 Xuehui Yu1,2†
Xuehui Yu1,2† Lamei Yuan1,2,3,4†
Lamei Yuan1,2,3,4† Sheng Deng5
Sheng Deng5 Hong Xia6
Hong Xia6 Xiaolong Tu6
Xiaolong Tu6 Xiong Deng2
Xiong Deng2 Xiangjun Huang7
Xiangjun Huang7 Xiao Cao2
Xiao Cao2 Hao Deng1,2,3,4*
Hao Deng1,2,3,4*- 1Health Management Center, The Third Xiangya Hospital, Central South University, Changsha, China
- 2Center for Experimental Medicine, The Third Xiangya Hospital, Central South University, Changsha, China
- 3Disease Genome Research Center, Central South University, Changsha, China
- 4Department of Neurology, The Third Xiangya Hospital, Central South University, Changsha, China
- 5Department of Pharmacy, Xiangya Hospital, Central South University, Changsha, China
- 6Department of Emergency, The Third Xiangya Hospital, Central South University, Changsha, China
- 7Department of General Surgery, The First Affiliated Hospital of Hunan University of Chinese Medicine, Changsha, China
The formation of left–right asymmetry of the visceral organs is a conserved feature of the human body, and the asymmetry specification of structure and function is precisely orchestrated by multiple regulatory mechanisms. The abnormal results of organ positioning situs arise from defective cilia structure or function during embryogenesis in humans. In this study, we recruited two unrelated Han-Chinese families with left–right asymmetry disorders. The combination of whole-exome sequencing and Sanger sequencing identified two compound heterozygous variants: c.4109C>T and c.9776C>T, and c.612C>G and c.8764C>T in the dynein axonemal heavy chain 17 gene (DNAH17) in two probands with left–right asymmetry disorders. We report for the first time a possible association between DNAH17 gene variants and left–right asymmetry disorders, which is known as a causal gene for asthenozoospermia. Altogether, the findings of our study may enlarge the DNAH17 gene variant spectrum in human left–right asymmetry disorders, pave a way to illustrate the potential pathogenesis of ciliary/flagellar disorders, and provide supplementary explanation for genetic counseling.
Introduction
The visceral organs of vertebrates have a strikingly conserved left–right (LR) asymmetry of the organ situs that is manifested in the chest (heart and lungs) and abdomen (stomach, spleen, liver, intestine, and colon) (McGrath et al., 2003; Blum et al., 2014). The normal organ asymmetry present across the LR axis of the body is called situs solitus (SS) (Sung et al., 2016). It is well recognized that the leftward flow of extracellular fluid at the node (i.e., nodal flow) plays a major role in normal LR axis determination during embryogenesis (McGrath et al., 2003; Pennekamp et al., 2015). Human LR asymmetry disorders have an estimated probability of more than 1 in 8000 live births and can be divided into two broad classes: situs inversus totalis (SIT) and situs ambiguous (SA). SIT is a malformation featuring a complete mirror image reversal of the organs and is usually not related to major influence on the patient’s health (Levin, 2004; Sung et al., 2016). In contrast, SA, also termed heterotaxy, is defined as any abnormal organ display that was not SS or SIT and is highly associated with human congenital heart disease (CHD) (Zhu et al., 2006; Best et al., 2019; Chen et al., 2020).
Abnormalities in the typical development of laterality usually occur as a result of genetic lesions, which form a number of human heritable disorders with significant clinical implications, including primary ciliary dyskinesia (PCD), nephronophthisis, Carpenter syndrome 2, and male infertility (Olbrich et al., 2002; Otto et al., 2003; Levin, 2004; Twigg et al., 2012; Ding et al., 2020). Variations in the genes related to the development and function of nodal cilia often lead to human laterality defects. Up to now, more than 82 genes have been reported to be related to LR asymmetry disorders, including cilia- and flagella-associated protein family members, coiled-coil domain-containing family members, dynein axonemal assembly factors, dynein axonemal light chains, dynein axonemal intermediate chains, and dynein axonemal heavy chains (DNAHs) (Osório et al., 2019; Al Mutairi et al., 2020; Bustamante-Marin et al., 2020a; Bustamante-Marin et al., 2020b; Cannarella et al., 2020; Chen et al., 2020; Cho et al., 2020; Ding et al., 2020; Heigwer et al., 2020; Sahabian et al., 2020; Sha et al., 2020a; Thomas et al., 2020; Wang et al., 2020; Yang and Qi, 2020; Abdelhamed et al., 2021; Derrick et al., 2021; Guo et al., 2021; Wang et al., 2021). Genes belonging to the DNAH family, such as DNAH1 (OMIM 603332), DNAH5 (OMIM 603335), DNAH6 (OMIM 603336), DNAH9 (OMIM 603330), and DNAH11 (OMIM 603339), are reported to be closely associated with cilia and/or flagella beating (Fliegauf et al., 2005; Hornef et al., 2006; Pifferi et al., 2010; Li et al., 2016). Variations in the dynein axonemal heavy chain 17 gene (DNAH17, OMIM 610063), encoding a component of outer dynein arms (ODAs) in the ciliary axonemes, have been reported to be associated with only flagella destabilization and asthenozoospermia (Whitfield et al., 2019; Zhang et al., 2020). There are, however, comparatively fewer studies that have investigated DNAH17 and multiple morphological abnormalities of the flagella and asthenozoospermia, perhaps limited by the number of LR asymmetry phenotype-associated patients; further research is needed.
In the present study, whole-exome sequencing (WES) combined with Sanger sequencing was used to identify the potential causal gene and variants in two Han-Chinese families with LR asymmetry disorders, and compound heterozygous variants (c.4109C>T and c.9776C>T; c.612C>G and c.8764C>T) in the DNAH17 gene were discovered.
Materials and Methods
Participants and Clinical Data
A 31-year-old healthy male and two unrelated Han-Chinese families were enrolled from the Third Xiangya Hospital. Central South University, and the First Affiliated Hospital of Hunan University of Chinese Medicine, Changsha, China. Available medical histories and examinations of the two probands were obtained. The entire study was approved by the Institutional Review Board of the Third Xiangya Hospital, Central South University, Changsha, China, and conducted following the tenets of the Declaration of Helsinki. Written informed consents were collected from all the participants or legal guardians.
DNA Extraction and WES
The standard phenol–chloroform extraction method was used to isolate genomic DNA (gDNA) from peripheral blood leucocytes (Yuan et al., 2015). The Qubit dsDNA HS Assay kit (Invitrogen, Thermo Fisher Scientific, Inc.) was used to quantify the gDNA samples. WES for the probands of the two pedigrees was performed by the BGI-Shenzhen, China, as previously described (Zheng et al., 2016; Hu et al., 2017). The qualified gDNA samples were randomly fragmented by using Covaris E220 (Covaris, Inc.), and 150-250 bp fragments were selected using the Agencourt AMpure XP Kit (Beckman Coulter, Inc.). After the process of end-repairing, A-tailing reactions, and adaptor ligation, the DNA fragments were amplified via ligation-mediated PCR. The obtained products were purified and hybridized to the exome array for enrichment. The exome capture is based on the Agilent SureSelect Human All Exon V6 platform, which covers about 99% of the human exonic regions. Captured fragments were then circularized, and DNA nanoballs were produced by rolling circle amplification, which were loaded on BGISEQ-500 sequencing platforms (BGI-Shenzhen, China), according to the quality control standards and operation procedures (Huang et al., 2017).
