 Austin A. Saugstad
Austin A. Saugstad Natasha Petry2,3
Natasha Petry2,3- 1Kansas City University, College of Osteopathic Medicine, Kansas City, MO, United States
- 2Sanford Health Imagenetics, Sioux Falls, SD, United States
- 3Department of Pharmacy Practice, College of Health Professions, North Dakota State University, Fargo, ND, United States
- 4University of South Dakota, Sanford School of Medicine, Department of Internal Medicine, Sioux Falls, SD, United States
As our understanding of genomics and genetic testing continues to advance, the personalization of medical decision making is progressing simultaneously. By carefully crafting medical care to fit the specific needs of the individual, patients can experience better long-term outcomes, reduced toxicities, and improved healthcare experiences. Genetic tests are frequently ordered to help diagnose a clinical presentation and even to guide surveillance. Through persistent investigation, studies have begun to delineate further therapeutic implications based upon unique relationships with genetic variants. In this review, a pre-emptive approach is taken to understand the existing evidence of relationships between specific genetic variants and available therapies. The review revealed an array of diverse relationships, ranging from well-documented clinical approaches to investigative findings with potential for future application. Therapeutic agents identified in the study ranged from highly specific targeted therapies to agents possessing similar risk factors as a genetic variant. Working in conjunction with national standardized treatment approaches, it is critical that physicians appropriately consider these relationships when developing personalized treatment plans for their patients.
Introduction
Advances in both availability and affordability of genetic screening, in conjunction with continued study of therapeutic interactions, has made the utilization of genetic data in clinical practice of the utmost importance. As the usage of genetic information to guide medical decision making continues to increase in frequency, patients are beginning to receive more precise clinical care. Through the utilization of patient-specific genetic information in the lens of physician-guided clinical context, patients are experiencing improved long-term outcomes, reduced toxicities, and fewer adverse effects (van der Wouden et al., 2019; Bernard et al., 2021).
The purpose of this review is to build upon and contribute to the future utilization of genetics in the clinical setting. This study takes a pre-emptive approach, reviewing evidence of key relationships between known cancer predisposing germline genetic variants and various therapies. Our review examines 23 individual genes each with an association to one of 15 different familial cancer syndromes, in concordance with the 2017 ACMG59 recommendation list (Kalia et al., 2017). As summarized in Table 1, our review uncovers a wide array of unique therapeutic interactions between individual patient genetics and pharmacologic interventions. Evidence identified in this review is specific to germline variants and does not include findings involving somatic or therapy-induced genetic variants. In total, our review presents 54 relationships, providing clinicians and researchers with a foundational platform for the future and contributing a useful summary of existing gene-drug relationships for cancer predisposing germline variants.
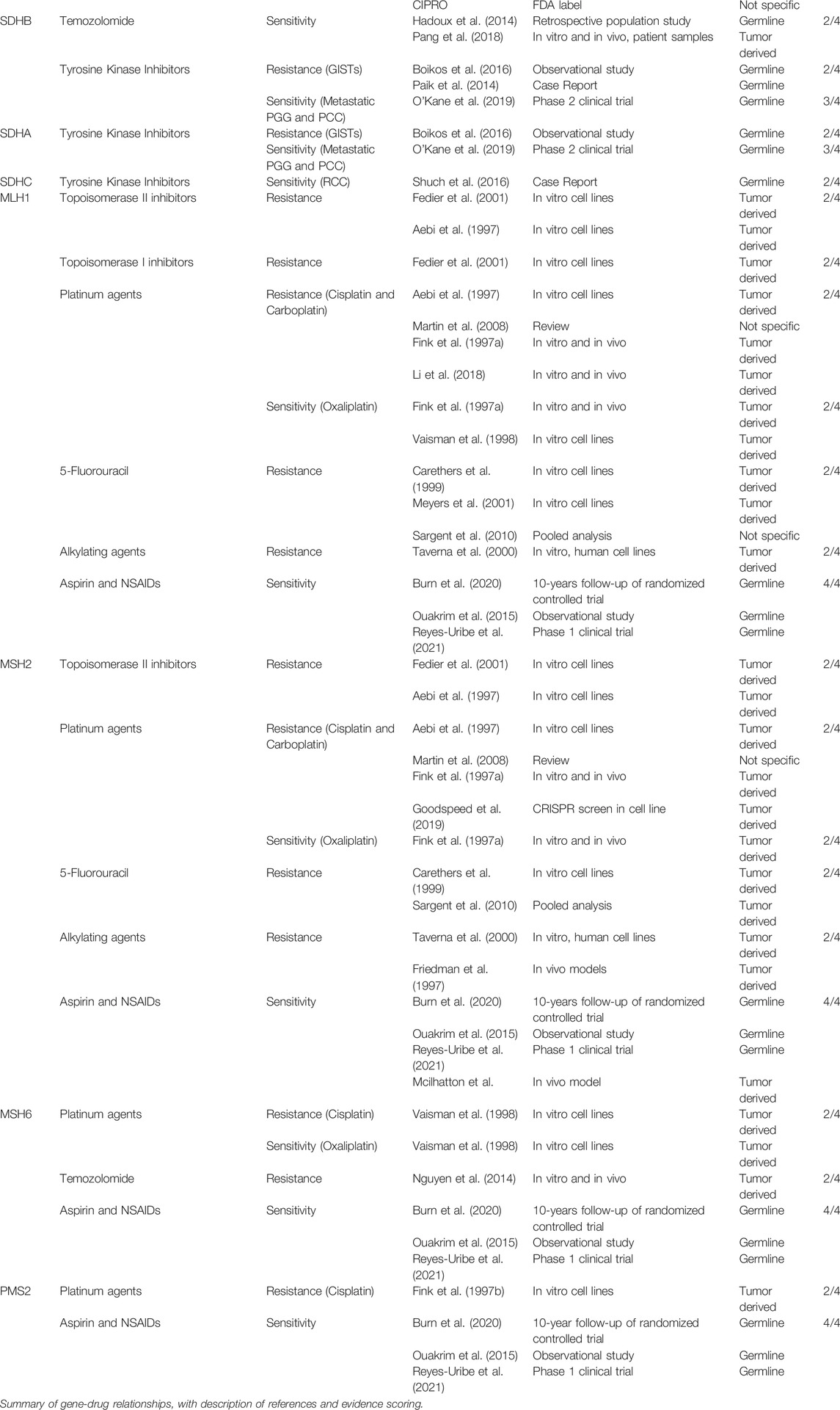
TABLE 1. Gene-drug relationships.
In this review, each of the 23 heritable cancer genes will be introduced and followed by discussion of the associated relationships, including those that are clinically actionable today and other potential investigative therapies. Our results are summarized into three categories: direct gene-drug relationships, shared risk outcomes, and clinical context decision making. First, we report on the extensive work conducted to understand the molecular mechanisms underlying gene variants in conferring sensitivity or resistance to pharmacologic therapies. Secondly, we identified gene variants and therapies sharing similar adverse clinical outcomes and may possess additive risk. Lastly, we touch on two different clinical contexts that may shape a response to a given gene-drug relationship.
Methods
Focused Literature Review
For this comprehensive literature review, multiple sources were utilized to provide a complete compilation of available information. A search for each of the 23 genes included in the study was completed on Clinical Pharmacogenomics Information Consortium (CPIC), The Pharmacogenomics Knowledge Base (PharmGKB), Online Mendelian Inheritance in Man (OMIM®), GeneReviews, UpToDate, and Access Medicine. Secondly a PubMed search was completed for each gene in combination with multiple keywords, including (gene) and (pharmacogenomics) (gene) and (therapy) (gene) and (adverse medication) (gene variant) and (therapy) (clinical syndrome) and (pharmacogenomics). In addition to utilizing the United States Food and Drug Administration Table of Pharmacogenomic Biomarkers in Drug Labeling, an additional review of FDA drug labels was conducted using their available online search tool (https://nctr-crs.fda.gov/fdalabel/ui/search). A PubMed search for similarly related topics was also included to provide appropriate discussion in the context of our study. Each significant finding was classified into one of three categories: direct drug-gene sensitivity or resistance, shared risk outcome, or clinical context consideration. The entirety of the review process was conducted from January 2021 through August 2021.
Clinical Implementation Scale
To succinctly summarize the evidence identified for each of the relationships listed, a four-tier scoring system, Clinical Implementation Scale (CIS), was created by our team. Each relationship was given a score out of 4, with 4/4 representing the most supported evidence (meta-analyses, multiple clinical trials) and possesses the greatest clinical utility today. Scores of 3/4 represent advisable evidence (few clinical trials, supported in vitro/vivo). Scores of 2/4 represent potential or prospective findings that require further validation (in vitro and in vivo findings). Lastly, evidence receiving a score of 1/4 indicate shared relationships with few to no definitive studies, though still possess a shared risk or considerable outcome. Our CIS scoring system is visualized below:
4/4 Actionable → clinical relationship Meta-analyses, multiple clinical trials
3/4 Advisable → association is present One/few clinical trials, supported cell data
2/4 Prospective → needs confirmation In vitro/in vivo cell data
1/4 Unknown → shared risk Limited/no studies investigating relationship
Results
APC
Operating within the WNT/beta-catenin (canonical WNT) signaling pathway, altered APC protein expression leads to inappropriate “gatekeeping” of cell homeostasis resulting in overactivation of WNT signal transmission and successive accumulation of beta-catenin in the cytoplasm of cells. Upon translocation to the nucleus, beta-catenin exerts its effect as a gene transcription activator leading to cancer progression (Online Mendelian Inheritance in Man OMIM®, 2021a; Krausova and Korinek, 2014). Germline mutations in the APC gene cause Familial Adenomatous Polyposis (FAP), an autosomal dominant colon cancer predisposition syndrome affecting as many as 3.2 people per 100,000 individuals. FAP is known clinically for the development of hundreds to thousands of adenomatous polyps within the colon and is inherited with nearly complete penetrance. Colectomy is considered first line therapy in FAP and is an absolute indication in confirmed colon cancer or in presence of distressing symptoms. Currently, there are no FDA-approved chemopreventive therapies for the treatment of FAP (Jasperson et al., 1998).
EGFR-Inhibitors
Current FDA-approved medications for the treatment of metastatic colorectal cancer include two monoclonal antibodies (cetuximab, panitumumab) that selectively inhibit EGFR (epidermal growth factor receptor). A member of the Type I receptor tyrosine kinases, EGFR is a transmembrane glycoprotein known to operate in multiple signaling pathways including vascular endothelial growth factor production, cell growth, and induction of apoptosis. Cetuximab and panitumumab competitively inhibit epidermal growth factor binding, preventing the growth and survival of EGFR-expressing tumor cells (FDA, 2004). Clinical usage of the medication has been limited due to drug resistance to wild-type RAS subpopulations. Upon review of additional tumor gene subpopulations, APC gene status was identified as a key prognostic indicator of EGFR-inhibitor efficacy. APC and TP53 doubly mutated tumors showed the greatest sensitivity to EGFR-inhibitors, while mutated APC tumors also possessed a significant sensitivity compared to wild-type APC tumors, increasing the potential for wider clinical implementation in metastatic colorectal cancer therapy (Yang et al., 2019).
Tankyrase (TNKS)-Inhibitors
Members of the PARP family, tankyrase enzymes one and two play a key role in WNT signaling and beta-catenin transcriptional activation. Recognized as therapeutic targets, inhibition of these enzymes leads to reduced beta-catenin translocation and subsequent cell death in cells harboring APC mutations. Recent work by Schatoff et al. (2019) identified variable response rates to TNKS-inhibitors based upon tumor APC genotype. ‘Early’ truncating APC mutations were found to be highly resistant to TNKS inhibition, while variants within the mutation cluster region (amino acids 1250–1580) revealed sensitivity. These results differ from earlier studies (Tanaka et al., 2017), which proposed all APC variant cells were sensitive to TNKS-inhibitors. TNKS-inhibitors may offer patients possessing germline APC variants a unique therapeutic option beyond colon resection.
Aspirin and NSAIDs
Both NSAIDs and selective COX-2 inhibitors (COXIBs) are known to reduce the incidence and mortality of numerous cancers. Known primarily as critical enzymes in the eicosanoid pathway, Cyclooxygenase-1 (COX-1) and COX-2 complete the two catalytic steps required for the conversion of arachidonic acid into prostaglandin H2. Aspirin covalently and irreversibly binds to COX-1 and COX-2, while other NSAIDs reversibly inhibit these enzymes preventing the downstream signal from inducing inflammation, fever, and prothrombotic events (Kemp Bohan et al., 2020). Also found to contribute a role in cell cycle progression, increased COX-2 expression resulting from upregulation of the Wnt/B-catenin signaling pathway as a key contributing factor in the formation of gastrointestinal polyps. Thus, COX-2 has become a promising target for patients with FAP (McLean et al., 2008; Nuñez et al., 2011). Celecoxib was the first COXIB to be approved as adjuvant therapy for FAP patients by the Food and Drug Administration in 1999, but follow-up investigations revealed an array of toxicities with prolonged time courses, including gastrointestinal bleeding, renal dysfunction, cardiovascular death, myocardial infarction, and stroke (FDA, 2018; Kemp Bohan et al., 2020). Studies involving the use of aspirin as a chemopreventive therapeutic agent have yielded more promising results. In a randomized control trial, Burn et al. (2011) investigated a 600 mg aspirin daily regimen with and without starch in FAP patients. Though the study revealed no reduction in risk of colorectal polyps, there was a statistically significant reduction in polyp diameter in patients treated for at least 1 year. A recent randomized controlled trial in Japan reported FAP patients experienced safe and effective suppression of recurrence for colorectal polyps greater than 5.0 mm when treated with low dose aspirin (Ishikawa et al., 2021). Similarly, a recent meta-analysis estimates a nearly 20% reduction in mortality for all cancer patients who take aspirin (Elmwood et al., 2021). Additionally, for FAP patients experiencing duodenal polyps, a phase II study using a treatment regimen of sulindac and erlotinib resulted in a 71% reduction in duodenal polyp burden, while simultaneously reducing colorectal adenoma burden (Samadder et al., 2018). Aspirin represents a potentially viable option for chemopreventive treatment for FAP patients due to its suppressive effects on large polyp recurrence, mortality reduction, and well tolerated long-term safety profile.
BRCA1 and BRCA2
Operating within the homologous repair pathway, altered BRCA1/2 protein expression contributes to mismatched recruitment and protein complex formation leading to disrupted repair of double stranded DNA breaks (Cousineau et al., 2005). Mutations in the BRCA1 or BRCA2 genes result in the clinical presentation known as hereditary breast and ovarian cancer syndrome. It is estimated BRCA1 and BRCA2 mutation prevalence falls between 0.3% and 0.8%, while the prevalence of founder mutations may be even greater in certain populations (Paluch-Shimon et al., 2016). Estimated lifetime risk for developing breast cancer in individuals harboring these germline variants is upwards of 72% and 69% for BRCA1 and BRCA2 respectively. Similarly, the lifetime risk of developing ovarian cancer is up to 44% for BRCA1 and 17% for BRCA2 variants (Kuchenbaecker et al., 2017). Testing for unaffected BRCA1 and BRCA2 carriers has enhanced surveillance and prophylactic procedures (risk-reducing mastectomy and risk-reducing bilateral salpingo-oophorectomy) (Petrucelli et al., 1998).
