun et al., 2018; Colombo et al., 2021; Mansard et al., 2021).
In conclusion, the results of this study have broadened the mutational spectrum of USH and showed a high heterogeneity of this disease in the Lebanese population. The screening of more Lebanese USH families would be essential to elucidate the genetic characteristics of USH and the accurate prevalence of its three subtypes and causative genes in this population.
The original contributions presented in the study are publicly available. These data can be found at: SCV002097249—SCV002097254.
The studies involving human participants were reviewed and approved by the Institutional Review Board of Beirut Arab University approved the study (2017H-0030-HS-R-0208). The patients/participants provided their written informed consent to participate in this study.
SES framed the methodology. LJ and HJ validated. AA, CH, and SES conducted the formal analysis. LJ, SES, HA, and MI investigated. HA, WJ, and SES collected resources. LJ and HA wrote the original draft preparation. SES wrote the review and editing. HA and SES administered the project. HA, SES, and WJ funded the acquisition.
The present work was funded with support from the Lebanese University (HA, SES, and WJ), Beirut, Lebanon.
The authors HL and GHS are employed by 3billion Inc. (Seoul, South Korea). 3billion® (Seoul, Korea) provided the whole-exome sequencing free of charge of the affected individuals from families F6, F25, F40, F53, and F54.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank the participants and their families for their time and effort in the present study, and 3billion® (Seoul, Korea) for providing the whole-exome sequencing free of charge of the affected individuals from families F6, F25, F40, F53, and F54.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.864228/full#supplementary-material
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 7 (4), 248–249. doi:10.1038/nmeth0410-248
Ahmed, Z. M., Riazuddin, S., Ahmad, J., Bernstein, S. L., Guo, Y., Sabar, M. F., et al. (2003). PCDH15 Is Expressed in the Neurosensory Epithelium of the Eye and Ear and Mutant Alleles Are Responsible for Both USH1F and DFNB23. Hum. Mol. Genet. 12 (24), 3215–3223. doi:10.1093/hmg/ddg358
Akoury, E., El Zir, E., Mansour, A., Mégarbané, A., Majewski, J., and Slim, R. (2011). A Novel 5-bp Deletion in Clarin 1 in a Family with Usher Syndrome. Ophthalmic Genet. 32 (4), 245–249. doi:10.3109/13816810.2011.587083
Audo, I., Bujakowska, K. M., Léveillard, T., Mohand-Saïd, S., Lancelot, M.-E., Germain, A., et al. (2012). Development and Application of a Next-Generation-Sequencing (NGS) Approach to Detect Known and Novel Gene Defects Underlying Retinal Diseases. Orphanet J. rare Dis. 7, 8. doi:10.1186/1750-1172-7-8
Baralle, D., and Baralle, M. (2005). Splicing in Action: Assessing Disease Causing Sequence Changes. J. Med. Genet. 42 (10), 737–748. doi:10.1136/jmg.2004.029538
Bonnet, C., Riahi, Z., Chantot-Bastaraud, S., Smagghe, L., Letexier, M., Marcaillou, C., et al. (2016). An Innovative Strategy for the Molecular Diagnosis of Usher Syndrome Identifies Causal Biallelic Mutations in 93% of European Patients. Eur. J. Hum. Genet. 24 (12), 1730–1738. doi:10.1038/ejhg.2016.99