Read Mapping and Variant Analysis
After the process of the raw data filtering, the clean reads were mapped to the human reference genome (GRCh37/hg19) via the Burrows–Wheeler Aligner (BWA, v0.7.15) program (Li and Durbin, 2010). To make assurance of variant accuracy, local realignment and base quality recalibration were performed by using the genome analysis toolkit (GATK, v3.3.0, https://www.broadinstitute.org/gatk/guide/best-practices), following the removal of duplicate reads using Picard tools (v2.5.0, https://broadinstitute.github.io/picard/) (Van der Auwera et al., 2013). For the qualified data, strict quality control was guaranteed. HaplotypeCaller of GATK was used to call insertions and deletions (indels) and single nucleotide polymorphisms (SNPs). Next, SnpEff software (https://pcingola.github.io/SnpEff/) provided the variants with annotation. The annotation data and final variants were prepared for the downstream analysis (Pereira et al., 2020). All candidate variants were filtered against several public databases: the Single Nucleotide Polymorphism database (version 154, dbSNP154), National Heart, Lung and Blood Institute’s Exome Sequencing Project 6500 (NHLBI-ESP6500), 1000 Genomes Project (1000G), Exome Aggregation Consortium (ExAC), Genome Aggregation Database (gnomAD), and an in-house exome database of BGI-Shenzhen (Lim et al., 2013; Xia et al., 2017). Then, Sanger sequencing was applied to confirm the identified potential causal variants using an ABI 3500 sequencer (Applied Biosystems, Thermo Fisher Scientific, Inc.) (Guo et al., 2013; Xiao et al., 2018). Locus-specific polymerase chain reaction (PCR) amplification and sequencing primers were designed using the online Primer3 program (http://primer3.ut.ee/) and National Center for Biotechnology Information Basic Local Alignment Search Tool (NCBI BLAST, https://blast.ncbi.nlm.nih.gov/Blast.cgi) (Untergasser et al., 2012), and the paired primers are listed in Table 1.

TABLE 1. Detecting primers for the dynein axonemal heavy chain 17 gene variants.
Bioinformatics Analyses
Several bioinformatic prediction software programs were used to estimate whether a variant is related to protein structure or function. For in silico analyses, Protein Variation Effect Analyzer (PROVEAN, http://provean.jcvi.org/index.php), Polymorphism Phenotyping version 2 (PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/), and MutationTaster (https://www.mutationtaster.org/) were applied to get access to impacts on the protein structure and function (Adzhubei et al., 2010; Schwarz et al., 2014; Choi and Chan, 2015). NCBI BLAST was used to assess sequence conservation of the amino acid at variant positions among different species.
The protein structures of wild type and variant type were predicted via the online SWISS-MODEL tool (https://swissmodel.expasy.org/) and the visualized structures were further constructed via PyMOL software (version 2.3, Schrödinger, LLC, Portland, United States).
Results
Clinical Findings
The normal individual presented normal organ placement (Figure 1A). Two probands from unrelated Han-Chinese families presented randomization of LR asymmetry. The proband 1 from family 1 is a 50-year-old woman whose chest X-ray and B-mode ultrasonographic diagnosis revealed the mirror image reversal of normal organ placement and no signs of other cilia-related disorders (Figure 1B). The proband 2 from family 2, a 5-year-old boy, was diagnosed with dextrocardia and complex CHD, including pulmonary valve stenosis, complete transposition of the great arteries, and endocardial cushion defect, by chest X-ray (Figure 1C), cardiac ultrasound, and CT scan. He was prone to having colds and coughs since early childhood. In addition, the available medical history showed that cardiac murmurs with cyanosis were discovered in infancy. The two probands declined further examinations such as transmission electron microscopy (TEM) and high-speed video microscopy (HSVM). Other members of the two families refused to participate in relative inspection, as they insisted on not suffering any cilia-related symptoms.
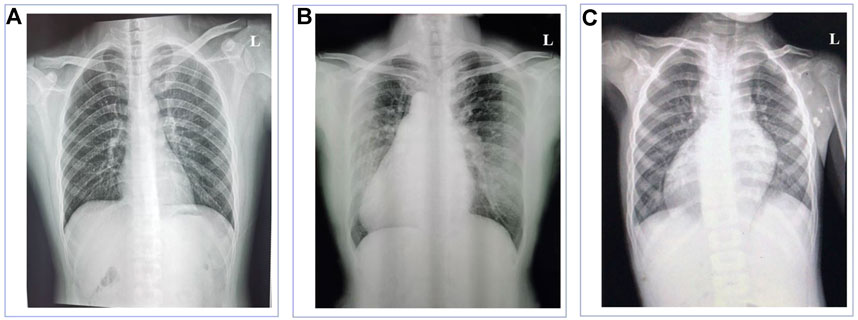
FIGURE 1. Chest X-ray images of the normal individual and patients with left-right asymmetry disorders. (A) Chest X-ray of the normal individual presented normal organ placement. (B) Chest X-ray of the proband in family 1 revealed dextrocardia. (C) Chest X-ray of the proband in family 2 revealed dextrocardia.
Genetic Findings
WES of the proband 1 and the proband 2 generated a total of 242.17 million and 255.35 million clean reads with an average of 99.94% successfully mapped to the human reference genome (GRCh37/hg19). On the target region, the mean sequencing depth of 264.94-fold (proband 1) and 276.52-fold (proband 2) guaranteed enough accuracy to call variants in 99.63% and 99.71% of the targeted bases covered by at least 10×, respectively. There were a total of 105,991 SNPs and 18,461 indels detected in proband 1, while a total of 106,426 SNPs and 18,992 indels were detected in proband 2. A variant filtering strategy referring to previous studies was utilized to identify potential causal variants in these patients (Zheng et al., 2016; Xiang et al., 2019). The following were considered: (i) variants recorded in dbSNP154, NHLBI-ESP6500, and 1000G with minor allele frequency (MAF) ≥1% were ruled out. (ii) The remaining variants were further filtered again in the in-house BGI exome database with 1,943 Han-Chinese controls without randomization of LR asymmetry, and variants with MAF ≥1% were ruled out. (iii) Variants predicted to be deleterious were reserved. (iv) Compound heterozygous or homozygous variants in known genes responsible for LR asymmetry disorders or other cilia-related disorders were prosecuted as potential candidate variants. With these criteria, only two compound heterozygous variants: c.4109C>T and c.9776C>T, and c.612C>G and c.8764C>T in the DNAH17 gene (NM_173628.4) were identified in two probands from unrelated families, respectively. Disease-causing variants in at least 82 of the known genes responsible for LR asymmetry disorder phenotypes were excluded in our patients, though gross deletion/duplication and complex rearrangement in these genes cannot be completely ruled out. These four variants are recorded in the dbSNP154 and has a low frequency in the global population of 1000G, ExAC, and gnomAD (Table 2), suggesting these two compound heterozygous variants are potential disorder-related variants. These four variants were further confirmed by Sanger sequencing (Figures 2A,B).