Oral Contraceptives
The protective role of oral contraceptives (OCPs) in the risk reduction of ovarian cancer has been well documented in the general population. Studies investigating the role of BRCA1/2 in response to oral contraceptives revealed a similar but weaker risk reduction compared to the general population. Despite the protective effects, BRCA1/2 germline carriers are in a unique clinical conundrum as the use of OCPs possesses a theoretical increased risk of breast cancer. Because studies are unable to fully exclude an increased risk of breast cancer, use of OCPs must be carefully considered within the clinical context (Narod et al., 2002; McGuire et al., 2004; Whittemore et al., 2004; Iodice et al., 2010; Huber et al., 2020).
Clomiphene Citrate
Used clinically as an ovulatory stimulant, clomiphene citrate possesses an FDA label warning regarding prolonged usage increasing the risk of borderline or invasive ovarian tumor development. Though no direct study comparing the risk of clomiphene citrate and BRCA1/2 germline variants exists, epidemiologic studies support these findings in the general population. Because BRCA1/2 carriers already possess an underlying risk for ovarian cancer, use of clomiphene citrate in these patients must be carefully monitored and considered on a patient-by-patient basis (FDA, 2012; Reigstad et al., 2017).
Estrogen-Based Hormone Replacement Therapy
In order to reduce the risk of breast and ovarian cancer, BRCA1/2 germline carriers are encouraged to undergo risk-reducing bilateral salpingo-oophorectomy (rrBSO) upon completion of childbearing, leading to the induction of surgical menopause. Following surgery patients may experience postmenopausal symptoms, an indication for hormone replacement therapy. Studies have revealed that BRCA1/2 carriers possess no change or an even further decrease in breast cancer risk while using short-term HRT to control symptoms following surgical menopause (Eisen et al., 2008; Domchek et al., 2011; Kotsopoulos et al., 2016; Gordhandas et al., 2019). In summary, HRT remains contraindicated prior to prophylactic rrBSO in BRCA1/2 carriers due to an increased breast cancer risk. Following prophylactic rrBSO, short term use of HRT is reasonable and has not been demonstrated to increase risk for breast cancer. Long term use of HRT following prophylactic rrBSO is not advised.
Platinum Compounds and TNBC
Great strides have been made in delineating the most effective treatment method for triple negative breast cancer, tumors which are estrogen receptor (ER), progesterone receptor (PR), and HER2 negative. Further investigation has also identified a key relationship for platinum-based chemotherapies in patients harboring BRCA1/2 germline variants who develop TNBC. Multiple clinical studies reveal improved sensitivity and pathologic complete response to neoadjuvant cisplatin or carboplatin (Silver et al., 2010; Byrski et al., 2014; Hahnen et al., 2017; Caramelo et al., 2019). Similarly, TNBC patients with BRCA1/2 germline variants experienced significantly improved response to carboplatin when compared to the standard of care docetaxel treatment regimen (Tutt et al., 2018). The use of platinum-based agents provides patients with BRCA1/2 germline variants with triple negative breast cancer new avenues for future treatment regimens.
PARP-Inhibitors (PARPi)
This class of medications is currently FDA approved for advanced, previously treated ovarian cancer with germline BRCA1/2 variants and previously treated metastatic HER2-negative BRCA1/2 variant breast cancer. Additionally, olaparib possesses approval for first-line maintenance therapy in BRCA variant ovarian cancer as well as approval for maintenance therapy of BRCA1/2 variant pancreatic cancer (Robson et al., 2017; Litton et al., 2018; FDA, 2020a; Madariaga et al., 2020). PARP enzymes are crucial in repair of single-stranded DNA breaks, thus inhibition of these enzymes allows these breaks to persist. BRCA1/2 carriers are incredibly sensitive to PARP-inhibitors, as single-strand breaks are coupled with the failed double-strand DNA repair leading to cell cycle arrest or cell death. (Farmer et al., 2005). Multiple clinical trials have investigated the efficacy of PARPi as monotherapy and in conjunction with other chemotherapies, with promising disease-free survival and progression free survival data (Madariaga et al., 2020). In a phase III trial, germline BRCA-variant pancreatic cancer patients who were treated with maintenance olaparib experienced prolonged progression-free survival compared to placebo (Golan et al., 2019). Promising safety and efficacy profiles have also been shown in rucaparib-based regimens for BRCA1/2 variant pancreatic cancer (Reiss et al., 2021). As clinical studies continue to delineate the role of PARPi in cancer therapy, this class of medications provides clinicians treating patients with germline BRCA1/2 variants a vast potential of clinical utility.
STK11
Functioning as the primary control mechanism for AMP-activated protein kinase (AMPK) family members, STK11 regulates critical cellular mechanisms including metabolism, homeostasis, apoptosis, and cell polarity (The Human Protein Atlas, 2021f). Peutz-Jeghers Syndrome (PJS) is an autosomal dominant disease resulting from heterozygous pathologic variants in STK11 possessing nearly 100% penetrance. PJS presents clinically with hamartomatous polyps dispersed throughout the gastrointestinal tract in association with pigmented mucocutaneous macules around the mouth, eyes, nostrils, buccal mucosa, and perianal region. Disease prevalence has been difficult to ascertain, occurring in an estimated range of one in every 25,000 to 280,000 individuals (Tchekmedyian et al., 2013). Due to STK11 role as a tumor suppressor, patients with PJS also possess a significant risk for developing an array of malignancies including colorectal, breast, gastric, small bowel, pancreatic, ovarian, cervical, lung, thyroid, and gonadal tumors (McGarrity et al., 2001; Syngal et al., 2015).
PD-1 axis Inhibitors
Programmed death ligand 1 (PD-L1) expression is currently the only FDA approved biomarker in advanced lung adenocarcinoma, acting as a prominent therapeutic target. STK11 variants have been shown to down regulate PD-L1 expression, rendering PD-1 axis inhibitors futile. Skoulidis et al. (2015) first identified STK11 tumor variants as a key initiator in resistance to PD-1 axis inhibitors while co-occurring with KRAS mutations. In follow-up to their findings, Skoulidis et al. (2018) formalized the specific role of STK11 in driving resistance in KRAS- mutated lung adenocarcinoma and hypothesized similar findings for STK11 variants regardless of KRAS status. Recent studies have expanded this hypothesis, as STK11 variants with wild-type KRAS also confer resistance (Laderian et al., 2020; Schoenfeld et al., 2020). Though further clinical investigation is needed, PD-1 axis inhibitors are likely to be less effective in patients possessing STK11 variants.
ERK Inhibitors
In 2018 Ulixertinib, a ERK1/2 inhibitor, was the first of its class to undergo Phase one study in MAPK mutant advanced solid tumors (Sullivan et al., 2018). This medication is highly potent and selective, resulting in reversible ATP-competitive inhibition of ERK one and 2. Using STK11 variant NSCLC cell lines and engineered mouse models, Caiola et al. (2020) found tumors with STK11 mutants expressed distinct sensitivity to ERK inhibitors. Though ERK inhibitors have yet to be studied clinically in patients with deleterious STK11 variants, these findings are encouraging for further investigation. ERK inhibitors may offer a promising therapeutic option for patients harboring STK11 variants in the future.
TSC1 and TSC2
Operating within the mTOR cellular signaling pathway, altered hamartin (TSC1) or tuberin (TSC2) protein expression leads to unregulated activation of mTORC1, a key controller of anabolic cell growth and proliferation (The Human Protein Atlas, 2021g). Tuberous Sclerosis Complex (TSC) is an autosomal dominant disease resulting from heterozygous pathologic variants in either TSC1 or TSC2. Present in approximately one in 5,800 individuals, TSC predisposes patients to a wide array of clinical features involving the skin, brain, kidneys, eyes, heart, and lungs. (Northrup et al., 1999).
Estrogen-Based Medications and Contraceptives
Evidence has shown increased levels of estrogen can contribute to lymphangioleiomyomatosis (LAM) induction or progression. Because individuals with pathogenic TSC1/2 variants have an increased baseline risk for LAM, it is advised that estrogen-containing products be limited or avoided in these patients (Yano, 2002; Oberstein et al., 2003). Though no direct clinical relationship has been defined between TSC variants and estrogen products, studies have revealed the promotion of invasiveness and survival of tuberin-null cells when treated with estrogens (Yu et al., 2004; Yu et al., 2009).
TP53
The “guardian of the genome”, p53 responds to cellular stressors by initiating cell cycle arrest to allow for DNA replication and repair. p53 also contributes to apoptosis, autophagy, senescence, differentiation, antioxidant stress response, and cellular metabolism. In non-stressed cells, p53 activity is repressed through negative feedback binding by ubiquitin ligase MDM2 (Toledo and Wahl, 2006; Bourdon, 2007; Vousden and Lane, 2007). Li-Fraumeni spectrum disease (LFSD) is a high-risk cancer predisposition syndrome with patients developing a range of both childhood and adult-onset malignancies. Inherited in an autosomal dominant pattern, LFSD results from pathologic variants in TP53 with 7–20% of mutations developing de novo (Schneider et al., 1999). Though not well established, one study estimates disease prevalence at one in every 3,555 to 5,476 individuals (de Andrade et al., 2019). Lifetime cancer risk in LFSD is greater than 70% for men and greater than 90% for women. Unlike other cancer syndromes LFSD is not localized to site-specific cancers, manifesting clinically as a heterogeneous collection of cancers with the most common including soft tissue sarcomas, adrenocortical carcinomas, breast cancer, central nervous system tumors, and osteosarcomas (Guha and Malkin, 2017).
Genotoxic Chemotherapy agents
Kasper et al. (2018) identified both genotoxic chemotherapy agents (etoposide) and radiotherapy significantly increased the risk of tumor development in a Li-Fraumeni spectrum disease mice model. Their study also recognized that non-genotoxic agents such as docetaxel, a mitotic spindle poison, did not contribute to further tumor formation. In patients possessing a pathologic TP53 variant, it is advised that surgical and ablative therapies be given priority while avoiding radiotherapy and genotoxic chemotherapies when possible (Frebourg et al., 2020).
Chemotherapy and Chronic Lymphocytic Leukemia
Multiple studies have identified TP53 mutations and 17p deletions as independent drivers of resistance to chemotherapeutic agents in CLL (Döhner et al., 1995; Hallek et al., 2010). Positive effective results were first observed in studies investigating the use of alemtuzumab (monoclonal antibody-CD52) in CLL patients with poor prognostic features including TP53 disruptions (Lozanski et al., 2004; Pettitt et al., 2012). This medication was approved as first line therapy for CLL before rebranding of the drug as a multiple sclerosis medication by the manufacturer led to the withdrawal of the license in 2012 (Hallek 2019; FDA, 2021). As a result, new and varied treatment regimens have been investigated with promising outcomes. CLL patients with TP53 dysfunction have seen much improved but not definitive disease control with small molecule agents such as venetoclax (BCL-2 inhibitor) and ibrutinib (Bruton tyrosine kinase inhibitor), especially when used in combination with rituximab and obinutuzumab or idelalisib (Farooqui et al., 2015; Roberts et al., 2016; Stilgenbauer et al., 2016; Hallek 2019). The use of chemotherapy agents as first line therapies in this patient group is not supported.
Chemotherapy and Mantle Cell Lymphoma
TP53 variants are negative prognostic indicators for clinical outcomes in patients with MCL (Eskelund et al., 2017). TP53 mutated MCL cells escape eradication by high dose cytarabine in combination with rituximab, a standard treatment for general MCL patients. Patients harboring germline TP53 mutations who develop MCL may be less likely to benefit from a cytarabine-rituximab combination therapy.
Carboplatin and Breast Cancer
In a large cohort study of Chinese women with breast cancer, Sheng et al. (2020) identified patients possessing TP53 germline variants were more likely to respond to carboplatin-based (with or without anthracycline) neoadjuvant therapy compared to anthracycline or taxane-based regimens. Though future clinical studies are needed, these cohort findings represent a reasonable justification for choosing carboplatin-based therapy regimens in TP53-variant breast cancer.
5-FU and Colorectal Cancer
5-FU is a synthetic pyrimidine antagonist included as first-line therapy in colorectal cancer regimens. Once transformed intracellularly, 5-FU metabolites disrupt RNA synthesis, initiate single and double-stranded breaks, and inhibit thymidylate synthase contributing to cellular apoptosis. As both germline and sporadic TP53 variants increase cellular ability to avoid apoptosis initiation, studies have identified TP53 null cells as resistant to 5-FU (Aghabozorgi et al., 2020). Patients with TP53 germline variants who develop colon cancer may be less likely to benefit from 5-FU based therapies.
PTEN
Operating as a negative regulator of the PI3K/AKT pathway, altered PTEN protein expression leads to uninhibited AKT phosphorylation and subsequent inability to initiate apoptosis (Pezzolesi et al., 2007). PTEN hamartoma tumor syndrome (PHTS) is an autosomal dominant disease resulting from heterozygous pathologic variants in PTEN. PHTS encompasses five distinct clinical phenotypes, with each phenotypic syndrome including hamartomatous tumor formation and a germline PTEN pathologic variant. The most common phenotype seen in PTEN variants is Cowden syndrome, consisting of multiple hamartomas found on skin and GI tract, vascular abnormalities, mucocutaneous features including trichilemmomas, papillomatous papules, lipomas, fibromas, and a severe risk for benign and malignant tumors of the thyroid, breast, and endometrium (Blumenthal and Dennis, 2008). As a result of the wide range and insidious onset of clinical manifestations, the estimated disease prevalence of one in every 200,000 is likely underestimated (Nelen et al., 1999; Yehia and Eng, 2001).
Tamoxifen/Raloxifene
Though patients with PTEN variants possess an increased risk for breast cancer development, there is no direct evidence to support the use of tamoxifen or raloxifene for breast cancer prevention. Additionally, women with PTEN germline mutations have up to 28% lifetime risk for endometrial cancer (Tischkowitz et al., 2020), for which these agents are known to increase the risk. Physicians may consider alternative agents in patients with PTEN variants in order to limit the potential increased risk of developing endometrial cancer (Yehia and Eng, 2001; Hobert and Eng, 2009).
Trastuzumab and Lapatinib
Studies have identified PTEN protein function as a critical component in the therapeutic efficacy of trastuzumab. PTEN is necessary for rapid AKT-dephosphorylation by trastuzumab, contributing to its antiproliferative effect. Evidence also exists for a PTEN-related role in the mechanism of lapatinib, as PTEN knockout cell lines showed decreased sensitivity to clinically therapeutic levels of lapatinib (Nagata et al., 2004; Berns et al., 2007; Eichhorn et al., 2008; Wang et al., 2011). Future clinical studies investigating the efficacy of these agents in patients with PTEN variants are needed, though initial lab evidence indicates these agents may be less effective.