Boughman, J. A., Vernon, M., and Shaver, K. A. (1983). Usher Syndrome: Definition and Estimate of Prevalence from Two High-Risk Populations. J. chronic Dis. 36 (8), 595–603. doi:10.1016/0021-9681(83)90147-9
Castiglione, A., and Möller, C. (2022). Usher Syndrome. Audiol. Res. 12 (1), 42–65. doi:10.3390/audiolres12010005
Colombo, L., Maltese, P. E., Castori, M., El Shamieh, S., Zeitz, C., Audo, I., et al. (2021). Molecular Epidemiology in 591 Italian Probands with Nonsyndromic Retinitis Pigmentosa and Usher Syndrome. Invest. Ophthalmol. Vis. Sci. 62 (2), 13. doi:10.1167/iovs.62.2.13
Dulon, D., Papal, S., Patni, P., Cortese, M., Vincent, P. F. Y., Tertrais, M., et al. (2018). Clarin-1 Gene Transfer Rescues Auditory Synaptopathy in Model of Usher Syndrome. J. Clin. Invest. 128 (8), 3382–3401. doi:10.1172/jci94351
Ebermann, I., Phillips, J. B., Liebau, M. C., Koenekoop, R. K., Schermer, B., Lopez, I., et al. (2010). PDZD7 Is a Modifier of Retinal Disease and a Contributor to Digenic Usher Syndrome. J. Clin. Invest. 120 (6), 1812–1823. doi:10.1172/jci39715
Edwards, A., Fishman, G. A., Anderson, R. J., Grover, S., and Derlacki, D. J. (1998). Visual Acuity and Visual Field Impairment in Usher Syndrome. Arch. Ophthalmol. 116 (2), 165–168. doi:10.1001/archopht.116.2.165
El-Amraoui, A., and Petit, C. (2005). Usher I Syndrome: Unravelling the Mechanisms that Underlie the Cohesion of the Growing Hair Bundle in Inner Ear Sensory Cells. J. Cel Sci 118 (Pt 20), 4593–4603. doi:10.1242/jcs.02636
Fokkema, I. F. A. C., den Dunnen, J. T., and Taschner, P. E. M. (2005). LOVD: Easy Creation of a Locus-specific Sequence Variation Database Using an "LSDB-In-A-Box" Approach. Hum. Mutat. 26 (2), 63–68. doi:10.1002/humu.20201
Frolenkov, G. I., Belyantseva, I. A., Friedman, T. B., and Griffith, A. J. (2004). Genetic Insights into the Morphogenesis of Inner Ear Hair Cells. Nat. Rev. Genet. 5 (7), 489–498. doi:10.1038/nrg1377
Fuster-García, C., García-García, G., Jaijo, T., Fornés, N., Ayuso, C., Fernández-Burriel, M., et al. (2018). High-throughput Sequencing for the Molecular Diagnosis of Usher Syndrome Reveals 42 Novel Mutations and Consolidates CEP250 as Usher-like Disease Causative. Sci. Rep. 8 (1), 17113. doi:10.1038/s41598-018-35085-0
Hartong, D. T., Berson, E. L., and Dryja, T. P. (2006). Retinitis Pigmentosa. The Lancet 368 (9549), 1795–1809. doi:10.1016/s0140-6736(06)69740-7
Hope, C. I., Bundey, S., Proops, D., and Fielder, A. R. (1997). Usher Syndrome in the City of Birmingham---prevalence and Clinical Classification. Br. J. Ophthalmol. 81 (1), 46–53. doi:10.1136/bjo.81.1.46
Jaffal, L., Joumaa, W. H., Assi, A., Helou, C., Cherfan, G., Zibara, K., et al. (2019). Next Generation Sequencing Identifies Five Novel Mutations in Lebanese Patients with Bardet-Biedl and Usher Syndromes. Genes (Basel) 10 (12), 1047. doi:10.3390/genes10121047
Jouret, G., Poirsier, C., Spodenkiewicz, M., Jaquin, C., Gouy, E., Arndt, C., et al. (2019). Genetics of Usher Syndrome: New Insights from a Meta-Analysis. Otol Neurotol 40 (1), 121–129. doi:10.1097/mao.0000000000002054
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The Mutational Constraint Spectrum Quantified From Variation in 141,456 Humans. Nature 581, 434–443. doi:10.1038/s41586-021-03758-y
Kent, W. J., Sugnet, C. W., Furey, T. S., Roskin, K. M., Pringle, T. H., Zahler, A. M., et al. (2002). The Human Genome Browser at UCSC. Genome Res. 12 (6), 996–1006. doi:10.1101/gr.229102
Kikuchi, S., Kameya, S., Gocho, K., El Shamieh, S., Akeo, K., Sugawara, Y., et al. (2015). Cone Dystrophy in Patient with Homozygous RP1L1 Mutation. Biomed. Res. Int. 2015, 545243. doi:10.1155/2015/545243
Kumar, P., Henikoff, S., and Ng, P. C. (2009). Predicting the Effects of Coding Non-synonymous Variants on Protein Function Using the SIFT Algorithm. Nat. Protoc. 4 (7), 1073–1081. doi:10.1038/nprot.2009.86
Le Quesne Stabej, P., Saihan, Z., Rangesh, N., Steele-Stallard, H. B., Ambrose, J., Coffey, A., et al. (2012). Comprehensive Sequence Analysis of Nine Usher Syndrome Genes in the UK National Collaborative Usher Study. J. Med. Genet. 49 (1), 27–36. doi:10.1136/jmedgenet-2011-100468
Liu, X.-Z., Hope, C., Walsh, J., Newton, V., Ke, X. M., Liang, C. Y., et al. (1998). Mutations in the Myosin VIIA Gene Cause a Wide Phenotypic Spectrum, Including Atypical Usher Syndrome. Am. J. Hum. Genet. 63 (3), 909–912. doi:10.1086/302026
Liu, X., Bulgakov, O. V., Darrow, K. N., Pawlyk, B., Adamian, M., Liberman, M. C., et al. (2007). Usherin Is Required for Maintenance of Retinal Photoreceptors and normal Development of Cochlear Hair Cells. Proc. Natl. Acad. Sci. U.S.A. 104 (11), 4413–4418. doi:10.1073/pnas.0610950104
Mansard, L., Baux, D., Vaché, C., Blanchet, C., Meunier, I., Willems, M., et al. (2021). The Study of a 231 French Patient Cohort Significantly Extends the Mutational Spectrum of the Two Major Usher Genes MYO7A and USH2A. Int. J. Mol. Sci. 22 (24), 13294. doi:10.3390/ijms222413294
Mathur, P., and Yang, J. (2015). Usher Syndrome: Hearing Loss, Retinal Degeneration and Associated Abnormalities. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis. 1852 (3), 406–420. doi:10.1016/j.bbadis.2014.11.020
McGee, J., Goodyear, R. J., McMillan, D. R., Stauffer, E. A., Holt, J. R., Locke, K. G., et al. (2006). The Very Large G-Protein-Coupled Receptor VLGR1: a Component of the Ankle Link Complex Required for the normal Development of Auditory Hair Bundles. J. Neurosci. 26 (24), 6543–6553. doi:10.1523/jneurosci.0693-06.2006
Millán, J. M., Aller, E., Jaijo, T., Blanco-Kelly, F., Gimenez-Pardo, A., and Ayuso, C. (2011). An Update on the Genetics of Usher Syndrome. J. Ophthalmol. 2011, 417217. doi:10.1155/2011/417217
Ness, S. L., Ben-Yosef, T., Bar-Lev, A., Madeo, A. C., Brewer, C. C., Avraham, K. B., et al. (2003). Genetic Homogeneity and Phenotypic Variability Among Ashkenazi Jews with Usher Syndrome Type III. J. Med. Genet. 40 (10), 767–772. doi:10.1136/jmg.40.10.767
Pakarinen, L., Tuppurainen, K., Laippala, P., Mantyjarvi, M., and Puhakka, H. (1995). The Ophthalmological Course of Usher Syndrome Type III. Int. Ophthalmol. 19 (5), 307–311. doi:10.1007/bf00130927
Pan, L., and Zhang, M. (2012). Structures of Usher Syndrome 1 Proteins and Their Complexes. Physiology 27 (1), 25–42. doi:10.1152/physiol.00037.2011
Petit, C. (2001). Usher Syndrome: from Genetics to Pathogenesis. Annu. Rev. Genom. Hum. Genet. 2, 271–297. doi:10.1146/annurev.genom.2.1.271
Reddy, R., Fahiminiya, S., El Zir, E., Mansour, A., Megarbane, A., Majewski, J., et al. (2014). Molecular Genetics of the Usher Syndrome in Lebanon: Identification of 11 Novel Protein Truncating Mutations by Whole Exome Sequencing. PloS one 9 (9), e107326. doi:10.1371/journal.pone.0107326