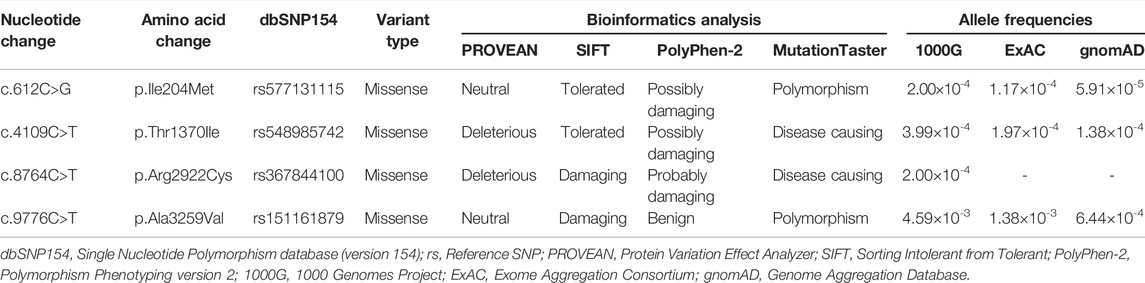
TABLE 2. In silico analysis of the dynein axonemal heavy chain 17 gene variants.
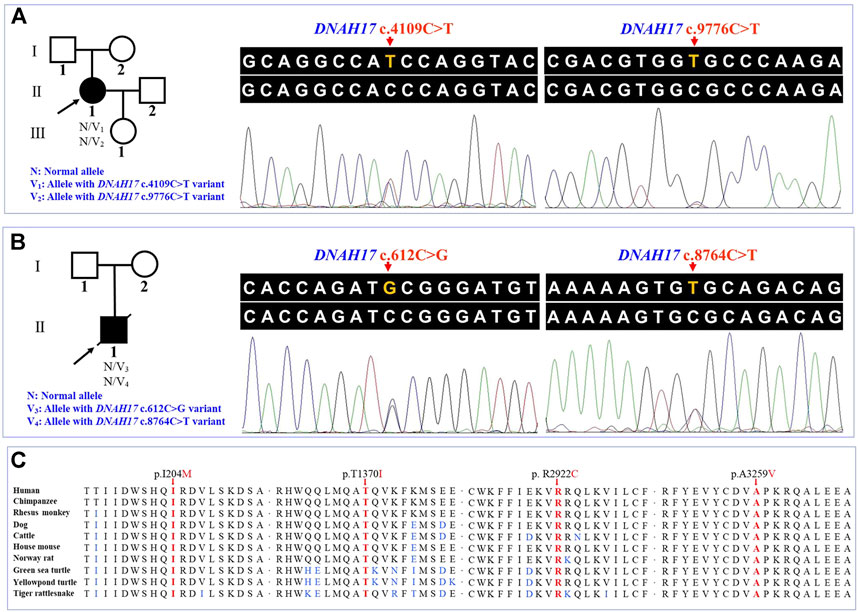
FIGURE 2. Pedigrees and sequence analysis of the two unrelated Han-Chinese patients with left-right asymmetry disorders. (A) and (B) Pedigrees with left-right asymmetry disorders and Sanger sequencing results. The proband, shown as a solid symbol, is indicated by an arrow, and the deceased family member is shown with a slash in the pedigree tree. (C) Sequence alignment of the dynein axonemal heavy chain 17 among different species, with the affected amino acids indicated by the arrows. DNAH17, the dynein axonemal heavy chain 17 gene.
Variant Bioinformatics Analysis
The c.8764C>T (p.Arg2922Cys) variant was predicted to be “deleterious,” “damaging,” “probably damaging,” and “disease causing” by PROVEAN, Sorting Intolerant from Tolerant (SIFT), PolyPhen-2, and MutationTaster, respectively. For the other three variants, c.612C>G (p.Ile204Met), c.4109C>T (p.Thr1370Ile), and c.9776C>T (p.Ala3259Val), at least one of four prediction programs showed that the variants were potentially deleterious (Table 2). Alignment of the protein sequences across different species was shown by a phylogenetic analysis (Figure 2C), indicating that the variant sites were conserved in mammals and reptiles, further supporting that these variants are disorder-related variants. Structural modeling showed the conformational alteration in the context of protein (Figure 3).
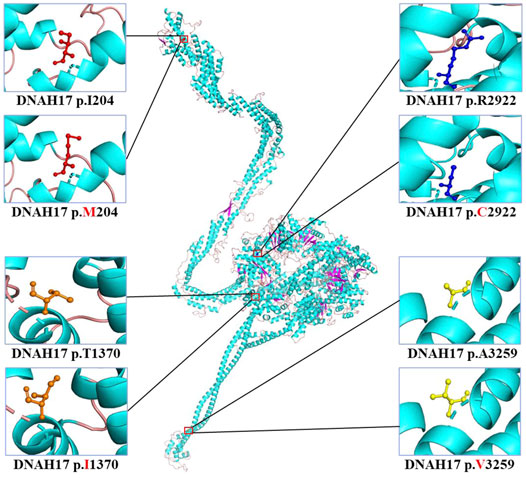
FIGURE 3. Cartoon model of the dynein axonemal heavy chain 17 protein structure visualized by PyMOL based on the SWISS-MODEL. The isoleucine (I) and mutated methionine (M) at position 204; threonine (T) and mutated isoleucine (I) at position 1370; arginine (R) and mutated cysteine (C) at position 2922; and alanine (A) and mutated valine (V) at position 3259 are indicated with ball-and-stick models.
Discussion
The formation of LR asymmetry of the visceral organs is a conserved feature of human body, which is precisely orchestrated by multiple regulatory mechanisms (Blum et al., 2014; Sung et al., 2016; Hobbs et al., 2018). The process initiating in the node and occurring during embryogenesis can be divided into four steps: i) symmetry breaking caused by the leftward nodal flow, (ii) transmission of asymmetric signals to the left lateral plate mesoderm (LPM), (iii) cascades of Nodal and Lefty2 expression in the left LPM, and (iv) situs-specific morphogenesis (Ramsdell and Yost, 1998; Nonaka et al., 2005; Okada et al., 2005; Yoshimoto et al., 2012).