CDK 4/6 Inhibitors
Currently used as part of the standard of care in estrogen receptor-positive HER2-negative advanced breast cancer, CDK4 and CDK6 inhibitors function by preventing complex formation with D-cyclins leading to tumor suppressor retinoblastoma protein (Rb) dephosphorylation and subsequent cell cycle arrest. Both in vitro and in vivo models of PTEN loss revealed increased levels of rephosphorylated Rb and cell cycle progression when treated with CDK4/6 inhibitors. Though the use of CDK4/6 inhibitors in patients with PTEN variants needs to be studied further in clinical trials, current evidence does not support successful use of these agents in this patient population (Costa et al., 2020).
PI3K-Alpha inhibitors
Typically reserved for advanced breast cancer following CDK4/6 inhibitor resistance, PI3K-alpha inhibitors have been observed clinically and in cell line data to be ineffective when used in PTEN-null cells (Juric et al., 2015; Costa et al., 2020). Other agents may be more effective in patients with PTEN variants who develop advanced breast cancer.
Each of the described relationships above were identified in studies involving somatic/tumor driven PTEN variants and did not involve specific investigation of germline variants. Because breast cancer is one of the most common phenotypes in patients presenting with PHTS, we chose to include the discussions above. The in vitro and in vivo evidence supports the direct mechanistic link underlying how PTEN loss confers resistance to these drugs. Though future clinical evidence is needed relating specifically to germline variant patients, these medications may be less effective in PHTS patients.
VHL
Operating within the oxygen-sensing pathway, von Hippel-Lindau tumor suppressor protein (pVHL) is recognized as a primary regulator of hypoxia-inducible genes via ubiquitinylation and subsequent degradation of HIF-alpha during times of adequate oxygenation. Disease-causing mutations in VHL trigger a hypoxia-analogous condition within cells, leading to excessive levels of HIF-alpha and downstream transcriptional activation of glycolysis and erythropoiesis. Additionally, pVHL also plays a role in a multitude of critical cellular mechanisms including tumorigenesis, cilia formation, post-translational gene expression, apoptosis, and extracellular matrix formation (The Human Protein Atlas, 2021a). von-Hippel Lindau syndrome (VHL) is an autosomal dominant disease resulting from heterozygous pathologic variants of VHL, with 20% of these variants developing as de novo mutations. Present in approximately one in 36,000 individuals, VHL predisposes patients to an array of both benign and malignant tumors, most notably clear-cell renal cell carcinoma, pancreatic neuroendocrine tumors, and hemangioblastomas in the retina and throughout the central nervous system. Clinical findings and severity are extremely variable, even among families with an identical pathologic variant. Though variable, VHL pathologic variants are highly penetrant, as nearly all patients with a confirmed variant are symptomatic by 65 years of age with the mean age of initial presentation of 26 years old (van Leeuwaarde et al., 2018). In addition to known clinical findings, further complications may also occur as angioma and hypertension increase the risk of subarachnoid hemorrhage and cerebellar hemangioblastoma or hypernephroma-like tumors in the kidney predispose patients to polycythemia (Online Mendelian Inheritance in Man OMIM®, 2021b).
HIF-2a Inhibitors
Ubiquitously upregulated in VHL-variant tumors, HIF-2a is a key proximal signal stimulating tumor growth and has rapidly become a target of interest for potential systemic therapies for VHL patients. Pre-clinical and clinical studies have produced encouraging results, revealing safety and efficacy in patients with advanced clear cell renal cell carcinoma (Chen et al., 2016; Courtney et al., 2020; Choueiri et al., 2021). A phase II study was recently published investigating the objective response of VHL patients with renal cell carcinoma to belzutifan (MK-6482) treatment regimen. Jonasch et al. (2021) reported an objective partial response in 49% of patients, with an additional 49% of patients with stable disease. 96% of all patients experienced progression-free survival at 24 months. Remarkably, the study also reports partial or complete responses in additional VHL-associated neoplasms present within the study population. 77% of patients with pancreatic lesions, 91% of patients with pancreatic neuroendocrine tumors, and 30% of patients with central nervous system hemangioblastomas experienced a response following treatment with belzutifan. Additionally, all retinal hemangioblastomas present in the study population were graded as showing improvement. With only grade I and II adverse events reported, belzutifan has the ability to transform care for VHL patients providing a potential treatment option beyond surgical excision. This systemic therapy targets the underlying pathophysiology of the disease and thus far has shown to effectively halt or reduce VHL-associated tumors throughout the body with minimal side effects.
MUTYH
The mutant Y homolog E. coli (MUTYH) gene encodes adenine DNA glycosylase necessary for the single-base excision repair and proper initiation of single-strand DNA breaks following oxidative damage leading to apoptosis (The Human Protein Atlas, 2021b). MUTYH Associated-Polyposis (MAP) is an autosomal recessive disease resulting from biallelic germline pathologic variants in MUTYH presenting as homozygotes or compound heterozygotes and occurs in an estimated one in every 20,000 to 60,000 individuals (Al-Tassan et al., 2002; Cleary et al., 2009; Win et al., 2017). Patients with MAP possess a significantly increased risk for developing colorectal polyps and cancer throughout their lifetime. Patients frequently develop between 10 and 100 polyps by the fifth or sixth decade of life, most commonly as adenoma-type, though hyperplastic and serrated polyps have also been identified (Boparai et al., 2008; Grover et al., 2012). It is estimated that 43%–63% of patients develop malignancy by 60 years old, with a median age of onset at 48 years old. MAP has also been reported to increase risk of other malignancies including bladder, ovarian, duodenal, breast, hepatobiliary, gastric, endometrial, and skin cancers (Vogt et al., 2009; Win et al., 2016).
Alkylating Agents
Fry et al. (2008) demonstrated that MUTYH knockout cells were less responsive upon exposure to a DNA alkylating agent. Due to MUTYH role in DNA base repair, alkylating agents are unable to successfully confer cytotoxicity through the formation of DNA crosslinks, escaping cell death. Though typically reserved for use in salvage regimens, alkylating agents play an important role in the treatment of ovarian cancer (Hutchcraft et al., 2021). Further study is necessary to determine whether individuals with ovarian cancer and biallelic germline pathologic variants in MUTYH demonstrate this potential resistance to alkylating agents.
RET
The RET gene specifically encodes the RET (Rearranged during Transfection) protein, a transmembrane receptor tyrosine kinase belonging to the cadherin superfamily necessary for cell growth, differentiation, migration, and survival (The Human Protein Atlas, 2021c; Takahashi et al., 1998). Medullary thyroid carcinoma (MTC) is a malignant tumor originating in the parafollicular C-cells of the thyroid resulting from pathologic RET variants, marked by drastic increases in calcitonin levels. MTC may be sporadic or hereditary and presents in a range of clinical syndromes known collectively as MEN2. A 2004 study estimates MEN2 occurs in one in every 35,000 individuals (DeLellis et al., 2004). Three clinical subtypes make up MEN2: multiple endocrine neoplasia type 2A and 2B (MEN2A and MEN2B, respectively) and familial MTC (FMTC). Patients with germline RET variants are encouraged to avoid dopamine D2 agonists and beta-adrenergic antagonists until pheochromocytoma is sufficiently ruled out, as these agents may trigger adverse events (Eng, 1999).
GLP-1 Agonists
Used to improve glycemic control in patients with type II diabetes mellitus, glucagon-like peptide 1 (GLP-1) agonists possess a black-box warning for risk of causing thyroid C-cell tumors. Though the clinical effects in humans are unknown, C-cell tumors developed in dose-dependent and duration-dependent manner in both genders of mice and rats but not primates (Bjerre Knudsen et al., 2010). This class of medications is contraindicated in patients with a personal or family history of MTC or MEN2 syndromes (FDA, 2010). Though studies document a GLP-1 receptor dependent role in C-cell hyperplasia and no association with RET activation, the FDA label remains (Madsen et al., 2012).
Tyrosine Kinase Inhibitors
In 2011, the FDA approved the multi-target TKI vandetanib for symptomatic or progressive MTC with unresectable locally advanced or metastatic disease. The phase III study, which showed an objective response in 46.4% of hereditary RET-variant MTC, was the beginning of a cascade of investigation into this class of medications (Wells et al., 2012; FDA, 2014). Similarly cabozantinib, also a multi-target TKI, was approved by the FDA for the treatment of progressive, metastatic MTC (Elisei et al., 2013; FDA, 2014). A phase II study involving the use of sunitinib in the treatment of advanced/metastatic paraganglioma and pheochromocytoma found one patient with MEN2A and a germline RET variant experienced a 64% tumor volume reduction (O'Kane et al., 2019). Follow up studies involving these and other multi-target TKIs (sunitinib, sorafenib, lenvatinib, anlotinib) have produced a common negative trend, as many of these medications increase progression-free survival without reduction in tumor size and many patients go on to develop disease resistance over time while experiencing a myriad of toxicities (Okafor et al., 2021). As a result, the development and evaluation of highly selective RET-specific TKIs (selpercatinib and pralsetinib) are on the forefront of clinical investigation and are showing promise in preliminary studies. In their phase I/II study, Wirth et al. (2020) revealed a 69% response rate in patients with RET-variant MTC who had undergone prior treatment with vandetanib or cabozantinib and a 73% response rate in patients with no prior TKI therapy history when treated with selpercatinib. Subbian et al. (2021) reported similar findings for RET-variant MTC patients using a pralsetinib treatment regimen, as 60% of formerly treated patients experienced a response and 71% of patients without prior treatment experienced a response. Both drugs possess a much more tolerable side effect profile than the multi-target TKIs as a result of their specificity. Though isolated reports of acquired tumor resistance drive concerns regarding the long-term efficacy of RET-selective agents, future follow-up studies will be needed to evaluate the viability of prolonged treatment courses. RET-selective TKIs offer patients harboring RET-variant related disease a much needed option for systemic therapy, providing both substantial tumor responses and a tolerable toxicity profile.
SDHAF2, SDHB, SDHC, SDHD
Succinate dehydrogenase (SDH) is a multi-protein complex functioning in both the tricarboxylic acid cycle and mitochondrial electron transport chain. Four genes required for appropriate assembly and function of SDH include SDHAF2, SDHB, SDHC, and SDHD. Hereditary paraganglioma-pheochromocytoma syndrome (HPP) is a tumor predisposition syndrome caused by germline heterozygous pathogenic variants in SDHAF2, SDHB, SDHC, SDHD, MAX, SDHA, and TMEM127. HPP is inherited in an autosomal dominant manner in all genes except for SDHD and SDHAF2 which are maternally imprinted (Else et al., 2008). Patients with these genetic variants present clinically with a paraganglioma or pheochromocytoma and are more likely to be multifocal, recurrent, metastatic and occur at an earlier age in comparison to sporadic tumor formation (Lenders et al., 2014). Patients primarily experience symptoms related to catecholamine excess or by mass effect depending on the tumor’s location and ability to secrete catecholamines (Else et al., 2008). Patient phenotypes vary according to which gene is altered. SDHB germline variants are most notably associated with the highest morbidity and mortality, commonly developing extra-adrenal sympathetic paragangliomas with the greatest risk for metastatic disease (Andrews et al., 2018).
Temozolomide
Used in the management of metastatic paragangliomas and pheochromocytomas, evidence has shown patients with SDHB variants respond remarkably well to temozolomide-based regimens (Hadoux et al., 2014). Recent studies in SDHB-knockout mouse models showed improved cytotoxicity, reduced metastatic lesions, and prolonged overall survival when treated with temozolomide coupled with a PARP-inhibitor (Pang et al., 2018). The use of temozolomide should be highly considered in patients with SDHB variants who develop metastatic paragangliomas and pheochromocytomas.
Tyrosine Kinase Inhibitors
TKIs can effectively treat KIT or PDGFRA mutated gastrointestinal stromal tumors (GISTs). However, SDHx-deficient GISTs display a poor response when treated with imatinib and limited response when treated with sunitinib (Boikos et al., 2016). There has been minimal investigation into alternative TKIs (Ibrahim and Chopra, 2020). Though these tumors possess a low overall mortality, SDHx-deficient GISTs are prone to progress and recur (Weldon et al., 2017). On the other end of the spectrum, case reports have shown quality responses in SDHx-null renal cell carcinoma to sunitinib and pazopanib (Paik et al., 2014; Shuch et al., 2016). Similarly, the role of TKIs in the treatment of advanced/metastatic paraganglioma and pheochromocytoma is encouraging, with evidence of a patient harboring an SDHB variant responding to sunitinib-based therapy (O'Kane et al., 2019). While further studies are needed to support the findings in both disease states, TKIs offer patients with SDHx-variant RCC or paraganglioma/pheochromocytoma a potential future therapeutic option when few others exist.
Because of the small sample size most of these studies generalize findings to include all SDHx variants. Specifically, Boikos study included SDHB and SDHA, Shuch included SDHC, Paik studied SHDB, and O’Kane reviewed SDHB and SDHA.
BMPR1A and SMAD4
Bone Morphogenetic Protein Receptor Type 1A (BMPR1A) is a member of the TGF-beta superfamily of receptors, which upon ligand binding forms a receptor complex of transmembrane serine/threonine kinases, triggering downstream SMAD protein activation (Howe et al., 2001). SMAD4 plays a crucial role in transcription regulation and once activated, SMAD family proteins form complexes to bind to and control transcription of target genes (The Human Protein Atlas, 2021d). Juvenile polyposis syndrome (JPS) is an autosomal dominant disease resulting from heterozygous loss of function pathogenic variants in SMAD4 or BMPR1A. JPS presents clinically as a predisposition for developing multiple hamartomatous polyps throughout the gastrointestinal tract. The occurrence of JPS is largely unknown, with an estimated prevalence ranging from one in every 16,000 to 100,000 (Larsen Haidle and Howe, 2003). A variation of this syndrome is present with mixed clinical findings of both JPS and hereditary hemorrhagic telangiectasia including epistaxis, telangiectasia, arteriovenous malformations, and digital clubbing. Patients with SMAD4 pathogenic variants have also been found to develop thoracic aortic aneurysms and aortic dissections (Wain et al., 2014). On the other hand, gain of function mutations in SMAD4 have been associated with a different clinical presentation, a complex connective tissue disorder known as Myhre syndrome (Caputo et al., 2012). Symptoms of Myhre syndrome frequently include extensive proliferative fibrosis and intellectual disability, and with phenotypes displaying varying involvement of the cardiovascular, respiratory, gastrointestinal, and skin systems (Starr et al., 2021).