Saouda, M., Mansour, A., Bou Moglabey, Y., El Zir, E., Mustapha, M., Chaib, H., et al. (1998). The Usher Syndrome in the Lebanese Population and Further Refinement of the USH2A Candidate Region. Hum. Genet. 103 (2), 193–198. doi:10.1007/s004390050806
Schwarz, J. M., Cooper, D. N., Schuelke, M., and Seelow, D. (2014). MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 11 (4), 361–362. doi:10.1038/nmeth.2890
Stenson, P. D., Mort, M., Ball, E. V., Shaw, K., Phillips, A. D., and Cooper, D. N. (2014). The Human Gene Mutation Database: Building a Comprehensive Mutation Repository for Clinical and Molecular Genetics, Diagnostic Testing and Personalized Genomic Medicine. Hum. Genet. 133 (1), 1–9. doi:10.1007/s00439-013-1358-4
Sun, T., Xu, K., Ren, Y., Xie, Y., Zhang, X., Tian, L., et al. (2018). Comprehensive Molecular Screening in Chinese Usher Syndrome Patients. Invest. Ophthalmol. Vis. Sci. 59 (3), 1229–1237. doi:10.1167/iovs.17-23312
Toualbi, L., Toms, M., and Moosajee, M. (2020). USH2A-retinopathy: From Genetics to Therapeutics. Exp. Eye Res. 201, 108330. doi:10.1016/j.exer.2020.108330
Verpy, E., Leibovici, M., Zwaenepoel, I., Liu, X.-Z., Gal, A., Salem, N., et al. (2000). A Defect in Harmonin, a PDZ Domain-Containing Protein Expressed in the Inner Ear Sensory Hair Cells, Underlies Usher Syndrome Type 1C. Nat. Genet. 26 (1), 51–55. doi:10.1038/79171
Wafa, T. T., Faridi, R., King, K. A., Zalewski, C., Yousaf, R., Schultz, J. M., et al. (2021). Vestibular Phenotype‐genotype Correlation in a Cohort of 90 Patients with Usher Syndrome. Clin. Genet. 99 (2), 226–235. doi:10.1111/cge.13868
Weston, M. D., Luijendijk, M. W. J., Humphrey, K. D., Möller, C., and Kimberling, W. J. (2004). Mutations in the VLGR1 Gene Implicate G-Protein Signaling in the Pathogenesis of Usher Syndrome Type II. Am. J. Hum. Genet. 74 (2), 357–366. doi:10.1086/381685
Yan, D., and Liu, X. Z. (2010). Genetics and Pathological Mechanisms of Usher Syndrome. J. Hum. Genet. 55 (6), 327–335. doi:10.1038/jhg.2010.29
Zallocchi, M., Meehan, D. T., Delimont, D., Askew, C., Garige, S., Gratton, M. A., et al. (2009). Localization and Expression of Clarin-1, the Clrn1 Gene Product, in Auditory Hair Cells and Photoreceptors. Hear. Res. 255 (1-2), 109–120. doi:10.1016/j.heares.2009.06.006
Zerbino, D. R., Achuthan, P., Akanni, W., Amode, M. R., Barrell, D., Bhai, J., et al. (2018). Ensembl 2018. Nucleic Acids Res. 46 (D1), D754–D761. doi:10.1093/nar/gkx1098
Keywords: Usher syndrome, mutation, genetics, developing countries, rare disease, next-generation sequencing
Citation: Jaffal L, Akhdar H, Joumaa H, Ibrahim M, Chhouri Z, Assi A, Helou C, Lee H, Seo GH, Joumaa WH and El Shamieh S (2022) Novel Missense and Splice Site Mutations in USH2A, CDH23, PCDH15, and ADGRV1 Are Associated With Usher Syndrome in Lebanon. Front. Genet. 13:864228. doi: 10.3389/fgene.2022.864228
Received: 28 January 2022; Accepted: 28 March 2022;
Published: 16 May 2022.
Edited by:
Tamar Ben-Yosef, Technion Israel Institute of Technology, IsraelReviewed by:
Junjiang Fu, Southwest Medical University, ChinaCopyright © 2022 Jaffal, Akhdar, Joumaa, Ibrahim, Chhouri, Assi, Helou, Lee, Seo, Joumaa and El Shamieh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Said El Shamieh, c2FpZC5zaGFtaWVoQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.