Rotational movement of motile monocilia in nodal cells creates the nodal flow and activates the asymmetric signaling, while the sperm flagella with similar structure are responsible for cell motility (McGrath et al., 2003; Shiraishi and Ichikawa, 2012; Pennekamp et al., 2015). Most motile cilia and sperm flagella share a highly conserved 9+2 axonemal structure (nine outer microtubule doublets surrounding one central microtubule pair), which are comprised of microtubules, motor dynein arms and the associated structures, exhibiting motile and sensory functions (Zhou et al., 2012; Ishikawa, 2017). Most immotile cilia have a 9+0 axoneme, lacking the central microtubule pair (Fliegauf et al., 2007). The inner and outer dynein arms (IDAs and ODAs), comprised of heavy, intermediate, and light dynein chains, are vital to motility of motile cilia and sperm flagella with 9+2 axonemes (King, 2016; Viswanadha et al., 2017; Lee and Ostrowski, 2021). Human LR asymmetry disorders are thought to be attributed to defective cilia structure or function during embryonic development (Blum et al., 2014; Shinohara and Hamada, 2017). Thus, exploring gene variants targeted establishment and function of nodal cilia during early embryogenesis may help the diagnosis and gene therapy of LR asymmetry disorder.
The DNAH17 gene, located at chromosome 17q25.3, is a large gene composed of 81 exons and encodes an axonemal dynein heavy chain of ODA. DNAHs, also named heavy chains (HCs), include 13 members (DNAH1-3, 5-12, 14, and 17) in humans (Pazour et al., 2006; Inaba, 2011). In the known axial filament complex, the ODAs play a major role in the beating of cilia and flagella through the ATPase activity of their HCs (Whitfield et al., 2019). Dynein HCs are large proteins that turn the energy of ATP into force supporting the sliding of outer microtubule doublets, which generates the beating of cilia (Pazour et al., 2006). To date, variants in most genes of DNAHs in humans have been reported to be associated with diseases related to cilia or flagella. A common autosomal recessive disorder caused by those variants is PCD, which is characterized by recurrent respiratory tract infections, laterality defects, and/or infertility, with highly genetic and clinical heterogeneity (Fliegauf et al., 2005; Hornef et al., 2006; Pifferi et al., 2010; Li et al., 2016). In the ultrastructure, sperm flagella are similar to cilia, underpinning the common relationship between male infertility with PCD and subfertility in women with PCD due to deficient ciliary function in the oviducts (Lucas et al., 2014). Of interest, variants in DNAH1 and DNAH9 genes, reported to be associated with PCD, have been depicted in patients with only male infertility, resulted from asthenozoospermia, without other ciliary disorders (Ben Khelifa et al., 2014; Fassad et al., 2018).
In this study, the proband from family 1 presented with SIT without any other cilia-related symptoms, and compound heterozygous variants c.4109C>T (p.Thr1370Ile) and c.9776C>T (p.Ala3259Val) in the DNAH17 gene were identified using a combination of WES and Sanger sequencing. The second DNAH17 compound heterozygous variants, c.612C>G (p.Ile204Met) and c.8764C>T (p.Arg2922Cys), were found in the proband from family 2. The boy presented with dextrocardia and CHD. Cardiac murmurs with cyanosis and recurrent cough were discovered in infancy, suggesting that he may suffer with the cilia-related symptoms. Detailed clinical characteristics of the available family members with DNAH17 variants are presented in Table 3. It seems that at least 10% (2/20) DNAH17 compound heterozygous or homozygous carriers have LR asymmetry disorders. There are only a few studies on DNAH17, and none was found on studying the gene function due to the large size of DNAH17 molecular mass (510 kDa). In addition, the current knowledge on the crucial part of DNAH17 playing in flagella destabilization and asthenozoospermia may depend on genetic or environmental factors such as the mutation type, organism or context (Whitfield et al., 2019; Zhang et al., 2020). Actually, different DNAHs members have been shown to play an important role in cilia/flagella formation and cilia/flagella regulation. DNAH1 and DNAH9, homologs of DNAH17, were located to ciliary axonemes and sperm flagella, and responsible for cilia/flagella-related phenotypes (Bartoloni et al., 2002; Olbrich et al., 2002; Ben Khelifa et al., 2014; Fassad et al., 2018). Due to a common highly conserved 9+2 axonemal structure of cilia and sperm flagella, different DNAH17 mutations may independently cause flagella destabilization, asthenozoospermia or LR asymmetry disorders in specific organelle sharing a common axonemal machinery. Though DNAH17 expression was detected in testis, brain, lung and other tissues (https://www.ncbi.nlm.nih.gov/gene/8632), the protein was only observed in the flagella by assays of immunoblotting and immunofluorescence staining in various human somatic cell lines, human respiratory epithelial cells and sperm cells (Fagerberg et al., 2014; Whitfield et al., 2019; Zhang et al., 2020). It implies that DNAH17 expression may be influenced by cell type-specific spatial localization and the switch point in the development of the nodal flow during early embryogenesis. The lack of typical symptoms, such as nasosinusitis and bronchiectasis, in the two patients may be due to absent or low expression of DNAH17 in specific tissues after the completion of the embryonic development. Biallelic variant types of DNAH17, genetic background, and epigenetic modification, as well as environmental factors, may potentially affect the phenotypic manifestation. The possible genotype-phenotype association should be warranted with more DNAH17-mutated carriers discovered. Our observation of the potential relationship between DNAH17 and LR asymmetry disorders may extend the field-of-view for new actor of DNAH17 in the development of human diseases.