5-Fluorouracil
First recognized as a poor prognostic factor in colorectal cancer, more recent studies have identified a correlation between SMAD4 expression and 5-FU sensitivity. In patients with colorectal cancer treated with 5-FU, those with SMAD4 loss displayed a significant decrease in both overall survival and progression free survival (Alhopuro et al., 2005; Papageorgis et al., 2011; Wasserman et al., 2019). For patients harboring SMAD4 variants and requiring adjuvant chemotherapy, options other than 5-FU may likely be more effective.
Cetuximab
Multiple studies have established an association between SMAD4 protein expression and sensitivity to cetuximab therapy. Lin et al. (2019) showed SMAD4 knockdown tumor cell lines were more viable and inhibited cell apoptosis when treated with cetuximab in comparison to cells with overexpression of SMAD4. Similar findings were observed in somatic SMAD4 loss of head and neck squamous cell carcinoma, conferring resistance to cetuximab therapy (Ozawa et al., 2017). Other agents are likely to be more successful than cetuximab in treating germline SMAD4 variant pathologies.
Irinotecan
Wong et al. (2020), while also conferring resistance of SMAD4-negative cell lines to 5-FU, found these same lines also conferred resistance to irinotecan. Conversely, the study found no resistance to therapeutic oxaliplatin levels. Patients with colorectal cancer harboring SMAD4 pathogenic variants may obtain more benefit from adjuvant chemotherapies that do not include irinotecan.
Fluoroquinolones
Patients harboring SMAD4 variants are known to possess an increased risk for collagen-associated disorders, including aortic aneurysm or aortic dissection (Meng et al., 2019; Yu et al., 2019). Though no direct relationship has been established between fluoroquinolones and SMAD4 pathogenic variants, both are associated with an increased risk for aortic aneurysms and dissections. Because patients with pathogenic SMAD4 variants commonly have one or more features of hereditary hemorrhagic telangiectasia (HHT), these patients may potentially increase their risk for developing severe aortic symptoms when taking fluoroquinolones.
MLH1, MSH2, MSH6, PMS2
Key in initiating DNA repair, MSH2 and MSH6 form the heterodimer MutS alpha which recognizes and binds to mismatched dsDNA. Following binding, MutS alpha recruits a second heterodimer MutL alpha formed by MLH1 and PMS2. The coupled complex activates the endonuclease activity of PMS2 to induce single strand breaks and recruits DNA polymerase III to the site of DNA mismatch (The Human Protein Atlas, 2021e). Lynch Syndrome is an autosomal dominant disease caused by heterozygous germline mutations in MLH1, MSH2, MSH6, PMS2 or an EPCAM deletion, occurring in one of every 279 individuals (Win et al., 2017). Characterized primarily by an increased risk for colorectal cancer, Lynch Syndrome also increases the risk of cancers of the endometrium, ovaries, stomach, small bowel, urinary tract, brain, pancreas, and prostate (Dominguez-Valentin et al., 2020). Patients present with unique clinical phenotypes correlating to each of the associated Lynch Syndrome genes. MLH1 variants possess the greatest risk for colorectal cancer, with evidence supporting a younger age of onset and increased risk for developing multiple primary tumors (Pinto et al., 2018). Conversely, MSH2 variants are frequently associated with extracolonic cancers and are more likely to present as the Muir-Torre version of Lynch Syndrome (Everett et al., 2014).
Topoisomerase Inhibitors
Fedier et al. (2001) found MLH1 and MSH2- deficient cell lines conferred resistance to topoisomerase II inhibitors (epirubicin, doxorubicin, and mitoxantrone). MLH1 loss has also been demonstrated to display resistance to topoisomerase I poisons. Etoposide resistance was also identified in previous studies in using MLH1 and MSH2 deficient colorectal and endometrial carcinoma cell lines respectively (Aebi et al., 1997). For patients possessing germline MLH1 or MSH2, use of topoisomerase-blocking agents are less likely to produce cell death.
Platinum Agents
Binding to DNA and creating DNA adducts, platinum agents produce crosslinks disrupting the DNA helix. These agents rely on a proper functioning mismatch repair system to identify damaged DNA and induce apoptosis. Studies have documented the role of MLH1 and MSH2 loss in the development of cisplatin and carboplatin resistance in colorectal carcinoma, as well as PMS2 loss conferring low level cisplatin resistance (Aebi et al., 1997; Fink et al., 1997a; Fink et al., 1997b; Martin et al., 2008). Similar resistance to cisplatin was also found in MLH1-silenced endometrial cell lines and MSH2-silenced bladder cancer cell lines (Li et al., 2018; Goodspeed et al., 2019). On the other hand, evidence has shown colorectal cancer cells possessing disrupted MLH1 and MSH6 confer similar sensitivity to oxaliplatin as wild-type cells (Fink et al., 1997a; Vaisman et al., 1998). When initiating chemotherapy regimens that include platinum agents, patients harboring MLH1 and MSH2 variants may be more successful with oxaliplatin over cisplatin or carboplatin-based regimens.
5-Fluorouracil
Though not all studies agree, most clinical findings indicate MLH1 and MSH2 deficiencies contribute to poorer outcomes with 5-FU treatment for colorectal cancer (Ribic et al., 2003; Sargent et al., 2010). Similar findings have been reported in multiple cell line studies, supporting evidence of resistance to 5-FU (Carethers et al., 1999; Meyers et al., 2001). Typically used as the standard of care for stage II and III colorectal cancer, 5-FU based regimen may not be as effective in patients with MLH1 and MSH2 variants.
Alkylating agents
Multiple studies have evaluated and identified high degrees of resistance from MLH1, MSH2, and MSH6 deficient tumor cell lines to temozolomide (Friedman et al., 1997; Taverna et al., 2000; Nguyen et al., 2014). Evidence for other alkylating agents such as cyclophosphamide and busulfan have also been implicated, though results are not consistent. Though further studies are needed to specifically address the role of these agents in patients with germline variants, the aforementioned studies present evidence of resistance in patient tumor samples indicating that these agents may be less likely to be effective.
Aspirin and NSAIDs
Like FAP patients, Lynch Syndrome patients have also seen quality outcomes when treated with aspirin and NSAIDs. In a long-term follow-up to their 2011 CAPP2 study, Burn et al. (2020) showed that aspirin provides significant protective effects against colorectal cancer in Lynch Syndrome patients for an upwards of 20 years. Daily aspirin use for at least 2 years produced a reduced hazard ratio of 0.65 and 0.56 for intention-to-treat and per-protocol analysis, respectively. Ouakrim et al. (2015) produced related results when investigating both aspirin and ibuprofen. In their study, Lynch Syndrome patients treated with aspirin for 1 month were estimated to experience a 60% decrease in colon cancer risk and a 65% decrease in risk when treated with 1 month of ibuprofen. A recent phase I study found naproxen was safe for chronic use as a primary and secondary chemopreventative intervention at both low and high doses in Lynch syndrome patients. Additionally, a co-occurring tissue-specific mouse model showed naproxen modulated tumor growth and prolonged survival (Reyes-Uribe et al., 2021). Currently underway are two additional randomized clinical trials, CAPP3 and AAS-Lynch, which share a primary endpoint of comparing the effectiveness of varying aspirin doses in preventing colorectal cancer in Lynch Syndrome patients (Soualy et al., 2020).
Discussion
This investigation into the relationships existing between genetic data and therapeutic outcomes is the tip of the iceberg in the continued pursuit of personalized medical decision making. The genes presented above represent many of the most common cancer predisposition syndromes, each with potentially actionable pharmacologic considerations. In total, 54 relationships were identified and scored according to our confidence scale (Table 1). Our work goes hand-in-hand with two National Institute of Health initiatives already underway: CPIC and PharmGKB. Both initiatives provide peer-reviewed, evidence-based translation of research findings into clinically actionable decision making in gene-drug pairs. Our review works to build on the understanding of gene-drug relationships by also considering the disease component, as clinical phenotypes also shape potential interactions.
Currently, CPIC has published clinical decision-making guidelines for more than 440 unique gene-drug relationships, largely consisting of drug metabolizers and transport genes. Of note, CPIC has also developed guidelines for germline variants of RYR1 and CACNA1S, presenting clinically as malignant hyperthermia (inherited, familiar syndrome). The publication of these genes marks a giant leap forward in developing gene variant-specific clinical decision support. Our study hopes to contribute to this area of expertise, as CPIC does not yet have any existing guidelines created for the disease genes investigated in our study. There is also great potential for investigation into other non-cancerous germline familial syndromes, specifically relating to cardiology and inborn errors of metabolism.
Similarly, of the 100 different pharmacologic agents listed in the United States Food and Drug Administration Table of Pharmacogenomic Biomarkers in Drug Labeling, only eight drugs include labeling for any of the 23 genes included in this study (also described in Table 2). By taking a broad and pre-emptive approach, our study was able to include relationships at the gene level, make connections to other FDA drug label associations, and provide discussion on difficult clinical context decision making.
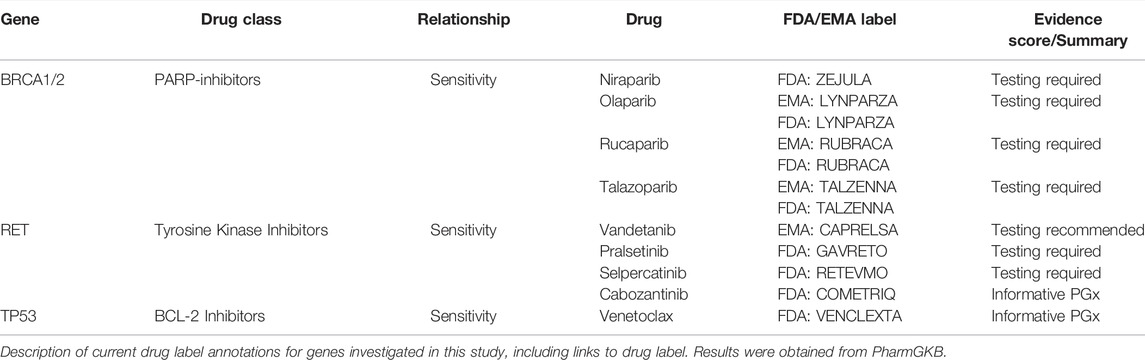
TABLE 2. PharmGKB drug label annotations.
As the field of pharmacogenetics continues to progress, clinical use of genomic information has made precision medicine an everyday reality. Yet, much of the available clinically actionable pharmacologic cancer specific evidence pertains to somatic or secondary treatment resistant tumor markers. In 2014, Wheeler et al. was a leader in reporting specifically on cancer pharmacogenomics, delineating evidence for gene-drug relationships including genes TMPT, UGT1A1, CYP2D6, and SLC O 1B1. Pasternak et al. (2017) investigated how germline variants affect disease outcome and severity as well as drug response. Their work summarized available evidence to provide discussion on eight individual pharmacogenes and their related drug implications. Lauschke et al. (2017) also provided a similar overview of genomic biomarkers as predictors of drug response and included approved targeted cancer drugs. Wellmann et al. (2018) investigated similar ends through a different approach, reviewing 125 oncology drugs for positive germline pharmacogenomic associations. Their work resulted in 12 individual clinical decision support summaries, six for prior established FDA pharmacogenomic associations and six for novel relationships. Our study set out to build upon these studies from a unique and alternative perspective, comprehensively review existing literature on an individual gene-by-gene basis. Our thorough investigation specifically focused on germline variants possessing a cancer-predisposition and included discussion of both current and prospective relationships.
Our study also included a comprehensive review of the MEN1 and RB1 genes, which yielded limited evidence of pharmacologic relationships with these germline variants. As mentioned below, evidence shows each of these genes possess an increased sensitivity to radiation which may help guide clinical decision-making. Further investigation into potential therapies, clinical outcomes, and pharmacologic sensitivities for patients with these conditions is warranted.
One complex relationship shared among many of the cancer predisposition genes reviewed in this study involves the role of radiation-based imaging and radiation-based therapy. Studies revealing poor outcomes following radiation exposure exist for patients with germline mutations in TSC1/2, TP53, MUTYH, SMAD4, MLH1, and RB1, with additional recommendations for conservative approaches taken in MEN1, STK11, and all pediatric populations (Wong et al., 1997; Matsumura et al., 1998; Evans et al., 2006; Tokairin et al., 2006; Kleinerman et al., 2007; Kleinerman 2009; Lenders et al., 2014; Wang et al., 2018; Lavergne et al., 2020). Limiting radiation exposure is a well understood clinical practice. Our findings offer evidence that patient populations with specific underlying germline variants are more sensitive to radiation exposure than the general public, requiring careful clinical consideration and observation for secondary primary tumor development following exposure. Many recommendations for surveillance scans with MRI or nuclear-based imaging over radiation-based exist. The use of radiation therapy is not uniform, as clinical usage varies upon patient presentation, tumor pathology, and tumor location. In many circumstances, surgical management must first be ruled out prior to the use of radiation therapy in cancer predisposition patients.
An additional but separate finding related to our study involves the delicate relationship between multiple pharmacologic agents and pheochromocytoma. As a catecholamine producing tumor, pheochromocytomas have the ability to produce severe adverse hemodynamic effects in the presence of surgical anesthesia, beta-receptor antagonists, dopamine receptor antagonists, sympathomimetics, antidepressants, and corticosteroid hormones (Eisenhofer et al., 2007). FDA labels for metoclopramide and prochlorperazine each include contraindications for use in the setting of a pheochromocytoma, as case clinical reports the development of hypertensive crisis in these patients. Though this relationship is not specifically related to a germline variant, RET, SDHB, SDHC, SDHD, and SDHAF2 each include a predisposition of developing pheochromocytoma and thus must be considered. Evaluation and ruling out of pheochromocytoma prior to initiating therapy with any of the agent discussed can prevent serious adverse events in these patients.
The greatest strength of our study was the uniqueness of our approach. We believe our study was the first of its kind to specifically seek out both prospective and current evidence for gene-drug relationships on a gene-by-gene basis. This approach allowed us to thoroughly investigate each of the 23 oncologic genes listed in the ACMG59, to ultimately provide clinicians with a succinct and comprehensive discussion on the potential gene-drug relationships for their patients. There is also great potential to build upon our study, as similar gene-drug relationships exist in cancer predisposition genes beyond the scope of the ACMG59 list. The NCCN guidelines include additional genes, such as PALB2, which has been investigated alongside BRCA1/2. Clinical trials recognize germline PALB2 variants as a sensitive target for PARP-inhibitors in platinum-sensitive advanced pancreatic cancer (Reiss et al., 2021).
We also recognize our study possesses some limitations. Due to the rarity of many of the investigated germline variants, few large-scale clinical trials exist. A large majority of the included studies involve cohort investigations of regionally based populations. Additionally, as mentioned above, some clinical relationships are expanded from evidence involving tumor-collected variant studies. These studies were included on the basis that the germline variant syndrome possesses an increased risk of developing a similar clinical phenotype as the study, and thus by harboring a similar genetic variant is likely to experience a related outcome.
Conclusion and Future Approaches
The future of genetic information in the clinical setting is an exciting and challenging opportunity. Through further investigation into the wide expanse of genetics, we can continue to drive forward the ability to generate highly individualized DNA-level care for all patients. By continuing to work collaboratively with evidence-based organizations, we can help provide clinicians with the critical information they need to best treat their patients. Future studies will be needed to validate and further delineate therapies and outcomes specific to genetic variants. Clinical trials differentiating outcomes based upon genetic variants within the study population will be needed to build upon existing evidence. Similarly, as further data is uncovered, clinical decision support documentation will be needed to provide clear and definitive language for providers.
Author Contributions
All authors contributed to the initial conception and design of the study. AS conducted the background research and drafted the manuscript. All authors contributed to the manuscript revision, read, and have approved the final version.
Funding
This work was supported by a generous gift from T Denny Sanford, establishing Sanford Imagenetics (merging Internal Medicine and Genetics).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors wish to acknowledge Katherine Kessel for her helpful assistance with formatting references.
References
Aebi, S., Fink, D., Gordon, R., Kim, H. K., Zheng, H., Fink, J. L., et al. (1997). Resistance to Cytotoxic Drugs in DNA Mismatch Repair-Deficient Cells. Clin. Cancer Res. 3, 1763–1767.
Aghabozorgi, A. S., Sarabi, M. M., Jafarzadeh-Esfehani, R., Koochakkhani, S., Hassanzadeh, M., Kavousipour, S., et al. (2020). Molecular Determinants of Response to 5-Fluorouracil-Based Chemotherapy in Colorectal Cancer: The Undisputable Role of Micro-ribonucleic Acids. Wjgo 12, 942–956. doi:10.4251/wjgo.v12.i9.942
Al-Tassan, N., Chmiel, N. H., Maynard, J., Fleming, N., Livingston, A. L., Williams, G. T., et al. (2002). Inherited Variants of MYH Associated with Somatic G: C-->T: A Mutations in Colorectal Tumors. Nat. Genet. 30 (2), 227–232. doi:10.1038/ng828
Alhopuro, P., Alazzouzi, H., Sammalkorpi, H., Dávalos, V., Salovaara, R., Hemminki, A., et al. (2005). SMAD4 Levels and Response to 5-fluorouracil in Colorectal Cancer. Clin. Cancer Res. 11, 6311–6316. doi:10.1158/1078-0432.CCR-05-0244
Andrews, K. A., Ascher, D. B., Pires, D. E. V., Barnes, D. R., Vialard, L., Casey, R. T., et al. (2018). Tumour Risks and Genotype-Phenotype Correlations Associated with Germline Variants in Succinate Dehydrogenase Subunit Genes SDHB, SDHC and SDHD. J. Med. Genet. 55, 384–394. doi:10.1136/jmedgenet-2017-105127
Bernard, P. S., Wooderchak-Donahue, W., Wei, M., Bray, S. M., Wood, K. C., Parikh, B., et al. (2021). Potential Utility of Pre-emptive Germline Pharmacogenetics in Breast Cancer. Cancers 13, 1219. doi:10.3390/cancers13061219
Berns, K., Horlings, H. M., Hennessy, B. T., Madiredjo, M., Hijmans, E. M., Beelen, K., et al. (2007). A Functional Genetic Approach Identifies the PI3K Pathway as a Major Determinant of Trastuzumab Resistance in Breast Cancer. Cancer Cell. 12, 395–402. doi:10.1016/j.ccr.2007.08.030
Bjerre Knudsen, L., Madsen, L. W., Andersen, S., Almholt, K., de Boer, A. S., Drucker, D. J., et al. (2010). Glucagon-like Peptide-1 Receptor Agonists Activate Rodent Thyroid C-Cells Causing Calcitonin Release and C-Cell Proliferation. Endocrinology 151, 1473–1486. doi:10.1210/en.2009-1272
Blumenthal, G. M., and Dennis, P. A. (2008). PTEN Hamartoma Tumor Syndromes. Eur. J. Hum. Genet. 16, 1289–1300. doi:10.1038/ejhg.2008.162
Boikos, S. A., Pappo, A. S., Killian, J. K., LaQuaglia, M. P., Weldon, C. B., George, S., et al. (2016). Molecular Subtypes of KIT/PDGFRA Wild-type Gastrointestinal Stromal Tumors: A Report from the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA Oncol. 2, 922–928. doi:10.1001/jamaoncol.2016.0256
Boparai, K. S., Dekker, E., Van Eeden, S., Polak, M. M., Bartelsman, J. F., Mathus-Vliegen, E. M., et al. (2008). Hyperplastic Polyps and Sessile Serrated Adenomas as a Phenotypic Expression of MYH-Associated Polyposis. Gastroenterology 135, 2014–2018. doi:10.1053/j.gastro.2008.09.020
Bourdon, J.-C. (2007). p53 and its Isoforms in Cancer. Br. J. Cancer 97, 277–282. doi:10.1038/sj.bjc.6603886
Burn, J., Bishop, D. T., Chapman, P. D., Elliott, F., Bertario, L., Dunlop, M. G., et al. (2011). A Randomized Placebo-Controlled Prevention Trial of Aspirin And/or Resistant Starch in Young People with Familial Adenomatous Polyposis. Cancer Prev. Res. 4, 655–665. doi:10.1158/1940-6207.CAPR-11-0106
Burn, J., Sheth, H., Elliot, F., Reed, L., Macrae, F., Mecklin, J., et al. (2020). Cancer Prevention with Aspirin in Hereditary Colorectal Cancer (Lynch Syndrome), 10-year Follow-Up and Registry-Based 20-year Data in the CAPP2 Study: a Double-Blind, Randomised, Placebo-Controlled Trial. Lancet 395, 1855–1863. doi:10.1016/S0140-6736(20)30366-4
Byrski, T., Huzarski, T., Dent, R., Marczyk, E., Jasiowka, M., Gronwald, J., et al. (2014). Pathologic Complete Response to Neoadjuvant Cisplatin in BRCA1-Positive Breast Cancer Patients. Breast Cancer Res. Treat. 147, 401–405. doi:10.1007/s10549-014-3100-x
Caiola, E., Iezzi, A., Tomanelli, M., Bonaldi, E., Scagliotti, A., Colombo, M., et al. (2020). LKB1 Deficiency Renders NSCLC Cells Sensitive to ERK Inhibitors. J. Thorac. Oncol. 15, 360–370. doi:10.1016/j.jtho.2019.10.009
Caputo, V., Cianetti, L., Niceta, M., Carta, C., Ciolfi, A., Bocchinfuso, G., et al. (2012). A Restricted Spectrum of Mutations in the SMAD4 Tumor-Suppressor Gene Underlies Myhre Syndrome. Am. J. Hum. Genet. 90, 161–169. doi:10.1016/j.ajhg.2011.12.011
Caramelo, O., Silva, C., Caramelo, F., Frutuoso, C., and Almeida-Santos, T. (2019). The Effect of Neoadjuvant Platinum-Based Chemotherapy in BRCA Mutated Triple Negative Breast Cancers -Systematic Review and Meta-Analysis. Hered. Cancer Clin. Pract. 17, 11. doi:10.1186/s13053-019-0111-y
Carethers, J. M., Chauhan, D. P., Fink, D., Nebel, S., Bresalier, R. S., Howell, S. B., et al. (1999). Mismatch Repair Proficiency and In Vitro Response to 5-Fluorouracil. Gastroenterology 117, 123–131. doi:10.1016/s0016-5085(99)70558-5
Chen, W., Hill, H., Christie, A., Kim, M. S., Holloman, E., Pavia-Jimenez, A., et al. (2016). Targeting Renal Cell Carcinoma with a HIF-2 Antagonist. Nature 539, 112–117. doi:10.1038/nature19796
Choueiri, T. K., Bauer, T. M., Papadopoulos, K. P., Plimack, E. R., Merchan, J. R., McDermott, D. F., et al. (2021). Inhibition of Hypoxia-Inducible Factor-2α in Renal Cell Carcinoma with Belzutifan: a Phase 1 Trial and Biomarker Analysis. Nat. Med. 27, 802–805. doi:10.1038/s41591-021-01324-7
Cleary, S. P., Cotterchio, M., Jenkins, M. A., Kim, H., Bristow, R., Green, R., et al. (2009). Germline MutY Human Homologue Mutations and Colorectal Cancer: a Multisite Case-Control Study. Gastroenterology 136 (4), 1251–1260. doi:10.1053/j.gastro.2008.12.050
Costa, C., Wang, Y., Ly, A., Hosono, Y., Murchie, E., Walmsley, C. S., et al. (2020). PTEN Loss Mediates Clinical Cross-Resistance to CDK4/6 and PI3Kα Inhibitors in Breast Cancer. Cancer Discov. 10, 72–85. doi:10.1158/2159-8290.CD-18-0830
Courtney, K. D., Ma, Y., de Leon, A. D., Christie, A., Xie, Z., Woolford, L., et al. (2020). HIF-2 Complex Dissociation, Target Inhibition, and Acquired Resistance with PT2385, a First-In-Class HIF-2 Inhibitor in Clear Cell Renal Cell Carcinoma. Clin Cancer Res. 26(4), 793-803. doi:10.1158/1078-0432.CCR-19-1459
Cousineau, I., Abaji, C., and Belmaaza, A. (2005). BRCA1 Regulates RAD51 Function in Response to DNA Damage and Suppresses Spontaneous Sister Chromatid Replication Slippage: Implications for Sister Chromatid Cohesion, Genome Stability, and Carcinogenesis. Cancer Res. 65, 11384–11391. doi:10.1158/0008-5472.CAN-05-2156
de Andrade, K. C., Frone, M. N., Wegman-Ostrosky, T., Khincha, P. P., Kim, J., Amadou, A., et al. (2019). Variable Population Prevalence Estimates of Germline TP53 Variants: A GnomAD-Based Analysis. Hum. Mutat. 40, 97–105. doi:10.1002/humu.23673
DeLellis, R. A., Lloyd, R. V., Heitz, P. U., and Eng, C. (2004). “Pathology and Genetics: Tumours of the Endocrine Organs,” in World Health Organization Classification of Tumours Series (Lyon, France: IARC Press), Vol. 8.
Döhner, H., Fischer, K., Bentz, M., Hansen, K., Benner, A., Cabot, G., et al. (1995). p53 Gene Deletion Predicts for Poor Survival and Non-response to Therapy with Purine Analogs in Chronic B-Cell Leukemias. Blood 85, 1580–1589. doi:10.1182/blood.v85.6.1580.bloodjournal8561580
Domchek, S. M., Friebel, T., Neuhausen, S. L., Lynch, H. T., Singer, C. F., Eeles, R. A., et al. (2011). “Is Hormone Replacement Therapy (HRT) Following Risk-Reducing Salpingo-Oophorectomy (RRSO) in BRCA1 (B1)- and BRCA2 (B2)-Mutation Carriers Associated with an Increased Risk of Breast Cancer,” in ASCO Annual Meeting, Chicago, IL, June 3–7, 2011. Abstract 1501. doi:10.1200/jco.2011.29.15_suppl.1501
Dominguez-Valentin, M., Sampson, J. R., Seppälä, T. T., Ten Broeke, S. W., Plazzer, J. P., Nakken, S., et al. (2020). Cancer Risks by Gene, Age, and Gender in 6350 Carriers of Pathogenic Mismatch Repair Variants: Findings from the Prospective Lynch Syndrome Database. Genet. Med. 22, 15–25. doi:10.1038/s41436-019-0596-9
Eichhorn, P. J. A., Gili, M., Scaltriti, M., Serra, V., Guzman, M., and Nijkamp, W. (2008). Phosphatidylinositol 3-Kinase Hyperactivation Results in Lapatinib Resistance that Is Reversed by the mTOR/phosphatidylinositol 3-kinase Inhibitor NVP-Bez235. Cancer Res. 68, 9221–9230. doi:10.1158/0008-5472.CAN-08-1740
Eisen, A., Lubinski, J., Gronwald, J., Moller, P., Lynch, H. T., Klijn, J., et al. (2008). Hormone Therapy and the Risk of Breast Cancer in BRCA1 Mutation Carriers. JNCI J. Natl. Cancer Inst. 100, 1361–1367. doi:10.1093/jnci/djn313
Eisenhofer, G., Rivers, G., Rosas, A. L., Quezado, Z., Manger, W. M., and Pacak, K. (2007). Adverse Drug Reaction in Patients with Phaeochromocytoma – Incidence, Prevention and Management. Drug Saf. 30, 1031–1062. doi:10.2165/00002018-200730110-00004
Elisei, R., Schlumberger, M. J., Müller, S. P., Schöffski, P., Brose, M. S., Shah, M. H., et al. (2013). Cabozantinib in Progressive Medullary Thyroid Cancer. J. Clin. Oncol. 31, 3639–3646. doi:10.1200/JCO.2012.48.4659
Else, T., Greenberg, S., and Fishbein, L. (2008). “Hereditary Paraganglioma-Pheochromocytoma Syndromes,” in GeneReviews. Editors M. P. Adams, H. H. Ardinger, and R. A. Pagon (Seattle, WA: University of Washington), 1993–2021. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1548/(Accessed July 21, 2021).
Elwood, P. C., Morgan, G., Delon, C., Protty, M., Galante, J., Pickering, J., et al. (2021). Aspirin and Cancer Survival: a Systematic Review and Meta-Analyses of 118 Observational Studies of Aspirin and 18 Cancers. Ecancermedicalscience 15, 1258. doi:10.3332/ecancer.2021.1258
Eng, C. (1999). “Multiple Endocrine Neoplasia Type 2,” in GeneReviews. Editors M. P. Adams, H. H. Ardinger, and R. A. Pagon (Seattle, WA: University of Washington), 1993–2021. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1257/(Accessed July 14, 2021).
Eskelund, C. W., Dahl, C., Hansen, J. W., Westman, M., Kolstad, A., Pedersen, L. B., et al. (2017). TP53 Mutations Identify Younger Mantle Cell Lymphoma Patients Who Do Not Benefit from Intensive Chemoimmunotherapy. Blood 130, 1903–1910. doi:10.1182/blood-2017-04-779736
Evans, D. G., Birch, J. M., Ramsden, R. T., Sharif, S., and Baser, M. E. (2006). Malignant Transformation and New Primary Tumours after Therapeutic Radiation for Benign Disease: Substantial Risks in Certain Tumour Prone Syndromes. J. Med. Genet. 43, 289–294. doi:10.1136/jmg.2005.036319
Everett, J. N., Raymond, V. M., Dandapani, M., Marvin, M., Kohlmann, W., Chittenden, A., et al. (2014). Screening for Germline Mismatch Repair Mutations Following Diagnosis of Sebaceous Neoplasm. JAMA Dermatol 150, 1315–1321. doi:10.1001/jamadermatol.2014.1217
Farmer, H., McCabe, N., Lord, C. J., Tutt, A. N. J., Johnson, D. A., Richardson, T. B., et al. (2005). Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature 434, 917–921. doi:10.1038/nature03445
Farooqui, M. Z. H., Valdez, J., Martyr, S., Aue, G., Saba, N., Niemann, C. U., et al. (2015). Ibrutinib for Previously Untreated and Relapsed or Refractory Chronic Lymphocytic Leukaemia with TP53 Aberrations: a Phase 2, Single-Arm Trial. Lancet Oncol. 16, 169–176. doi:10.1016/S1470-2045(14)71182-9
FDA (2014). CAPRELSA® (Vandetanib) Tablets for Oral Use. Silver Spring, MD: U.S Food and Drug Administration.
FDA (2012). CLOMID® (Clomiphene Citrate Tablets USP). Silver Spring, MD: U.S Food and Drug Administration.
FDA (2020b). COMETRIQ® (Cabozantinib) Capsules. Silver Spring, MD: U.S Food and Drug Administration.
FDA (2021). LEMTRADA® (Alemtuzumab) Injection, for Intravenous Use. Silver Spring, MD: U.S Food and Drug Administration.
FDA (2010). VICTOZA® (Liraglutide) Injection, for Subcutaneous Use. Silver Spring, MD: U.S Food and Drug Administration.
Fedier, A., Schwarz, V. A., Walt, H., Carpini, R. D., Haller, U., and Fink, D. (2001). Resistance to Topoisomerase Poisons Due to Loss of DNA Mismatch Repair. Int. J. Cancer 93, 571–576. doi:10.1002/ijc.1356
Fink, D., Nebel, S., Aebi, S., Nehme, A., and Howell, S. (1997b). Loss of DNA Mismatch Repair Due to Knockout of MSH2 or PMS2 Results in Resistance to Cisplatin and Carboplatin. Int. J. Oncol. 11, 539–542. doi:10.3892/ijo.11.3.539
Fink, D., Zheng, H., Nebel, S., Norris, P. S., Aebi, S., Lin, T. P., et al. (1997a). In Vitro and In Vivo Resistance to Cisplatin in Cells that Have Lost DNA Mismatch Repair. Cancer Res. 57, 1841–1845.
Frebourg, T., Lagercrantz, S. B., Bajalica Lagercrantz, S., Oliveira, C., Magenheim, R., and Evans, D. G. (2020). Guidelines for the Li-Fraumeni and Heritable TP53-Related Cancer Syndromes. Eur. J. Hum. Genet. 28, 1379–1386. doi:10.1038/s41431-020-0638-4
Friedman, H. S., Johnson, S. P., Dong, Q., Schold, S. C., Rasheed, B. K., Bigner, S. H., et al. (1997). Methylator Resistance Mediated by Mismatch Repair Deficiency in a Glioblastoma Multiforme Xenograft. Cancer Res. 57, 2933–2936.
Fry, R. C., Svensson, J. P., Valiathan, C., Wang, E., Hogan, B. J., Bhattacharya, S., et al. (2008). Genomic Predictors of Interindividual Differences in Response to DNA Damaging Agents. Genes. Dev. 22, 2621–2626. doi:10.1101/gad.1688508
Golan, T., Hammel, P., Reni, M., Van Cutsem, E., Macarulla, T., Hall, M. J., et al. (2019). Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 381, 317–327. doi:10.1056/NEJMoa1903387
Goodspeed, A., Jean, A., and Costello, J. C. (2019). A Whole-Genome CRISPR Screen Identifies a Role of MSH2 in Cisplatin-Mediated Cell Death in Muscle-Invasive Bladder Cancer. Eur. Urol. 75, 242–250. doi:10.1016/j.eururo.2018.10.040
Gordhandas, S., Norquist, B. M., Pennington, K. P., Yung, R. L., Laya, M. B., and Swisher, E. M. (2019). Hormone Replacement Therapy after Risk Reducing Salpingo-Oophorectomy in Patients with BRCA1 or BRCA2 Mutations; a Systematic Review of Risks and Benefits. Gynecol. Oncol. 153, 192–200. doi:10.1016/j.ygyno.2018.12.014
Grover, S., Kastrinos, F., Steyerberg, E. W., Cook, E. F., Dewanwala, A., Burbidge, L. A., et al. (2012). Prevalence and Phenotypes of APC and MUTYH Mutations in Patients with Multiple Colorectal Adenomas. JAMA 308, 485–492. doi:10.1001/jama.2012.8780
Guha, T., and Malkin, D. (2017). Inherited TP53 Mutations and the Li-Fraumeni Syndrome. Cold Spring Harb. Perspect. Med. 7, 7. doi:10.1101/cshperspect.a026187
Hadoux, J., Favier, J., Scoazec, J. Y., Leboulleux, S., Al Ghuzlan, A., Caramella, C., et al. (2014). SDHB Mutations Are Associated with Response to Temozolomide in Patients with Metastatic Pheochromocytoma or Paraganglioma. Int. J. Cancer 135, 2711–2720. doi:10.1002/ijc.28913
Hahnen, E., Lederer, B., Hauke, J., Loibl, S., Kröber, S., Schneeweiss, A., et al. (2017). Germline Mutation Status, Pathological Complete Response, and Disease-free Survival in Triple-Negative Breast Cancer. JAMA Oncol. 3, 1378–1385. doi:10.1001/jamaoncol.2017.1007
Hallek, M. (2019). Chronic Lymphocytic Leukemia: 2020 Update on Diagnosis, Risk Stratification and Treatment. Am. J. Hematol. 94, 1266–1287. doi:10.1002/ajh.25595
Hallek, M., Fischer, K., Fingerle-Rowson, G., Fink, A., Busch, R., Mayer, J., et al. (2010). Addition of Rituximab to Fludarabine and Cyclophosphamide in Patients with Chronic Lymphocytic Leukaemia: a Randomised, Open-Label, Phase 3 Trial. Lancet 376, 1164–1174. doi:10.1016/S0140-6736(10)61381-5
Hobert, J. A., and Eng, C. (2009). PTEN Hamartoma Tumor Syndrome: An Overview. Genet. Med. 11, 687–694. doi:10.1097/GIM.0b013e3181ac9aea
Howe, J. R., Bair, J. L., Sayed, M. G., Anderson, M. E., Mitros, F. A., Petersen, G. M., et al. (2001). Germline Mutations of the Gene Encoding Bone Morphogenetic Protein Receptor 1A in Juvenile Polyposis. Nat. Genet. 28, 184–187. doi:10.1038/88919
Huber, D., Seitz, S., Kast, K., Emons, G., and Ortmann, O. (2020). Use of Oral Contraceptives in BRCA Mutation Carriers and Risk for Ovarian and Breast Cancer: a Systematic Review. Arch. Gynecol. Obstet. 301, 875–884. doi:10.1007/s00404-020-05458-w
Hutchcraft, M. L., Gallion, H. H., and Kolesar, J. M. (2021). MUTYH as an Emerging Predictive Biomarker in Ovarian Cancer. Diagn. (Basel) 11, 84. doi:10.3390/diagnostics11010084
Ibrahim, A., and Chopra, S. (2020). Succinate Dehydrogenase-Deficient Gastrointestinal Stromal Tumors. Arch. Pathol. Lab. Med. 144, 655–660. doi:10.5858/arpa.2018-0370-RS
Iodice, S., Barile, M., Rotmensz, N., Feroce, I., Bonanni, B., Radice, P., et al. (2010). Oral Contraceptive Use and Breast or Ovarian Cancer Risk in BRCA1/2 Carriers: a Meta-Analysis. Eur. J. Cancer 46, 2275–2284. doi:10.1016/j.ejca.2010.04.018
Ishikawa, H., Mutoh, M., Sato, Y., Doyama, H., Tajika, M., Tanaka, S., et al. (2021). Chemoprevention with Low-Dose Aspirin, Mesalazine, or Both in Patients with Familial Adenomatous Polyposis without Previous Colectomy (J-FAPP Study IV): a Multicentre, Double-Blind, Randomised, Two-By-Two Factorial Design Trial. Lancet Gastroenterology Hepatology 6, 474–481. doi:10.1016/S2468-1253(21)00018-2
Jasperson, K. W., Patel, S. G., and Ahnen, D. J. (1998). “APC-associated Polyposis Conditions,” in GeneReviews. Editors M. P. Adams, H. H. Ardinger, and R. A. Pagon (Seattle, WA: University of Washington), 1993–2021. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1345/(Accessed June 10, 2021).
Jonasch, E., Donskov, F., Iliopoulos, O., Rathmell, W. K., Narayan, V. K., Maughan, B. L., et al. (2021). Belzutifan for Renal Cell Carcinoma in von Hippel–Lindau Disease. N. Engl. J. Med. 385, 2036–2046. doi:10.1056/NEJMoa2103425
Juric, D., Castel, P., Griffith, M., Griffith, O. L., Won, H. H., Ellis, H., et al. (2015). Convergent Loss of PTEN Leads to Clinical Resistance to a PI(3)Kα Inhibitor. Nature 518, 240–244. doi:10.1038/nature13948
Kalia, S. S., Adelman, K., Bale, S. J., Chung, W. K., Eng, C., Evans, J. P., et al. (2017). Recommendations for Reporting of Secondary Findings in Clinical Exome and Genome Sequencing, 2016 Update (ACMG SF v2.0): a Policy Statement of the American College of Medical Genetics and Genomics. Genet. Med. 19, 249–255. doi:10.1038/gim.2016.190
Kasper, E., Angot, E., Colasse, E., Nicol, L., Sabourin, J.-C., Adriouch, S., et al. (2018). Contribution of Genotoxic Anticancer Treatments to the Development of Multiple Primary Tumours in the Context of Germline TP53 Mutations. Eur. J. Cancer 101, 254–262. doi:10.1016/j.ejca.2018.06.011
Kemp Bohan, P. M., Mankaney, G., Vreeland, T. J., Chick, R. C., Hale, D. F., Cindass, J. L., et al. (2021). Chemoprevention in Familial Adenomatous Polyposis: Past, Present and Future. Fam. Cancer 20, 23–33. doi:10.1007/s10689-020-00189-y
Kleinerman, R. A. (2009). Radiation-Sensitive Genetically Susceptible Pediatric Sub-populations. Pediatr. Radiol. 39 (Suppl. 1), S27–S31. doi:10.1007/s00247-008-1015-6
Kleinerman, R. A., Tucker, M. A., Abramson, D. H., Seddon, J. M., Tarone, R. E., and Fraumeni, J. F. (2007). Risk of Soft Tissue Sarcomas by Individual Subtype in Survivors of Hereditary Retinoblastoma. J. Natl. Cancer Inst. 99, 24–31. doi:10.1093/jnci/djk002
Kotsopoulos, J., Huzarski, T., Gronwald, J., Moller, P., Lynch, H. T., Neuhausen, S. L., et al. (2016). Hormone Replacement Therapy after Menopause and Risk of Breast Cancer in BRCA1 Mutation Carriers: a Case-Control Study. Breast Cancer Res. Treat. 155, 365–373. doi:10.1007/s10549-016-3685-3
Krausova, M., and Korinek, V. (2014). Wnt Signaling in Adult Intestinal Stem Cells and Cancer. Cell. Signal. 26, 570–579. doi:10.1016/j.cellsig.2013.11.032
Kuchenbaecker, K. B., Hopper, J. L., Barnes, D. R., Phillips, K.-A., Mooij, T. M., Roos-Blom, M.-J., et al. (2017). Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 317 (23), 2402–2416. doi:10.1001/jama.2017.7112
Laderian, B., Mundi, P., Fojo, T., and E. Bates, S. (2020). Emerging Therapeutic Implications of STK11 Mutation: Case Series. Oncologist 25, 733–737. doi:10.1634/theoncologist.2019-0846
Larsen Haidle, J., and Howe, J. R. (2003). “Juvenile Polyposis Syndrome,” in GeneReviews. Editors M. P. Adams, H. H. Ardinger, and R. A. Pagon (Seattle, WA: University of Washington), 1993–2021. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1469/(Accessed July 16, 2021).
Lauschke, V. M., Milani, L., and Ingelman-Sundberg, M. (2017). Pharmacogenomic Biomarkers for Improved Drug Therapy-Recent Progress and Future Developments. AAPS J. 20, 4. doi:10.1208/s12248-017-0161-x
Lavergne, V., Sabnis, A., Tupule, A., Davidson, P. R., Kline, C., Matthay, K., et al. (2020). Germline MUTYH Mutation in a Pediatric Cancer Survivor Developing a Secondary Malignancy. J. Pediatr. Hematol. Oncol. 42, e647–e654. doi:10.1097/MPH.0000000000001668
Lenders, J. W., Duh, Q. Y., Eisenhofer, G., Gimenez-Roqueplo, A. P., Grebe, S. K., Murad, M. H., et al. (2014). Endocrine Society. Pheochromocytoma and Paraganglioma: an Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 99, 1915–1942. doi:10.1210/jc.2014-1498
Li, Y., Zhang, S., Wang, Y., Peng, J., Fang, F., and Yang, X. (2018). MLH1 Enhances the Sensitivity of Human Endometrial Carcinoma Cells to Cisplatin by Activating the MLH1/c-Abl Apoptosis Signaling Pathway. BMC Cancer 18, 1294. doi:10.1186/s12885-018-5218-4
Lin, Z., Zhang, L., Zhou, J., and Zheng, J. (2019). Silencing Smad4 Attenuates Sensitivity of Colorectal Cancer Cells to Cetuximab by Promoting Epithelial Mesenchymal Transition. Mol. Med. Rep. 20, 3735–3745. doi:10.3892/mmr.2019.10597
Litton, J. K., Rugo, H. S., Ettl, J., Hurvitz, S. A., Gonçalves, A., Lee, K.-H., et al. (2018). Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 379, 753–763. doi:10.1056/NEJMoa1802905
Lozanski, G., Heerema, N. A., Flinn, I. W., Smith, L., Harbison, J., Webb, J., et al. (2004). Alemtuzumab Is an Effective Therapy for Chronic Lymphocytic Leukemia with P53 Mutations and Deletions. Blood 103, 3278–3281. doi:10.1182/blood-2003-10-3729
Madariaga, A., Bowering, V., Ahrari, S., Oza, A. M., and Lheureux, S. (2020). Manage Wisely: Poly (ADP-Ribose) Polymerase Inhibitor (PARPi) Treatment and Adverse Events. Int. J. Gynecol. Cancer 30, 903–915. doi:10.1136/ijgc-2020-001288
Madsen, L. W., Knauf, J. A., Gotfredsen, C., Pilling, A., Sjögren, I., Andersen, S., et al. (2012). GLP-1 Receptor Agonists and the Thyroid: C-Cell Effects in Mice Are Mediated via the GLP-1 Receptor and Not Associated with RET Activation. Endocrinology 153, 1538–1547. doi:10.1210/en.2011-1864
Martin, L. P., Hamilton, T. C., and Schilder, R. J. (2008). Platinum Resistance: the Role of DNA Repair Pathways. Clin. Cancer Res. 14, 1291–1295. doi:10.1158/1078-0432.CCR-07-2238
Matsumura, H., Takimoto, H., Shimada, N., Hirata, M., Ohnishi, T., and Hayakawa, T. (1998). Glioblastoma Following Radiotherapy in a Patient with Tuberous Sclerosis. Neurol. Med. Chir. (Tokyo) 38, 287–291. doi:10.2176/nmc.38.287
McGarrity, T. J., Amos, C. I., and Baker, M. J. (2001). “Peutz-Jeghers Syndrome,” in GeneReviews. Editors M. P. Adams, H. H. Ardinger, and R. A. Pagon, (Seattle, WA: University of Washington), 1993–2021. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1266/. (Accessed June 28, 2021).
McGuire, V., Felberg, A., Mills, M., Ostrow, K. L., DiCioccio, R., John, E. M., et al. (2004). Relation of Contraceptive and Reproductive History to Ovarian Cancer Risk in Carriers and Noncarriers of BRCA1 Gene Mutations. Am. J. Epidemiol. 160, 613–618. doi:10.1093/aje/kwh284
McLean, M. H., Murray, G. I., Fyfe, N., Hold, G. L., Mowat, N. A. G., and El-Omar, E. M. (2008). COX-2 Expression in Sporadic Colorectal Adenomatous Polyps Is Linked to Adenoma Characteristics. Histopathology 52, 806–815. doi:10.1111/j.1365-2559.2008.03038.x
Meng, L., Huang, J., Jia, Y., Huang, H., Qiu, F., and Sun, S. (2019). Assessing Fluoroquinolone-Associated Aortic Aneurysm and Dissection: Data Mining of the Public Version of the FDA Adverse Event Reporting System. Int. J. Clin. Pract. 73, 5. doi:10.1111/ijcp.13331
Meyers, M., Wagner, M. W., Hwang, H. S., Kinsella, T. J., and Boothman, D. A. (2001). Role of the Hmlh1 DNA Mismatch Repair Protein in Fluoropyrimidine-Mediated Cell Death and Cell Cycle Responses. Cancer Res. 61, 5193–5201.
Nagata, Y., Lan, K.-H., Zhou, X., Tan, M., Esteva, F. J., Sahin, A. A., et al. (2004). PTEN Activation Contributes to Tumor Inhibition by Trastuzumab, and Loss of PTEN Predicts Trastuzumab Resistance in Patients. Cancer Cell. 6, 117–127. doi:10.1016/j.ccr.2004.06.022
Narod, S. A., Dubé, M. P., Klijn, J., Lubinski, J., Lynch, H. T., Ghadirian, P., et al. (2002). Oral Contraceptives and the Risk of Breast Cancer in BRCA1 and BRCA2 Mutation Carriers. J. Natl. Cancer Inst. 94, 1773–1779. doi:10.1093/jnci/94.23.1773
Nelen, M. R., Kremer, H., Konings, I. B., Schoute, F., Essen, A. J. v., Koch, R., et al. (1999). Novel PTEN Mutations in Patients with Cowden Disease: Absence of Clear Genotype-Phenotype Correlations. Eur. J. Hum. Genet. 7, 267–273. doi:10.1038/sj.ejhg.5200289
Nguyen, S. A., Stechishin, O. D., Luchman, H. A., Lun, X. Q., Senger, D. L., Robbins, S. M., et al. (2014). Novel MSH6 Mutations in Treatment-Naïve Glioblastoma and Anaplastic Oligodendroglioma Contribute to Temozolomide Resistance Independently of MGMT Promoter Methylation. Clin. Cancer Res. 20, 4894–4903. doi:10.1158/1078-0432.CCR-13-1856
Northrup, H., Koenig, M. K., Pearson, D. A., and Au, K. S. (1999). “Tuberous Sclerosis Complex,” in GeneReviews. Editors M. P. Adams, H. H. Ardinger, and R. A. Pagon (Seattle, WA: University of Washington), 1993–2021. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1220/(Accessed June 29, 2021).
Nuñez, F., Bravo, S., Cruzat, F., Montecino, M., and De Ferrari, G. V. (2011). Wnt/β-Catenin Signaling Enhances Cyclooxygenase-2 (COX2) Transcriptional Activity in Gastric Cancer Cells. PloS One 6, e18562. doi:10.1371/journal.pone.0018562
O'Kane, G. M., Ezzat, S., Joshua, A. M., Bourdeau, I., Leibowitz-Amit, R., Olney, H. J., et al. (2019). A Phase 2 Trial of Sunitinib in Patients with Progressive Paraganglioma or Pheochromocytoma: the SNIPP Trial. Br. J. Cancer 120, 1113–1119. doi:10.1038/s41416-019-0474-x
Oberstein, E. M., Fleming, L. E., Gómez-Marin, O., and Glassberg, M. K. (2003). Pulmonary Lymphangioleiomyomatosis (LAM): Examining Oral Contraceptive Pills and the Onset of Disease. J. Women's Health 12, 81–85. doi:10.1089/154099903321154176
Okafor, C., Hogan, J., Raygada, M., Thomas, B. J., Akshintala, S., Glod, J. W., et al. (2021). Update on Targeted Therapy in Medullary Thyroid Cancer. Front. Endocrinol. 12, 708949. doi:10.3389/fendo.2021.708949
Online Mendelian Inheritance in Man OMIM® (2021a). APC Regulator of WNT Signaling Pathway; APC. MIM Number: {* 611731}. https://www.omim.org/entry/611731?search=apc&highlight=apc (Accessed June 10, 2021).
Online Mendelian Inheritance in Man OMIM® (2021b). VON HIPPEL-LINDAU SYNDROME; VHLS. MIM Number: {* 193300}. Available at: https://www.omim.org/entry/193300?search=vhl&highlight=vhl (Accessed July 29, 2021).
Ouakrim, D. A., Dashti, S. G., Chau, R., Buchanan, D. D., Clendenning, M., Rosty, C., et al. (2015). Aspirin, Ibuprofen, and the Risk for Colorectal Cancer in Lynch Syndrome. J Natl Cancer Inst. 107, Djv170. doi:10.1093/jnci/djv170
Ozawa, H., Ranaweera, R. S., Izumchenko, E., Makarev, E., Zhavoronkov, A., Fertig, E. J., et al. (2017). SMAD4 Loss Is Associated with Cetuximab Resistance and Induction of MAPK/JNK Activation in Head and Neck Cancer Cells. Clin. Cancer Res. 23, 5162–5175. doi:10.1158/1078-0432.CCR-16-1686
Paik, J. Y., Toon, C. W., Benn, D. E., High, H., Hasovitz, C., Pavlakis, N., et al. (2014). Renal Carcinoma Associated with Succinate Dehydrogenase B Mutation: a New and Unique Subtype of Renal Carcinoma. J. Clin. Oncol. 32, e10–e13. doi:10.1200/JCO.2012.47.2647
Paluch-Shimon, S., Cardoso, F., Sessa, C., Balmana, J., Cardoso, M. J., Gilbert, F., et al. (2016). Prevention and Screening in BRCA Mutation Carriers and Other Breast/ovarian Hereditary Cancer Syndromes: ESMO Clinical Practice Guidelines for Cancer Prevention and Screening. Ann. Oncol. 27 (Suppl. 5), v103–v110. doi:10.1093/annonc/mdw327
Pang, Y., Lu, Y., Caisova, V., Liu, Y., Bullova, P., Huynh, T. T., et al. (2018). Targeting NAD+/PARP DNA Repair Pathway as a Novel Therapeutic Approach to SDHB-Mutated Cluster I Pheochromocytoma and Paraganglioma. Clin. Cancer Res. 24, 3423–3432. doi:10.1158/1078-0432.CCR-17-3406
Papageorgis, P., Cheng, K., Ozturk, S., Gong, Y., Lambert, A. W., Abdolmaleky, H. M., et al. (2011). Smad4 Inactivation Promotes Malignancy and Drug Resistance of Colon Cancer. Cancer Res. 71, 998–1008. doi:10.1158/0008-5472.CAN-09-3269
Pasternak, A. L., Ward, K. M., Luzum, J. A., Ellingrod, V. L., and Hertz, D. L. (2017). Germline Genetic Variants with Implications for Disease Risk and Therapeutic Outcomes. Physiol. Genomics 49, 567–581. doi:10.1152/physiolgenomics.00035.2017
Petrucelli, N., Daly, M. B., and Pal, T. (1998). “BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer,” in GeneReviews. Editors M. P. Adams, H. H. Ardinger, and R. A. Pagon (Seattle, WA: University of Washington), 1993–2021. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1247/(Accessed June 14, 2021).
Pettitt, A. R., Jackson, R., Carruthers, S., Dodd, J., Dodd, S., Oates, M., et al. (2012). Alemtuzumab in Combination with Methylprednisolone Is a Highly Effective Induction Regimen for Patients with Chronic Lymphocytic Leukemia and Deletion of TP53: Final Results of the National Cancer Research Institute CLL206 Trial. Jco 30, 1647–1655. doi:10.1200/jco.2011.35.9695
Pezzolesi, M. G., Zbuk, K. M., Waite, K. A., and Eng, C. (2007). Comparative Genomic and Functional Analyses Reveal a Novel Cis-Acting PTEN Regulatory Element as a Highly Conserved Functional E-Box Motif Deleted in Cowden Syndrome. Hum. Molec. Genet. 16, 1058–1071. doi:10.1093/hmg/ddm053
Pinto, D., Pinto, C., Guerra, J., Pinheiro, M., Santos, R., Vedeld, H. M., et al. (2018). Contribution of MLH1 Constitutional Methylation for Lynch Syndrome Diagnosis in Patients with Tumor MLH1 Downregulation. Cancer Med. 7, 433–444. doi:10.1002/cam4.1285
Reigstad, M. M., Storeng, R., MyklebustÅ., T. Å., Oldereid, N. B., Omland, A. K., Robsahm, T. E., et al. (2017). Cancer Risk in Women Treated with Fertility Drugs According to Parity Status-A Registry-Based Cohort Study. Cancer Epidemiol. Biomarkers Prev. 26, 953–962. doi:10.1158/1055-9965.EPI-16-0809
Reiss, K. A., Mick, R., O'Hara, M. H., Teitelbaum, U., Karasic, T. B., Schneider, C., et al. (2021). Phase II Study of Maintenance Rucaparib in Patients with Platinum-Sensitive Advanced Pancreatic Cancer and a Pathogenic Germline or Somatic Variant in BRCA1, BRCA2, or PALB2. Jco 39, 2497–2505. doi:10.1200/JCO.21.00003
Reyes-Uribe, L., Wu, W., Gelincik, O., Bommi, P. V., Francisco-Cruz, A., Solis, L. M., et al. (2021). Naproxen Chemoprevention Promotes Immune Activation in Lynch Syndrome Colorectal Mucosa. Gut 70, 555–566. doi:10.1136/gutjnl-2020-320946
Ribic, C. M., Sargent, D. J., Moore, M. J., Thibodeau, S. N., French, A. J., Goldberg, R. M., et al. (2003). Tumor Microsatellite-Instability Status as a Predictor of Benefit from Fluorouracil-Based Adjuvant Chemotherapy for Colon Cancer. N. Engl. J. Med. 349, 247–257. doi:10.1056/NEJMoa022289
Roberts, A. W., Davids, M. S., Pagel, J. M., Kahl, B. S., Puvvada, S. D., Gerecitano, J. F., et al. (2016). Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 374, 311–322. doi:10.1056/NEJMoa1513257
Robson, M., Im, S.-A., Senkus, E., Xu, B., Domchek, S. M., Masuda, N., et al. (2017). Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 377, 523–533. doi:10.1056/NEJMoa1706450
Samadder, N. J., Kuwada, S. K., Boucher, K. M., Byrne, K., Kanth, P., Samowitz, W., et al. (2018). Association of Sulindac and Erlotinib vs Placebo with Colorectal Neoplasia in Familial Adenomatous Polyposis. JAMA Oncol. 4, 671–677. doi:10.1001/jamaoncol.2017.5431
Sargent, D. J., Marsoni, S., Monges, G., Thibodeau, S. N., Labianca, R., Hamilton, S. R., et al. (2010). Defective Mismatch Repair as a Predictive Marker for Lack of Efficacy of Fluorouracil-Based Adjuvant Therapy in Colon Cancer. J. Clin. Oncol. 28, 3219–3226. doi:10.1200/JCO.2009.27.1825
Schatoff, E. M., Goswami, S., Zafra, M. P., Foronda, M., Shusterman, M., Leach, B. I., et al. (2019). Distinct Colorectal Cancer-Associated APC Mutations Dictate Response to Tankyrase Inhibition. Cancer Discov. 9, 1358–1371. doi:10.1158/2159-8290.CD-19-0289
Schneider, K., Zelley, K., Nichols, K. E., and Garber, J. (1999). “Li-Fraumeni Syndrome,” in GeneReviews. Editors M. P. Adams, H. H. Ardinger, and R. A. Pagon (Seattle, WA: University of Washington), 1993–2021. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1311/(Accessed July 2, 2021).
Schoenfeld, A. J., Rizvi, H., Bandlamudi, C., Sauter, J. L., Travis, W. D., Rekhtman, N., et al. (2020). Clinical and Molecular Correlates of PD-L1 Expression in Patients with Lung Adenocarcinomas. Ann. Oncol. 31, 599–608. doi:10.1016/j.annonc.2020.01.065
Sheng, S., Xu, Y., Guo, Y., Yao, L., Hu, L., Ouyang, T., et al. (2020). Prevalence and Clinical Impact of TP53 Germline Mutations in Chinese Women with Breast Cancer. Int. J. Cancer 146, 487–495. doi:10.1002/ijc.32424
Shuch, B., Agochukwu, N., Ricketts, C. J., Vocke, C. D., Gautam, R., Merino, M., et al. (2016). Vascular Endothelial Growth Factor Receptor-Targeted Therapy in Succinate Dehydrogenase C Kidney Cancer. J. Clin. Oncol. 34, e76–e79. doi:10.1200/JCO.2013.51.0214
Silver, D. P., Richardson, A. L., Eklund, A. C., Wang, Z. C., Szallasi, Z., Li, Q., et al. (2010). Efficacy of Neoadjuvant Cisplatin in Triple-Negative Breast Cancer. Jco 28, 1145–1153. doi:10.1200/jco.2009.22.4725
Skoulidis, F., Goldberg, M. E., Greenawalt, D. M., Hellmann, M. D., Awad, M. M., Gainor, J. F., et al. (2018). STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 8, 822–835. doi:10.1158/2159-8290.CD-18-0099
Skoulidis, F., Byers, L. A., Diao, L., Papadimitrakopoulou, V. A., Tong, P., Izzo, J., et al. (2015). Co-occurring Genomic Alterations Define Major Subsets of KRAS-Mutant Lung Adenocarcinoma with Distinct Biology, Immune Profiles, and Therapeutic Vulnerabilities. Cancer Discov. 5, 860–877. doi:10.1158/2159-8290.CD-14-1236
Soualy, A., Deutsch, D., Benallaoua, M., Ait-Omar, A., Mary, F., Helfen, S., et al. (2020). Effect of Chemoprevention by Low-Dose Aspirin of New or Recurrent Colorectal Adenomas in Patients with Lynch Syndrome (AAS-Lynch): Study Protocol for a Multicenter, Double-Blind, Placebo-Controlled Randomized Controlled Trial. Trials 21, 764. doi:10.1186/s13063-020-04674-8
Starr, L. J., Lindor, N. M., and Lin, A. E. (2021). “Myhre Syndrome,” in GeneReviews. Editors M. P. Adams, H. H. Ardinger, and R. A. Pagon (Seattle, WA: University of Washington), 1993–2021. Available at: https://www.ncbi.nlm.nih.gov/books/NBK425723/(Accessed July 17, 2021).
Stilgenbauer, S., Eichhorst, B., Schetelig, J., Coutre, S., Seymour, J. F., Munir, T., et al. (2016). Venetoclax in Relapsed or Refractory Chronic Lymphocytic Leukaemia with 17p Deletion: a Multicentre, Open-Label, Phase 2 Study. Lancet Oncol. 17, 768–778. doi:10.1016/S1470-2045(16)30019-5
Subbian, V., Hu, M. I., Wirth, L. J., Schuler, M., Mansfield, A. S., Curigliano, G., et al. (2021). Pralsetinib for Patients with Advanced or Metastatic RET-Altered Thyroid Cancer (ARROW): a Multi-Cohort, Open-Label, Registrational, Phase 1/2 Study. Lancet - Diabetes & Endocrinol. 9, 491–501. doi:10.1016/S2213-8587(21)00120-0
Subbian, V., Velcheti, V., Tuch, B. B., Ebata, K., Busaidy, N. L., Cabanillas, M. E., et al. (2018). Selective RET Kinase Inhibition for Patients with RET-Altered Cancers. Ann. Oncol. 29, 1869–1876. doi:10.1093/annonc/mdy137
Sullivan, R. J., Infante, J. R., Janku, F., Wong, D. J. L., Sosman, J. A., Keedy, V., et al. (2018). First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer Discov. 8, 184–195. doi:10.1158/2159-8290.CD-17-1119
Syngal, S., Brand, R. E., Church, J. M., Giardiello, F. M., Hampel, H. L., and Burt, R. W. (2015). ACG Clinical Guideline: Genetic Testing and Management of Hereditary Gastrointestinal Cancer Syndromes. Am. J. Gastroenterol. 110, 223–262. doi:10.1038/ajg.2014.435
Takahashi, M., Asai, N., Iwashita, T., Murakami, H., and Ito, S. (1998). Molecular Mechanisms of Development of Multiple Endocrine Neoplasia 2 by RET Mutations. J. Intern Med. 243, 509–513.
Tanaka, N., Mashima, T., Mizutani, A., Sato, A., Aoyama, A., Gong, B., et al. (2017). APC Mutations as a Potential Biomarker for Sensitivity to Tankyrase Inhibitors in Colorectal Cancer. Mol. Cancer Ther. 16, 752–762. doi:10.1158/1535-7163.MCT-16-0578
Taverna, P., Liu, L., Hanson, A. J., Monks, A., and Gerson, S. L. (2000). Characterization of MLH1 and MSH2 DNA Mismatch Repair Proteins in Cell Lines of the NCI Anticancer Drug Screen. Cancer Chemother. Pharmacol. 46, 507–516. doi:10.1007/s002800000186
Tchekmedyian, A., Amos, C. I., Bale, S. J., Zhu, D., Arold, S., Berrueta, J., et al. (2013). Findings from the Peutz-Jeghers Syndrome Registry of Uruguay. PLoS One 8, e79639. doi:10.1371/journal.pone.0079639
The Human Protein Atlas (2021e). MutL Homolog 1. Available at: https://www.proteinatlas.org/ENSG00000076242-MLH1 (Accessed July 23, 2021).
The Human Protein Atlas (2021b). MutY DNA Glycosylase. Available at: https://www.proteinatlas.org/ENSG00000132781-MUTYH (Accessed July 10, 2021).
The Human Protein Atlas (2021c). Ret Proto-Oncogene. Available at: https://www.proteinatlas.org/ENSG00000165731-RET (Accessed July 14, 2021).
The Human Protein Atlas (2021f). Serine/threonine Kinase 11. Available at: https://www.proteinatlas.org/ENSG00000118046-STK11 (Accessed June 20, 2021).
The Human Protein Atlas (2021d). SMAD Family Member 4. Available at: https://www.proteinatlas.org/ENSG00000141646-SMAD4 (Accessed July 16, 2021).
The Human Protein Atlas (2021g). TSC Complex Subunit 1. Available at: https://www.proteinatlas.org/ENSG00000165699-TSC1 (Accessed June 29, 2021).
The Human Protein Atlas (2021a). Von Hippel-Lindau Tumor Suppressor. Available at: https://www.proteinatlas.org/ENSG00000134086-VHL (Accessed Aug 1, 2021).
Tischkowitz, M., Colas, C., Pouwels, S., Hoogerbrugge, N., Bisseling, T., Bubien, V., et al. (2020). Cancer Surveillance Guideline for Individuals with PTEN Hamartoma Tumour Syndrome. Eur. J. Hum. Genet. 28, 1387–1393. doi:10.1038/s41431-020-0651-7
Tokairin, Y., Kakinuma, S., Arai, M., Nishimura, M., Okamoto, M., Ito, E., et al. (2006). Accelerated Growth of Intestinal Tumours after Radiation Exposure in Mlh1-Knockout Mice: Evaluation of the Late Effect of Radiation on a Mouse Model of HNPCC. Int. J. Exp. Pathol. 87, 89–99. doi:10.1111/j.0959-9673.2006.00464.x
Toledo, F., and Wahl, G. M. (2006). Regulating the P53 Pathway: In Vitro Hypotheses, In Vivo Veritas. Nat. Rev. Cancer 6, 909–923. doi:10.1038/nrc2012
Tutt, A., Tovey, H., Cheang, M. C. U., Kernaghan, S., Kilburn, L., Gazinska, P., et al. (2018). Carboplatin in BRCA1/2-Mutated and Triple-Negative Breast Cancer BRCAness Subgroups: the TNT Trial. Nat. Med. 24, 628–637. doi:10.1038/s41591-018-0009-7
Vaisman, A., Varchenko, M., and Umar, A. (1998). Defects in hMSH6, but Not in hMSH3, Correlate with Increased Resistance and Enhanced Replicative Bypass of Cisplatin, but Not Oxaliplatin, Adducts. Proc. Am. Assoc. Cancer Res. 39, 159.
van der Wouden, C. H., van Rhenen, M. H., Jama, W. O. M., Ingelman‐Sundberg, M., Lauschke, V. M., Konta, L., et al. (2019). Development of the PG x‐Passport: A Panel of Actionable Germline Genetic Variants for Pre‐Emptive Pharmacogenetic Testing. Clin. Pharmacol. Ther. 106, 866–873. doi:10.1002/cpt.1489
van Leeuwaarde, R. S., Ahmad, S., Links, T. P., and Giles, R. H. (2018). “Von Hippel-Lindau Syndrome,” in GeneReviews. Editors M. P. Adams, H. H. Ardinger, and R. A. Pagon (Seattle, WA: University of Washington), 1993–2021. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1469/(Accessed July 29, 2021).
Vogt, S., Jones, N., Christian, D., Engel, C., Nielsen, M., Kaufmann, A., et al. (2009). Expanded Extracolonic Tumor Spectrum in MUTYH-Associated Polyposis. Gastroenterology 137, 1976–1985. doi:10.1053/j.gastro.2009.08.052
Vousden, K. H., and Lane, D. P. (2007). p53 in Health and Disease. Nat. Rev. Mol. Cell. Biol. 8, 275–283. doi:10.1038/nrm2147
Wain, K. E., Ellingson, M. S., McDonald, J., Gammon, A., Roberts, M., Pichurin, P., et al. (2014). Appreciating the Broad Clinical Features of SMAD4 Mutation Carriers: a Multicenter Chart Review. Genet. Med. 16, 588–593. doi:10.1038/gim.2014.5
Wang, F., Xia, X., Yang, C., Shen, J., Mai, J., Kim, H. C., et al. (2018). SMAD4 Gene Mutation Renders Pancreatic Cancer Resistance to Radiotherapy through Promotion of Autophagy. Clin. Cancer Res. 24, 3176–3185. doi:10.1158/1078-0432.CCR-17-3435
Wang, L., Zhang, Q., Zhang, J., Sun, S., Guo, H., Jia, Z., et al. (2011). PI3K Pathway Activation Results in Low Efficacy of Both Trastuzumab and Lapatinib. BMC Cancer 11, 248. doi:10.1186/1471-2407-11-248
Wasserman, I., Lee, L. H., Ogino, S., Marco, M. R., Wu, C., Chen, X., et al. (2019). SMAD4 Loss in Colorectal Cancer Patients Correlates with Recurrence, Loss of Immune Infiltrate, and Chemoresistance. Clin. Cancer Res. 25, 1948–1956. doi:10.1158/1078-0432.CCR-18-1726
Weldon, C. B., Madenci, A. L., Boikos, S. A., Janeway, K. A., George, S., von Mehren, M., et al. (2017). Surgical Management of Wild-type Gastrointestinal Stromal Tumors: A Report from the National Institutes of Health Pediatric and Wildtype GIST Clinic. J. Clin. Oncol. 35, 523–528. doi:10.1200/JCO.2016.68.6733
Wellmann, R., Borden, B. A., Danahey, K., Nanda, R., Polite, B. N., Stadler, W. M., et al. (2018). Analyzing the Clinical Actionability of Germline Pharmacogenomic Findings in Oncology. Cancer 124, 3052–3065. doi:10.1002/cncr.31382
Wells, S. A., Robinson, B. G., Gagel, R. F., Dralle, H., Fagin, J. A., Santoro, M., et al. (2012). Vandetanib in Patients with Locally Advanced or Metastatic Medullary Thyroid Cancer: A Randomized, Double-Blind Phase III Trial. J. Clin. Oncol. 30, 134–141. doi:10.1200/JCO.2011.35.5040
Whittemore, A. S., Balise, R. R., Balise, R. R., Pharoah, P. D. P., DiCioccio, R. A., Oakley-Girvan, I., et al. (2004). Oral Contraceptive Use and Ovarian Cancer Risk Among Carriers of BRCA1 or BRCA2 Mutations. Br. J. Cancer 91, 1911–1915. doi:10.1038/sj.bjc.6602239
Win, A. K., Jenkins, M. A., Dowty, J. G., Antoniou, A. C., Lee, A., Giles, G. G., et al. (2017). Prevalence and Penetrance of Major Genes and Polygenes for Colorectal Cancer. Cancer Epidemiology, Biomarkers & Prevention. a Publ. Am. Assoc. Cancer Res. cosponsored by Am. Soc. Prev. Oncol. 26 (3), 404–412. doi:10.1158/1055-9965.EPI-16-0693
Win, A. K., Reece, J. C., Dowty, J. G., Buchanan, D. D., Clendenning, M., Rosty, C., et al. (2016). Risk of Extracolonic Cancers for People with Biallelic and Monoallelic Mutations in MUTYH. Int. J. Cancer 139, 1557–1563. doi:10.1002/ijc.30197
Wirth, L. J., Sherman, E., Robinson, B., Solomon, B., Kang, H., Lorch, J., et al. (2020). Efficacy of Selpercatinib in RET-Altered Thyroid Cancers. N. Engl. J. Med. 383, 825–835. doi:10.1056/NEJMoa2005651
Wong, C. K., Lambert, A. W., Ozturk, S., Papageorgis, P., Lopez, D., Shen, N., et al. (2020). Targeting RICTOR Sensitizes SMAD4-Negative Colon Cancer to Irinotecan. Mol. Cancer Res. 18, 414–423. doi:10.1158/1541-7786.MCR-19-0525
Wong, F. L., Boice, J. D., Abramson, D. H., Tarone, R. E., Kleinerman, R. A., Stovall, M., et al. (1997). Cancer Incidence after Retinoblastoma. Radiation Dose and Sarcoma Risk. JAMA 278, 1262–1267. doi:10.1001/jama.278.15.126210.1001/jama.1997.03550150066037
Yang, M., Schell, M. J., Loboda, A., Nebozhyn, M., Li, J., Teer, J. K., et al. (2019). Repurposing EGFR Inhibitor Utility in Colorectal Cancer in Mutant APC and TP53 Subpopulations. Cancer Epidemiol. Biomarkers Prev. 28, 1141–1152. doi:10.1158/1055-9965.EPI-18-1383
Yano, S. (2002). Exacerbation of Pulmonary Lymphangioleiomyomatosis by Exogenous Oestrogen Used for Infertility Treatment. Thorax 57, 1085–1086. doi:10.1136/thorax.57.12.1085
Yehia, L., and Eng, C. (2001). “PTEN Hamartoma Tumor Syndrome,” in GeneReviews. Editors M. P. Adams, H. H. Ardinger, and R. A. Pagon (Seattle, WA: University of Washington), 1993–2021. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1488/(Accessed July 8, 2021).
Yu, J., Astrinidis, A., Howard, S., and Henske, E. P. (2004). Estradiol and Tamoxifen Stimulate LAM-Associated Angiomyolipoma Cell Growth and Activate Both Genomic and Nongenomic Signaling Pathways. Am. J. Physiology-Lung Cell. Mol. Physiology 286, L694–L700. doi:10.1152/ajplung.00204.2003
Yu, J. J., Robb, V. A., Morrison, T. A., Ariazi, E. A., Karbowniczek, M., Astrinidis, A., et al. (2009). Estrogen Promotes the Survival and Pulmonary Metastasis of Tuberin-Null Cells. Proc. Natl. Acad. Sci. U.S.A. 106, 2635–2640. doi:10.1073/pnas.0810790106
Keywords: pharmacogenomics, pharmacogenetics, germline, gene-drug relationships, Cancer predisposition gene
Citation: Saugstad AA, Petry N and Hajek C (2022) Pharmacogenetic Review: Germline Genetic Variants Possessing Increased Cancer Risk With Clinically Actionable Therapeutic Relationships. Front. Genet. 13:857120. doi: 10.3389/fgene.2022.857120
Received: 18 January 2022; Accepted: 27 April 2022;
Published: 24 May 2022.
Edited by:
José A. G. Agúndez, University of Extremadura, SpainReviewed by:
Peter Hulick, NorthShore University HealthSystem, United StatesAttila Patocs, Semmelweis University, Hungary
Copyright © 2022 Saugstad, Petry and Hajek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Austin A. Saugstad, YXNhdWdzdGFkQGthbnNhc2NpdHkuZWR1