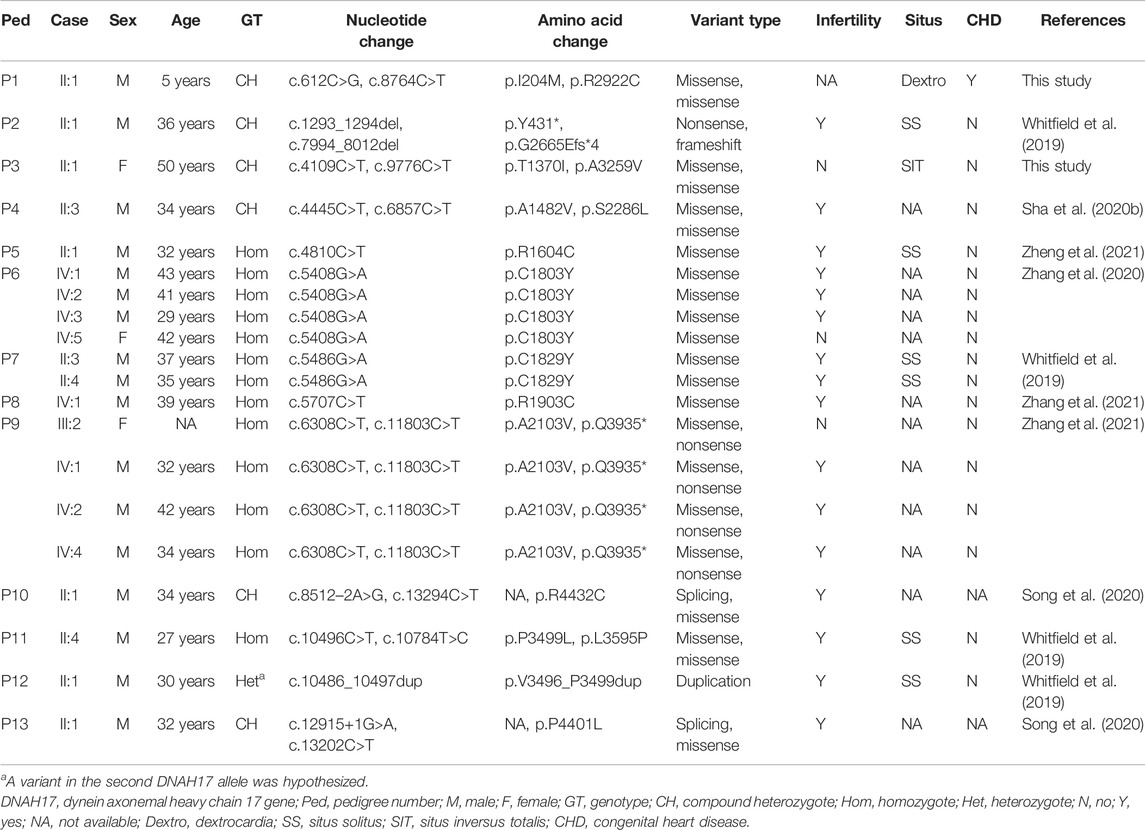
TABLE 3. Clinical data of the DNAH17 variant carriers in different families.
Taken together, our research identified compound heterozygous DNAH17 variants (c.4109C>T and c.9776C>T; c.612C>G and c.8764C>T) in families with LR asymmetry disorders, typical phenotypes of ciliary disorders, including SIT, dextrocardia, and CHD, albeit infertility cannot be excluded. To our knowledge, this is, the first report of relationships between DNAH17 variants and ciliogenesis, which expands the phenotypic spectrum and benefits genetic counseling. Combined with the reported DNAH17-associated asthenozoospermia, we proposed that DNAH17 compound heterozygous variants, or homozygous variants, may potentially cause a specific disease, the DNAH17-associated ciliary/flagellar disorder. The study may be limited by the lack of nasal epithelial brush biopsy samples for ciliary beating and ultrastructure analysis. Further constructing DNAH17 variant-targeted animal models and performing experimental therapies will facilitate an in-depth comprehension of cellular and molecular mechanisms of ciliary and flagellar defects, and contribute to rectification of the defects.
Data Availability Statement
The datasets presented in this article are not readily available because the data information is in a controlled state due to the national legislation, specifically the Ministry of Science and Technology of the People’s Republic of China. Data of this project can be accessed after an approval application by the China National GeneBank DataBase (CNGBdb). Please refer to CNGBdb: https://db.cngb.org/, or email:Q05HQmRiQGNuZ2Iub3Jnfor detailed application guidance. The project accession code CNP0002422 should be included in the application.
Ethics Statement
The studies involving human participants were reviewed and approved by the Institutional Review Board of the Third Xiangya Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
XY, LY, and HD conceived and designed this study. LY, SD, HX, XT, XH, XC, and HD collected the patient samples and clinical data. XY, LY, and XD performed the experiments. XY, LY, HX, and HD analyzed the data. XY, LY, and HD wrote the manuscript. The final version of the manuscript was read and approved by all authors.
Funding
This study was supported by the National Natural Science Foundation of China (Grant Nos. 81670216, 81873686, and 81800219), Natural Science Foundation of Hunan Province (Grant Nos. 2020JJ4830 and 2020JJ3057), Scientific Research Project of Health Commission of Hunan Province (Grant No. B2019174), Hunan Provincial Innovation Foundation for Postgraduate (Grant No. CX20210375), Fundamental Research Funds for the Central Universities of Central South University (Grant No. 2021zzts0403), Undergraduate Innovative Training Plan Program of Central South University (Grant No. XCX2021222), Wisdom Accumulation and Talent Cultivation Project of the Third Xiangya Hospital of Central South University (Grant No. YX202109), and Distinguished Professor of the Lotus Scholars Award Program of Hunan Province, China.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank all the enrolled individuals for their participation in the present study.
References
Abdelhamed, Z., Lukacs, M., Cindric, S., Ali, S., Omran, H., and Stottmann, R. W. (2021). Correction: A Novel Hypomorphic Allele of Spag17 Causes Primary Ciliary Dyskinesia Phenotypes in Mice. Dis. Model. Mech. 14, dmm048645. doi:10.1242/dmm.048645
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 7, 248–249. doi:10.1038/nmeth0410-248
Al Mutairi, F., Alkhalaf, R., Alkhorayyef, A., Alroqi, F., Yusra, A., Umair, M., et al. (2020). Homozygous Truncating NEK10 Mutation, Associated with Primary Ciliary Dyskinesia: a Case Report. BMC Pulm. Med. 20, 141. doi:10.1186/s12890-020-1175-1
Bartoloni, L., Blouin, J.-L., Pan, Y., Gehrig, C., Maiti, A. K., Scamuffa, N., et al. (2002). Mutations in the DNAH11 (Axonemal Heavy Chain Dynein Type 11) Gene Cause One Form of Situs Inversus Totalis and Most Likely Primary Ciliary Dyskinesia. Proc. Natl. Acad. Sci. U.S.A. 99, 10282–10286. doi:10.1073/pnas.152337699
Ben Khelifa, M., Coutton, C., Zouari, R., Karaouzène, T., Rendu, J., Bidart, M., et al. (2014). Mutations in DNAH1, Which Encodes an Inner Arm Heavy Chain Dynein, lead to Male Infertility from Multiple Morphological Abnormalities of the Sperm Flagella. Am. J. Hum. Genet. 94, 95–104. doi:10.1016/j.ajhg.2013.11.017
Best, S., Shoemark, A., Rubbo, B., Patel, M. P., Fassad, M. R., Dixon, M., et al. (2019). Risk Factors for Situs Defects and Congenital Heart Disease in Primary Ciliary Dyskinesia. Thorax 74, 203–205. doi:10.1136/thoraxjnl-2018-212104
Blum, M., Feistel, K., Thumberger, T., and Schweickert, A. (2014). The Evolution and Conservation of Left-Right Patterning Mechanisms. Development 141, 1603–1613. doi:10.1242/dev.100560
Bustamante-Marin, X. M., Horani, A., Stoyanova, M., Charng, W.-L., Bottier, M., Sears, P. R., et al. (2020a). Mutation of CFAP57, a Protein Required for the Asymmetric Targeting of a Subset of Inner Dynein Arms in Chlamydomonas, Causes Primary Ciliary Dyskinesia. Plos Genet. 16, e1008691. doi:10.1371/journal.pgen.1008691
Bustamante-Marin, X. M., Shapiro, A., Sears, P. R., Charng, W.-L., Conrad, D. F., Leigh, M. W., et al. (2020b). Identification of Genetic Variants in CFAP221 as a Cause of Primary Ciliary Dyskinesia. J. Hum. Genet. 65, 175–180. doi:10.1038/s10038-019-0686-1
Cannarella, R., Maniscalchi, E. T., Condorelli, R. A., Scalia, M., Guerri, G., La Vignera, S., et al. (2020). Ultrastructural Sperm Flagellum Defects in a Patient with CCDC39 Compound Heterozygous Mutations and Primary Ciliary Dyskinesia/situs Viscerum Inversus. Front. Genet. 11, 974. doi:10.3389/fgene.2020.00974
Chen, X., Deng, S., Xia, H., Yuan, L., Xu, H., Tang, S., et al. (2020). Identification of a CCDC114 Variant in a Han-Chinese Patient with Situs Inversus. Exp. Ther. Med. 20, 3336–3342. doi:10.3892/etm.2020.9059
Cho, E. H., Huh, H. J., Jeong, I., Lee, N. Y., Koh, W. J., Park, H. C., et al. (2020). A Nonsense Variant in NME5 Causes Human Primary Ciliary Dyskinesia with Radial Spoke Defects. Clin. Genet. 98, 64–68. doi:10.1111/cge.13742
Choi, Y., and Chan, A. P. (2015). PROVEAN Web Server: a Tool to Predict the Functional Effect of Amino Acid Substitutions and Indels. Bioinformatics 31, 2745–2747. doi:10.1093/bioinformatics/btv195
Derrick, C. J., Sánchez-Posada, J., Hussein, F., Tessadori, F., Pollitt, E. J. G., Savage, A. M., et al. (2021). Asymmetric Hapln1a Drives Regionalized Cardiac ECM Expansion and Promotes Heart Morphogenesis in Zebrafish Development. Cardiovasc. Res. 118, 226–240. doi:10.1093/cvr/cvab004
Ding, X., Fragoza, R., Singh, P., Zhang, S., Yu, H., and Schimenti, J. C. (2020). Variants in RABL2A Causing Male Infertility and Ciliopathy. Hum. Mol. Genet. 29, 3402–3411. doi:10.1093/hmg/ddaa230
Fagerberg, L., Hallström, B. M., Oksvold, P., Kampf, C., Djureinovic, D., Odeberg, J., et al. (2014). Analysis of the Human Tissue-specific Expression by Genome-wide Integration of Transcriptomics and Antibody-Based Proteomics. Mol. Cell Proteomics 13, 397–406. doi:10.1074/mcp.M113.035600
Fassad, M. R., Shoemark, A., Legendre, M., Hirst, R. A., Koll, F., le Borgne, P., et al. (2018). Mutations in Outer Dynein Arm Heavy Chain DNAH9 Cause Motile Cilia Defects and Situs Inversus. Am. J. Hum. Genet. 103, 984–994. doi:10.1016/j.ajhg.2018.10.016
Fliegauf, M., Benzing, T., and Omran, H. (2007). When Cilia Go Bad: Cilia Defects and Ciliopathies. Nat. Rev. Mol. Cel Biol 8, 880–893. doi:10.1038/nrm2278
Fliegauf, M., Olbrich, H., Horvath, J., Wildhaber, J. H., Zariwala, M. A., Kennedy, M., et al. (2005). Mislocalization of DNAH5 and DNAH9 in Respiratory Cells from Patients with Primary Ciliary Dyskinesia. Am. J. Respir. Crit. Care Med. 171, 1343–1349. doi:10.1164/rccm.200411-1583OC
Guo, T., Tu, C.-F., Yang, D.-H., Ding, S.-Z., Lei, C., Wang, R.-C., et al. (2021). Bi-allelic BRWD1 Variants Cause Male Infertility with Asthenoteratozoospermia and Likely Primary Ciliary Dyskinesia. Hum. Genet. 140, 761–773. doi:10.1007/s00439-020-02241-4
Guo, Y., Yang, H., Deng, X., Song, Z., Yang, Z., Xiong, W., et al. (2013). Genetic Analysis of the S100B Gene in Chinese Patients with Parkinson Disease. Neurosci. Lett. 555, 134–136. doi:10.1016/j.neulet.2013.09.037
Heigwer, J., Kutzner, J., Haeussler, M., Burkhalter, M. D., Draebing, T., Juergensen, L., et al. (2020). miR-103/107 Regulates Left-Right Asymmetry in Zebrafish by Modulating Kupffer's Vesicle Development and Ciliogenesis. Biochem. Biophysical Res. Commun. 527, 432–439. doi:10.1016/j.bbrc.2020.04.066
Hobbs, S. J., Nauwelaerts, S., Sinclair, J., Clayton, H. M., and Back, W. (2018). Sagittal Plane Fore Hoof Unevenness Is Associated with Fore and Hindlimb Asymmetrical Force Vectors in the Sagittal and Frontal Planes. PLoS One 13, e0203134. doi:10.1371/journal.pone.0203134
Hornef, N., Olbrich, H., Horvath, J., Zariwala, M. A., Fliegauf, M., Loges, N. T., et al. (2006). DNAH5 Mutations Are a Common Cause of Primary Ciliary Dyskinesia with Outer Dynein Arm Defects. Am. J. Respir. Crit. Care Med. 174, 120–126. doi:10.1164/rccm.200601-084OC
Hu, P., Wu, S., Yuan, L., Lin, Q., Zheng, W., Xia, H., et al. (2017). Compound Heterozygous POMT1 Mutations in a Chinese Family with Autosomal Recessive Muscular Dystrophy-Dystroglycanopathy C1. J. Cel. Mol. Med. 21, 1388–1393. doi:10.1111/jcmm.13068
Huang, J., Liang, X., Xuan, Y., Geng, C., Li, Y., Lu, H., et al. (2017). A Reference Human Genome Dataset of the BGISEQ-500 Sequencer. Gigascience 6, 1–9. doi:10.1093/gigascience/gix024
Inaba, K. (2011). Sperm Flagella: Comparative and Phylogenetic Perspectives of Protein Components. Mol. Hum. Reprod. 17, 524–538. doi:10.1093/molehr/gar034
Ishikawa, T. (2017). Axoneme Structure from Motile Cilia. Cold Spring Harb Perspect. Biol. 9, a028076. doi:10.1101/cshperspect.a028076
King, S. M. (2016). Axonemal Dynein Arms. Cold Spring Harb Perspect. Biol. 8, a028100. doi:10.1101/cshperspect.a028100
Lee, L., and Ostrowski, L. E. (2021). Motile Cilia Genetics and Cell Biology: Big Results from Little Mice. Cell. Mol. Life Sci. 78, 769–797. doi:10.1007/s00018-020-03633-5
Levin, M. (2004). The Embryonic Origins of Left-Right Asymmetry. Crit. Rev. Oral Biol. Med. 15, 197–206. doi:10.1177/154411130401500403
Li, H., and Durbin, R. (2010). Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 26, 589–595. doi:10.1093/bioinformatics/btp698
Li, Y., Yagi, H., Onuoha, E. O., Damerla, R. R., Francis, R., Furutani, Y., et al. (2016). DNAH6 and its Interactions with PCD Genes in Heterotaxy and Primary Ciliary Dyskinesia. Plos Genet. 12, e1005821. doi:10.1371/journal.pgen.1005821
Lim, B. C., Lee, S., Shin, J.-Y., Hwang, H., Kim, K. J., Hwang, Y. S., et al. (2013). Molecular Diagnosis of Congenital Muscular Dystrophies with Defective Glycosylation of Alpha-Dystroglycan Using Next-Generation Sequencing Technology. Neuromuscul. Disord. 23, 337–344. doi:10.1016/j.nmd.2013.01.007
Lucas, J. S., Burgess, A., Mitchison, H. M., Moya, E., Williamson, M., and Hogg, C.National Pcd Service UK (2014). Diagnosis and Management of Primary Ciliary Dyskinesia. Arch. Dis. Child. 99, 850–856. doi:10.1136/archdischild-2013-304831
McGrath, J., Somlo, S., Makova, S., Tian, X., and Brueckner, M. (2003). Two Populations of Node Monocilia Initiate Left-Right Asymmetry in the Mouse. Cell 114, 61–73. doi:10.1016/s0092-8674(03)00511-7
Nonaka, S., Yoshiba, S., Watanabe, D., Ikeuchi, S., Goto, T., Marshall, W. F., et al. (2005). De Novo formation of Left-Right Asymmetry by Posterior Tilt of Nodal Cilia. Plos Biol. 3, e268. doi:10.1371/journal.pbio.0030268
Okada, Y., Takeda, S., Tanaka, Y., Belmonte, J.-C. I., and Hirokawa, N. (2005). Mechanism of Nodal Flow: a Conserved Symmetry Breaking Event in Left-Right Axis Determination. Cell 121, 633–644. doi:10.1016/j.cell.2005.04.008
Olbrich, H., Häffner, K., Kispert, A., Völkel, A., Volz, A., Sasmaz, G., et al. (2002). Mutations in DNAH5 Cause Primary Ciliary Dyskinesia and Randomization of Left-Right Asymmetry. Nat. Genet. 30, 143–144. doi:10.1038/ng817
Osório, L., Wu, X., Wang, L., Jiang, Z., Neideck, C., Sheng, G., et al. (2019). ISM1 Regulates NODAL Signaling and Asymmetric Organ Morphogenesis during Development. J. Cel Biol 218, 2388–2402. doi:10.1083/jcb.201801081
Otto, E. A., Schermer, B., Obara, T., O'Toole, J. F., Hiller, K. S., Mueller, A. M., et al. (2003). Mutations in INVS Encoding Inversin Cause Nephronophthisis Type 2, Linking Renal Cystic Disease to the Function of Primary Cilia and Left-Right axis Determination. Nat. Genet. 34, 413–420. doi:10.1038/ng1217
Pazour, G. J., Agrin, N., Walker, B. L., and Witman, G. B. (2006). Identification of Predicted Human Outer Dynein Arm Genes: Candidates for Primary Ciliary Dyskinesia Genes. J. Med. Genet. 43, 62–73. doi:10.1136/jmg.2005.033001
Pennekamp, P., Menchen, T., Dworniczak, B., and Hamada, H. (2015). Situs Inversus and Ciliary Abnormalities: 20 Years Later, what Is the Connection? Cilia 4, 1. doi:10.1186/s13630-014-0010-9
Pereira, R., Oliveira, J., and Sousa, M. (2020). Bioinformatics and Computational Tools for Next-Generation Sequencing Analysis in Clinical Genetics. Jcm 9, 132. doi:10.3390/jcm9010132
Pifferi, M., Michelucci, A., Conidi, M. E., Cangiotti, A. M., Simi, P., Macchia, P., et al. (2010). New DNAH11 Mutations in Primary Ciliary Dyskinesia with Normal Axonemal Ultrastructure. Eur. Respir. J. 35, 1413–1416. doi:10.1183/09031936.00186209
Ramsdell, A. F., and Yost, H. J. (1998). Molecular Mechanisms of Vertebrate Left-Right Development. Trends Genet. 14, 459–465. doi:10.1016/s0168-9525(98)01599-6
Sahabian, A., von Schlehdorn, L., Drick, N., Pink, I., Dahlmann, J., Haase, A., et al. (2020). Generation of Two hiPSC Clones (MHHi019-A, MHHi019-B) from a Primary Ciliary Dyskinesia Patient Carrying a Homozygous Deletion in the NME5 Gene (c.415delA (p.Ile139Tyrfs*8)). Stem Cel Res. 48, 101988. doi:10.1016/j.scr.2020.101988
Schwarz, J. M., Cooper, D. N., Schuelke, M., and Seelow, D. (2014). MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 11, 361–362. doi:10.1038/nmeth.2890
Sha, Y., Wei, X., Ding, L., Ji, Z., Mei, L., Huang, X., et al. (2020a). Biallelic Mutations of CFAP74 May Cause Human Primary Ciliary Dyskinesia and MMAF Phenotype. J. Hum. Genet. 65 (65), 961–969. doi:10.1038/s10038-020-0790-2
Sha, Y., Wei, X., Ding, L., Mei, L., Huang, X., Lin, S., et al. (2020b). DNAH17 Is Associated with Asthenozoospermia and Multiple Morphological Abnormalities of Sperm Flagella. Ann. Hum. Genet. 84, 271–279. doi:10.1111/ahg.12369
Shinohara, K., and Hamada, H. (2017). Cilia in Left-Right Symmetry Breaking. Cold Spring Harb Perspect. Biol. 9, a028282. doi:10.1101/cshperspect.a028282
Shiraishi, I., and Ichikawa, H. (2012). Human Heterotaxy Syndrome. Circ. J. 76, 2066–2075. doi:10.1253/circj.CJ-12-0957
Song, B., Liu, C., Gao, Y., Marley, J. L., Li, W., Ni, X., et al. (2020). Novel Compound Heterozygous Variants in Dynein Axonemal Heavy Chain 17 Cause Asthenoteratospermia with Sperm Flagellar Defects. J. Genet. Genomics 47, 713–717. doi:10.1016/j.jgg.2020.07.004
Sung, Y. H., Baek, I.-J., Kim, Y. H., Gho, Y. S., Oh, S. P., Lee, Y. J., et al. (2016). PIERCE1 Is Critical for Specification of Left-Right Asymmetry in Mice. Sci. Rep. 6, 27932. doi:10.1038/srep27932
Thomas, L., Bouhouche, K., Whitfield, M., Thouvenin, G., Coste, A., Louis, B., et al. (2020). TTC12 Loss-Of-Function Mutations Cause Primary Ciliary Dyskinesia and Unveil Distinct Dynein Assembly Mechanisms in Motile Cilia versus Flagella. Am. J. Hum. Genet. 106, 153–169. doi:10.1016/j.ajhg.2019.12.010
Twigg, S. R. F., Lloyd, D., Jenkins, D., Elçioglu, N. E., Cooper, C. D. O., Al-Sannaa, N., et al. (2012). Mutations in Multidomain Protein MEGF8 Identify a Carpenter Syndrome Subtype Associated with Defective Lateralization. Am. J. Hum. Genet. 91, 897–905. doi:10.1016/j.ajhg.2012.08.027
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3-new Capabilities and Interfaces. Nucleic Acids Res. 40, e115. doi:10.1093/nar/gks596
Van der Auwera, G. A., Carneiro, M. O., Hartl, C., Poplin, R., Del Angel, G., Levy-Moonshine, A., et al. (2013). From FastQ Data to High Confidence Variant Calls: the Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinformatics 43, 11–33. doi:10.1-3310.1002/0471250953.bi1110s43
Viswanadha, R., Sale, W. S., and Porter, M. E. (2017). Ciliary Motility: Regulation of Axonemal Dynein Motors. Cold Spring Harb Perspect. Biol. 9, a018325. doi:10.1101/cshperspect.a018325
Wang, R., Yang, D., Guo, T., Lei, C., Chen, X., Kang, X., et al. (2021). Case Report: Identification of a Novel ODAD3 Variant in a Patient with Primary Ciliary Dyskinesia. Front. Genet. 12, 652381. doi:10.3389/fgene.2021.652381
Wang, Y., Tu, C., Nie, H., Meng, L., Li, D., Wang, W., et al. (2020). Novel DNAAF6 Variants Identified by Whole-Exome Sequencing Cause Male Infertility and Primary Ciliary Dyskinesia. J. Assist. Reprod. Genet. 37, 811–820. doi:10.1007/s10815-020-01735-4
Whitfield, M., Thomas, L., Bequignon, E., Schmitt, A., Stouvenel, L., Montantin, G., et al. (2019). Mutations in DNAH17, Encoding a Sperm-specific Axonemal Outer Dynein Arm Heavy Chain, Cause Isolated Male Infertility Due to Asthenozoospermia. Am. J. Hum. Genet. 105, 198–212. doi:10.1016/j.ajhg.2019.04.015
Xia, H., Hu, P., Yuan, L., Xiong, W., Xu, H., Yi, J., et al. (2017). A Homozygous MYO7A Mutation Associated to Usher Syndrome and Unilateral Auditory Neuropathy Spectrum Disorder. Mol. Med. Rep. 16, 4241–4246. doi:10.3892/mmr.2017.7053
Xiang, Q., Cao, Y., Xu, H., Guo, Y., Yang, Z., Xu, L., et al. (2019). Identification of Novel Pathogenic ABCA4 Variants in a Han Chinese Family with Stargardt Disease. Biosci. Rep. 39, BSR20180872. doi:10.1042/BSR20180872
Xiao, H., Yuan, L., Xu, H., Yang, Z., Huang, F., Song, Z., et al. (2018). Novel and Recurring Disease-Causing NF1 Variants in Two Chinese Families with Neurofibromatosis Type 1. J. Mol. Neurosci. 65, 557–563. doi:10.1007/s12031-018-1128-9
Yang, F., and Qi, J. (2020). miR-430a Regulates the Development of Left-Right Asymmetry by Targeting Sqt in the Teleost. Gene 745, 144628. doi:10.1016/j.gene.2020.144628
Yoshimoto, K., Mizoguchi, M., Hata, N., Murata, H., Hatae, R., Amano, T., et al. (2012). Complex DNA Repair Pathways as Possible Therapeutic Targets to Overcome Temozolomide Resistance in Glioblastoma. Front. Oncol. 2, 186. doi:10.3389/fonc.2012.00186
Yuan, L., Deng, X., Song, Z., Yang, Z., Ni, B., Chen, Y., et al. (2015). Genetic Analysis of the RAB39B Gene in Chinese Han Patients with Parkinson's Disease. Neurobiol. Aging 36, 2907.e11-2907.e12. doi:10.1016/j.neurobiolaging.2015.06.019
Zhang, B., Khan, I., Liu, C., Ma, A., Khan, A., Zhang, Y., et al. (2021). Novel Loss‐of‐function Variants in DNAH17 Cause Multiple Morphological Abnormalities of the Sperm Flagella in Humans and Mice. Clin. Genet. 99, 176–186. doi:10.1111/cge.13866
Zhang, B., Ma, H., Khan, T., Ma, A., Li, T., Zhang, H., et al. (2020). A DNAH17 Missense Variant Causes Flagella Destabilization and Asthenozoospermia. J. Exp. Med. 217, e20182365. doi:10.1084/jem.20182365
Zheng, R., Sun, Y., Jiang, C., Chen, D., Yang, Y., and Shen, Y. (2021). A Novel Mutation in DNAH17 Is Present in a Patient with Multiple Morphological Abnormalities of the Flagella. Reprod. BioMedicine Online 43, 532–541. doi:10.1016/j.rbmo.2021.05.009
Zheng, W., Zhang, J., Deng, X., Xiao, J., Yuan, L., Yang, Y., et al. (2016). Identification of a Premature Termination Mutation in the Proline-Rich Transmembrane Protein 2 Gene in a Chinese Family with Febrile Seizures. Mol. Neurobiol. 53, 835–841. doi:10.1007/s12035-014-9047-4
Zhou, J., Yang, F., Leu, N. A., and Wang, P. J. (2012). MNS1 Is Essential for Spermiogenesis and Motile Ciliary Functions in Mice. Plos Genet. 8, e1002516. doi:10.1371/journal.pgen.1002516
Keywords: DNAH17, left–right asymmetry disorders, whole-exome sequencing, gene variants, ciliary/flagellar disorders
Citation: Yu X, Yuan L, Deng S, Xia H, Tu X, Deng X, Huang X, Cao X and Deng H (2022) Identification of DNAH17 Variants in Han-Chinese Patients With Left–Right Asymmetry Disorders. Front. Genet. 13:862292. doi: 10.3389/fgene.2022.862292
Received: 09 February 2022; Accepted: 18 April 2022;
Published: 27 May 2022.
Edited by:
Babak Behnam, National Sanitation Foundation International, United StatesReviewed by:
Takashi Ishikawa, Paul Scherrer Institut, SwitzerlandAmir Amiri-Yekta, Royan Institute, Iran
Copyright © 2022 Yu, Yuan, Deng, Xia, Tu, Deng, Huang, Cao and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao Deng, aGRlbmcwMDhAMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship