 Ziqi Yu
Ziqi Yu Xiang Shen
Xiang Shen Chong Hu2
Chong Hu2- 1Munich Medical Research School, LMU Munich, Munich, Germany
- 2Department of Gastroenterology and Hepatology, The First Affiliated Hospital of Nanchang Medical College, Jiangxi Provincial People’s Hospital, Nanchang, China
Polycystic liver disease (PLD) is a rare autosomal dominant disorder including two genetically and clinically distinct forms: autosomal dominant polycystic kidney disease (ADPKD) and isolated polycystic liver disease (PCLD). The main manifestation of ADPKD is kidney cysts, while PCLD has predominantly liver presentations with mild or absent kidney cysts. Over the past decade, PRKCSH, SEC63, ALG8, and LRP5 have been candidate genes of PCLD. Recently, more candidate genes such as GANAB, SEC61B, and ALR9 were also reported in PCLD patients. This review focused on all candidate genes of PCLD, including the newly established novel candidate genes. In addition, we also discussed some other genes which might also contribute to the disease.
Introduction
PLD and PCLD
Polycystic liver disease (PLD) is a rare autosomal dominant disorder characterized by multiple fluid-filled cysts in the liver (Russell and Pinson, 2007; Boerrigter et al., 2021). It is normally diagnosed by CT scans, ultrasound, and magnetic resonance imaging (Qian, 2010). Most of the PLD patients (>80%) are asymptomatic with preserved synthetic function and parenchymal liver volume (Qian, 2010; Gevers and Drenth, 2013). However, due to hepatomegaly, some patients might present abdominal pain, nausea, dyspnoea, and early satiety (Everson and Taylor, 2005). Occasionally, hemorrhage or infection in cysts due to cystic rupture could cause severe abdominal pain, fever, and leukocytosis (Russell and Pinson, 2007). The risk factor of PLD includes gender (with the majority of female patients), usage of estrogens, and multiple pregnancies (Sherstha et al., 1997; Alvaro et al., 2006; Gevers and Drenth, 2013). Normally there are no specific laboratory abnormalities in patients with PLD. Only gamma-glutamyl transferase (gGT) was increased in 51% and alkaline phosphatase (AP) was elevated in 17% of the patients (Van Keimpema et al., 2011).
Surgical treatment of PLD includes aspiration and sclerotherapy, fenestration, hepatic resection, and liver transplantation. However, all therapies except for liver transplantation have risks of recurrence, and only liver transplantation is the curative way (Drenth et al., 2010). Drug therapy such as somatostatin analogs and mTOR inhibitors are normally used in treating PLD. Somatostatin analogs such as octreotide and lanreotide could significantly reduce approximately 4% the liver volumes after 1 year of treatment. The cessation of treatment would cause a recurrence of liver growth (Chrispijn et al., 2012). A single case series using mTOR inhibitor sirolimus showed a significant reduction in liver volume (Qian et al., 2008). However, other clinical randomized studies of mTOR inhibitors such as everolimus and sirolimus had no significant effect on cystogenesis (Serra et al., 2010; Walz et al., 2010; Chrispijn et al., 2013).
Genetically, PLD consists of two different types: isolated polycystic liver disease (PCLD) and PLD with autosomal dominant polycystic kidney disease (ADPKD). ADPKD, including PKD1 (OMIM #173900) and PKD2 (OMIM #613095), is the main hereditary kidney disease that causes chronic renal failure. More than half of the patients with ADPKD would present liver cysts in the later stage (Hoevenaren et al., 2008). However, PCLD is normally without or only presents mild kidney cysts, and the prognosis of PCLD is normally better than ADPKD (Chandok, 2012). In OMIM, four types of PCLD are included: PCLD1 (OMIM #174050) caused by heterozygous mutation in PRKCSH, PCLD2 (OMIM #617004) caused by heterozygous variant in SEC63, PCLD3 (OMIM #617874) caused by heterozygous mutant ALG8, and PCLD4 (OMIM#617875) caused by heterozygous mutation in LRP5. Among these four types, PCLD1 and PCLD2 are the most common types of PCLD. Mutations in PRKCSH and SEC63 could explain 20%–41% of the cases (Van Keimpema et al., 2011; Gevers and Drenth, 2013; Masyuk et al., 2018). ADPKD and PCLD are both autosomal dominant diseases, which means most of the patients may carry a de novo mutation or have a positive family history. For people with a positive PCLD family history, genetic testing is suggested, such as whole-exome sequencing as a precaution. It is also beneficial for the diagnosis, surveillance, and regulation of diseases. For patients already diagnosed with PCLD, genetic counseling is also recommended.
Moreover, Janssen et al. (2011) found that 76% of the cyst from patients carrying a germline heterozygous PRKCSH mutation acquired a somatic second mutation in the wild-type allele. The germline mutation serves as the “first hit” and the somatic as the “second hit,” resulting in the loss of heterozygosity (LOH) of PRKCSH, which might be the reason for cyst formation. Meanwhile, Janssen et al. (2012) also found that the LOH in SEC63 in patients with germline SEC63 mutations confirmed the possibility of the “second hit” in cytogenesis. This explanation could also explain the high variability in disease presentations in PCLD patients (van Aerts et al., 2018).
Genetics
PRKCSH
PRKCSH encodes protein kinase C substrate 80K-H, also called the beta-subunit of glucosidase II (Li et al., 2003), which is a highly conserved protein. This protein contains a signal peptide domain, a low-density lipoprotein class A (LDL) domain, an EF-hand domain, a glutamic acid-rich (GLUT) segment, a mannose-6-phosphate receptor-binding domain (M6R), and an ER retrieval signal His-Asp-Glu-Leu (HDEL) tetrapeptide. The noncatalytic beta-subunit can interact with the catalytic alpha-subunit of glucosidase II and facilitate the removal of 1,3-linked glucose from newly synthesized glycoprotein (Gao et al., 2010) (Figure 1). This process is essential for the folding and quality control of glycoprotein (Qian, 2010; Waanders et al., 2006). The linkage between PRKCSH and PCLD was established in 2000 and 2003 (Li et al., 2003; Reynolds et al., 2000). After that, more PCLD patients with mutations in PRKCSH were found. All reported mutations according to HGMD and ClinVar databases are summarized in Figure 2. According to the spectrum of mutations, we found that most of the mutations clustered in the LDL domain (11 out of 35 mutations, 31.4%) and the M6R domain (10 out of 35 mutations, 28.6%), while no mutation was found in the GLUT and the HDEL region. In regard to mutation types, 31.4% of the mutations are frameshift (11 out of 35), followed by splice (8 out of 35) and nonsense (8 out of 35) mutations, both accounting for 22.9% of the mutations. Loss-of-functional mutations, including nonsense, frameshift, and splice mutations, are 77.1% of the reported mutations. In the reports, the patients carrying mutant PRKCSH normally have mild PCLD with a positive family history or severe PCLD without a family history. PKD is normally absent or very mildly presented in the patients (Drenth et al., 2003; Li et al., 2003; Drenth et al., 2004; Peces et al., 2005; Waanders et al., 2006; Waanders et al., 2010; Besse et al., 2017). However, these studies were normally performed on patients who did not meet the clinical criteria of ADPKD. An overall genetic screening in PLD patients, including ADPKD and PCLD, could be performed to have a better view of the genetic distinction between ADPKD and PCLD.
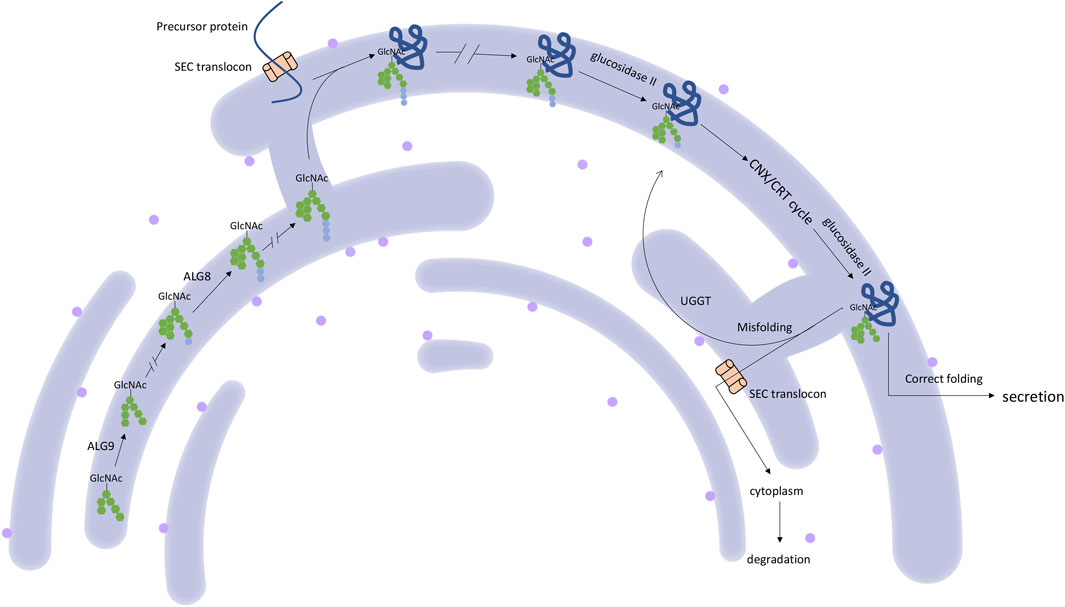
FIGURE 1. The schematic of the role of the PCLD genes in the protein synthesis process in ER.
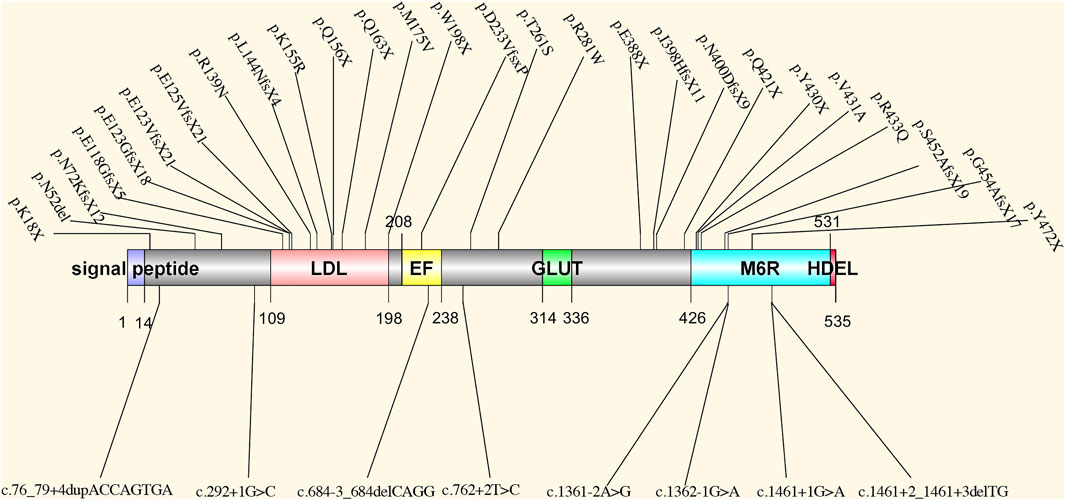
FIGURE 2. The spectrum of all reported mutations in PRKCSH. LDL: low-density lipoprotein class A domain; EF: EF-hand domain; GLUT: a glutamic acid-rich segment; M6R: mannose-6-phosphate receptor-binding domain; HDEL: ER retrieval signal His-Asp-Glu-Leu tetrapeptide.
SEC63
SEC63 encodes an endoplasmic reticulum (ER) protein. This protein contains a J-domain that can interact with the Hsp70 family of ATPase (Misselwitz et al., 1999). SEC63, together with SEC61 and SEC62, comprises a translocon (Qian, 2010), which plays a role in precursor proteins translocation in ER (Waanders et al., 2006). This translocational process is upstream of the PRKCSH protein quality control process (Schnell and Hebert, 2003) (Figure 1). The translocon complex is also thought to play a role in the Ca2+ channel (Lang et al., 2017). The relationship between SEC63 mutation and PCLD was first found in 2004. Seven loss-of-function mutations were identified in eight families with PCLD but without PRKCSH mutations (Davila et al., 2004). After that, more SEC63 mutations were detected in PCLD patients. All reported mutations collected by HGMD and ClinVar database are summarized in Figure 3. These mutations are evenly distributed in all gene parts with no hotspot and cluster region. By considering the mutation types, nonsense mutation with 36.1% (13 out of 36) is the most common type, followed by frameshift mutation with 25% (9 out of 36). More missense mutations were reported in SEC63 with 19.4% (7 out of 36), which is different from the mutation types of PRKCSH. However, loss-of-function mutations, including nonsense, frameshift, and splice, are still the mainstream with 75%. In these studies, most of the patients recruited in screening for SEC63 mutations were also PCLD patients with a positive family history or patients with severe and symptomatic PCLD (Davila et al., 2004; Waanders et al., 2006; Waanders et al., 2010; Kleffmann et al., 2012; Wills et al., 2016; Besse et al., 2017). Similar to patients with PRKCSH mutations, these patients also normally have no or very mild PKD.
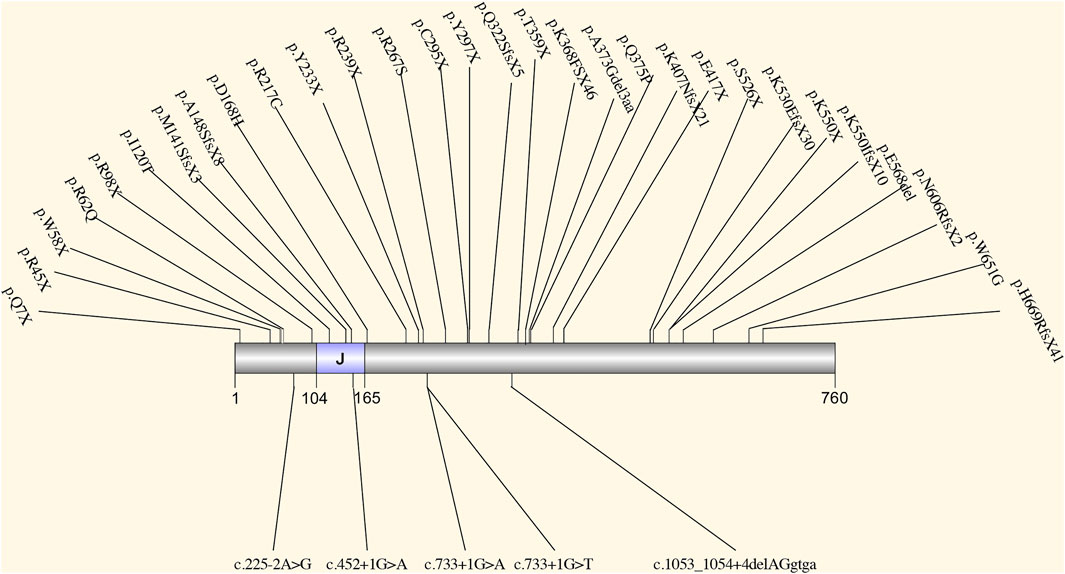
FIGURE 3. The spectrum of all reported mutations in SEC63. It contains a J-domain that can interact with the Hsp70 family of ATPase.
ALG8
ALG8 encodes a glycosyltransferase (Oriol et al., 2002) in ER that catalyzes the assembly of the second glucose residue to the lipid-linked oligosaccharide precursor (Aebi, 2013). It is a protein that plays a role in glycoprotein quality control in ER (van de Laarschot and Drenth, 2018). In 2017, (Besse et al., 2017) screened three heterozygous mutations (p. R364X, p. R179X, c.1038+1G>T) in the ALG8 gene in five patients with PCLD. Four of the five patients present with severe PCLD with very mild PKD, and one patient presents moderate liver cysts without kidney assay. In their study, they genetically engineered knock-out of Alg8 in mouse epithelial cell line and observed decreased levels of Pkd1 protein, defective posttranslational glycosylation, modification, and impaired trafficking of Pkd1 protein. These defects can be rescued by re-expression of wild-type Alg8 cDNA in the Alg8−/− cells (Besse et al., 2017).
LRP5
LRP5 is a single-pass transmembrane co-receptor for the canonical Wnt signaling (Sawakami et al., 2006). Wnt signaling cascade is required in many distinct developmental processes such as planar cell polarity, cell proliferation, fate decision, and stem/progenitor compartments regulation (Croce and McClay, 2008; Badders et al., 2009). The canonical Wnt signaling cascade is that after ligand binding to the Frizzled (Fzd) and the LRP5/6/arrow co-receptor complex, the cytoplasmic tail of co-receptor phosphorylated, leading to the recruitment of Axin and inhibition of GSK3β. This results in the inhibition of the phosphorylation of β-catenin and the dissociation of the β-catenin destruction complex, thus increasing the stability of β-catenin (Mao et al., 2001; Johnson et al., 2004; Nusse and Clevers, 2017; Lee-Law et al., 2019; Ren et al., 2021). Wnt proteins can also activate the Rho family (Wu et al., 2008), Ca2+ pathway (Kühl et al., 2000), PKCδ (Tu et al., 2007), and mTORC1 (Inoki et al., 2006), which consists of mTOR. mTOR was thought to play a role in cystogenesis (Chandok, 2012). Besides, a study also showed that PKD1 might be associated with Wnt signaling (Kim et al., 2016), indicating a possible linkage between PKD1 and LRP5.
The association between LRP5 and PCLD was first established in 2014 (Cnossen et al., 2014). This study screened four heterozygous mutations (i.e., p.V454M, p.R1188W, p.R1529S, and p.D1551N in LRP5). All the patients carrying LRP5 mutations are symptomatic PCLD patients. All four mutations led to a decreased Wnt signaling. Patients with mutations p.R1188W and p.R1529S present with moderate to severe PCLD without renal cysts, while patients carrying p.D1551N have PCLD with three renal cysts and one patient carrying p.V454M has mild PCLD with several bilateral renal cysts. However, no significant differences could be found in the disease characteristics between PCLD patients with or without LRP5 mutation (Cnossen et al., 2014).
GANAB
GANAB encodes the catalytic alpha-subunit of glucosidase II in the ER lumen, which interacts with the noncatalytic beta-subunit encoded by PRKCSH and removes the glucose from glycoprotein (Gao et al., 2010; Porath et al., 2016) (Figure 1). Loss of function in GANAB would cause decreased expression level of PKD1 (Besse et al., 2017), reduced maturation and localization of PKD1 and PKD2 protein (Porath et al., 2016), and impaired trafficking of PKD1 past the Golgi apparatus (Besse et al., 2017). Although PCLD caused by the deficiency in GANAB was not included in OMIM, several dependent groups have successfully screened variants in GANAB in patients with PCLD or ADPKD. All mutations in the HGMD database are summarized in Figure 4. In patients diagnosed as PCLD carrying GANAB mutation, mild-to-severe PKD with absent to moderate PKD could be observed (Porath et al., 2016; Besse et al., 2017; Besse et al., 2018; van de Laarschot et al., 2020). Compared to the PCLD patients with other gene mutations, a higher frequency of PKD could be observed in these patients. This might be because GANAB is also a causative gene of PKD, and PKD patients sometimes also have PLD presentations.
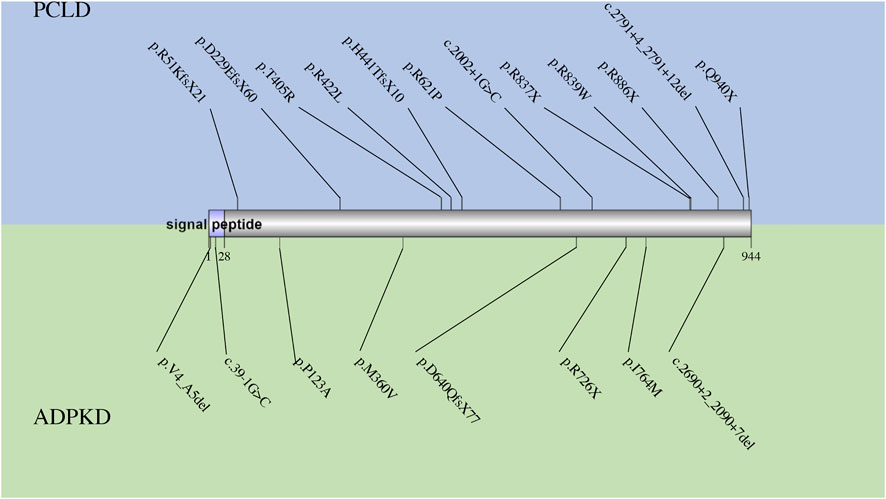
FIGURE 4. The spectrum of all reported mutations in GANAB. The mutations reported in PCLD are shown in blue, while those reported in ADPKD are shown in green.
SEC61B
Recently, SEC61B has been candidate gene in PCLD. This gene encodes the beta-subunit of SEC61 protein, which, together with SEC62 and SEC63, comprises the SEC61 translocon (Qian, 2010) (Figure 1). SEC61 has three subunits: alpha, beta, and gamma. Alpha-subunit consists of the SEC61 translocon channel pores, while gamma-subunit directly combines with alpha-subunit. However, the function of the beta-subunits is still under characterization (Lee-Law et al., 2019). However, research shows that the inactivation of SEC61B could result in a deprived PKD1 expression level (Besse et al., 2017). Till now, only two mutations in SEC61B (p.M1V and p. N36KfsX42) have been reported to cause PCLD. The patients carrying SEC61B mutation present moderate-to-severe PCLD without PKD (Besse et al., 2017).
ALG9
In 2019, ALG9 was also discovered as a novel candidate gene in PCLD. This gene encodes α-1,2-mannosyltransferase that catalyzes the addition of mannose to the N-glycan precursor in ER (Besse et al., 2019) (Figure 1). Similar to ALG8, loss of function in ALG9 also causes congenital disorder of glycosylation (CDG) (Höck et al., 2015; Besse et al., 2019), indicating an essential role of ALG9 in the quality control of glycoprotein. In the study considering ALG9 and PCLD, mutations p.W227X and p.R370K were found in ALG9. However, the two PCLD patients present moderate-to-severe PKD. To investigate whether ALG9 could cause PCLD, they genetically engineered ALG9 knock-out cell lines using CRISPR-Cas9 and observed a reduced manner of cell surface-expressed mature PKD1 with an altered N-glycosylation pattern (Besse et al., 2019).
Discussion
Because the SEC61 translocon complex is critical for the Ca2+ channel (Lang et al., 2017), PRKCSH is also shown to directly bind Ca2+ by the EF-hand domain and control the channel protein TRPV5 (Gkika et al., 2004). Wnt signaling could also play a role in Ca2+ pathway activation (Kühl et al., 2000). The Ca2+ pathway might also play a role in the cytogenesis of PCLD. Lower cytoplasmic Ca2+ level and ER Ca2+ stores were also found in cystic cells (Spirli et al., 2017), indirectly indicating a possible role of Ca2+ in PCLD. Moreover, decreased Ca2+ level is reported to cause elevation of cAMP and cAMP-dependent Ras/Raf/ERK pathway (Spirli et al., 2017). Increased cAMP can result in biliary cell proliferation and decreased apoptosis, which is also thought to be critical for cytogenesis in PCLD (Drenth et al., 2010).
Besides, except for LRP5, all candidate genes of PCLD are ER located and play a role in glycosylation, quality control, and translocation of glycoprotein across the ER (Cornec-Le Gall et al., 2018). Their roles in the glycosylation process are shown in Figure 1. Disordered glycosylation results in their retention in ER (Ushioda et al., 2013) and decreased protein activity, for example, protein PKD1. The bile duct also seems to have a lower tolerance for the PKD1 activity (Besse et al., 2017). This might explain the reason for the cytogenesis in the liver.
Therefore, not only genes involved in glycosylation and translocation of protein across ER but also genes involved in the Ca2+ pathway might be considered possible candidate genes in PCLD. Below, we enumerate some promising candidate genes in PCLD.
SEC61A1, SEC61A2, SEC61G, and SEC62
SEC61A1, SEC61A2, SEC61G, and SEC62 all encode the components of the SEC61 translocon complex. As mentioned before, this complex is critical in the translocation and quality control of glycoproteins. Because mutations in SEC61B and SEC63, which also encode the protein comprising SEC61 translocon, can result in disruption of the complex and cause PCLD. Other genes involved in the SEC61 complex might also play a role in PCLD.
ALG6
The same as ALG8, ALG6 also encodes a α-1,3-glucosyltransferase, ALG6 and ALG8 belongs to the same glucosyltransferase family, GT57 (Albuquerque-Wendt et al., 2019). However, ALG6 catalyzes the addition of the first glucose residue to the lipid-linked oligosaccharide chain of the N-glycan precursor, while ALG8 catalyzes the addition of the second glucose (Sun et al., 2005). Because the deficiency in ALG6 is reported the same as ALG8 and ALG9 causing CDG, it reveals an essential role of ALG6 in the quality control of glycoprotein.
LRP6
LRP6 and LRP5 shared 71% of identity in the amino acid level (Brown et al., 1998). They are always co-expressed at the embryonic stage (Pinson et al., 2000) and function together as the co-receptor of Fzd and mediate the Wnt signaling. Since the deficiency in LRP5 resulting in reduced Wnt signaling could be a pathogenic reason for PCLD, deficiency in LRP6 might also be related to PCLD.
Conclusion
PCLD consists of a heterogeneous group of disorders with different genetic mutations and disease presentations. Glycosylation, translocation, and quality control of protein in ER play a central role in the diseases. However, the established candidate genes can only explain less than half of the cases. More candidate genes of diseases should be explored and investigated in the future.
Author Contributions
ZY and JC has designed the study. ZY and XS has written the manuscript. CH, JZ and AW have made contributions to the revision.
Funding
This study was supported by the National Natural Science Foundation of China (81960111) and Natural Science Foundation of Jiangxi Province (20202BABL206013). ZY was supported by the China Scholarship Council (202008360174). XS was supported by the China Scholarship Council (201909110092).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank the reviewers of the manuscript, whose thoughtful and insightful comments helped improve this review and also thank the funding sources and all colleagues for useful discussion.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.846877/full#supplementary-material
Supplementary Table S1 | The precise information of all reported mutations in PRKCSH and SEC63 gene including chromosomal location and cohort size of the studies.
References
Aebi, M. (2013). N-linked Protein Glycosylation in the ER. Biochim. Biophys. Acta (Bba) - Mol. Cel Res. 1833, 2430–2437. doi:10.1016/j.bbamcr.2013.04.001
Albuquerque-Wendt, A., Hütte, H. J., Buettner, F. F. R., Routier, F. H., and Bakker, H. (2019). Membrane Topological Model of Glycosyltransferases of the GT-C Superfamily. Int. J. Mol. Sci. 20, 4842. doi:10.3390/ijms20194842
Alvaro, D., Barbaro, B., Franchitto, A., Onori, P., Glaser, S. S., Alpini, G., et al. (2006). Estrogens and Insulin-like Growth Factor 1 Modulate Neoplastic Cell Growth in Human Cholangiocarcinoma. Am. J. Pathol. 169, 877–888. doi:10.2353/ajpath.2006.050464
Badders, N. M., Goel, S., Clark, R. J., Klos, K. S., Kim, S., Bafico, A., et al. (2009). The Wnt Receptor, Lrp5, Is Expressed by Mouse Mammary Stem Cells and Is Required to Maintain the Basal Lineage. PLoS One 4, e6594. doi:10.1371/journal.pone.0006594
Besse, W., Chang, A. R., Luo, J. Z., Triffo, W. J., Moore, B. S., Gulati, A., et al. (2019). ALG9 Mutation Carriers Develop Kidney and Liver Cysts. Jasn 30, 2091–2102. doi:10.1681/asn.2019030298
Besse, W., Choi, J., Ahram, D., Mane, S., Sanna-Cherchi, S., Torres, V., et al. (2018). A Noncoding Variant in GANAB Explains Isolated Polycystic Liver Disease (PCLD) in a Large Family. Hum. Mutat. 39, 378–382. doi:10.1002/humu.23383
Besse, W., Dong, K., Choi, J., Punia, S., Fedeles, S. V., Choi, M., et al. (2017). Isolated Polycystic Liver Disease Genes Define Effectors of Polycystin-1 Function. J. Clin. Invest. 127, 3558. doi:10.1172/jci96729
Boerrigter, M. M., Bongers, E. M. H. F., Lugtenberg, D., Nevens, F., and Drenth, J. P. H. (2021). Polycystic Liver Disease Genes: Practical Considerations for Genetic Testing. Eur. J. Med. Genet. 64, 104160. doi:10.1016/j.ejmg.2021.104160
Brown, S. D., Twells, R. C. J., Hey, P. J., Cox, R. D., Levy, E. R., Soderman, A. R., et al. (1998). Isolation and Characterization ofLRP6,a Novel Member of the Low Density Lipoprotein Receptor Gene Family. Biochem. Biophysical Res. Commun. 248, 879–888. doi:10.1006/bbrc.1998.9061
Chandok, N. (2012). Polycystic Liver Disease: a Clinical Review. Ann. Hepatol. 11, 819–826. doi:10.1016/s1665-2681(19)31406-1
Chrispijn, M., Gevers, T. J. G., Hol, J. C., Monshouwer, R., Dekker, H. M., and Drenth, J. P. H. (2013). Everolimus Does Not Further Reduce Polycystic Liver Volume when Added to Long Acting Octreotide: Results from a Randomized Controlled Trial. J. Hepatol. 59, 153–159. doi:10.1016/j.jhep.2013.03.004
Chrispijn, M., Nevens, F., Gevers, T. J. G., Vanslembrouck, R., van Oijen, M. G. H., Coudyzer, W., et al. (2012). The Long-Term Outcome of Patients with Polycystic Liver Disease Treated with Lanreotide. Aliment. Pharmacol. Ther. 35, 266–274. doi:10.1111/j.1365-2036.2011.04923.x
Cnossen, W. R., te Morsche, R. H. M., Hoischen, A., Gilissen, C., Chrispijn, M., Venselaar, H., et al. (2014). Whole-exome Sequencing Reveals LRP5 Mutations and Canonical Wnt Signaling Associated with Hepatic Cystogenesis. Proc. Natl. Acad. Sci. U.S.A. 111, 5343–5348. doi:10.1073/pnas.1309438111
Cornec-Le Gall, E., Torres, V. E., and Harris, P. C. (2018). Genetic Complexity of Autosomal Dominant Polycystic Kidney and Liver Diseases. Jasn 29, 13–23. doi:10.1681/asn.2017050483
Croce, J. C., and McClay, D. R. (2008). Evolution of the Wnt Pathways. Methods Mol. Biol. 469, 3–18. doi:10.1007/978-1-60327-469-2_1
Davila, S., Furu, L., Gharavi, A. G., Tian, X., Onoe, T., Qian, Q., et al. (2004). Mutations in SEC63 Cause Autosomal Dominant Polycystic Liver Disease. Nat. Genet. 36, 575–577. doi:10.1038/ng1357
Drenth, J. P. H., Chrispijn, M., Nagorney, D. M., Kamath, P. S., and Torres, V. E. (2010). Medical and Surgical Treatment Options for Polycystic Liver Disease1. Hepatology 52, 2223–2230. doi:10.1002/hep.24036
Drenth, J. P. H., Tahvanainen, E., te Morsche, R. H. M., Tahvanainen, P., Kääriäinen, H., Höckerstedt, K., et al. (2004). Abnormal Hepatocystin Caused by Truncating PRKCSH Mutations Leads to Autosomal Dominant Polycystic Liver Disease. Hepatology 39, 924–931. doi:10.1002/hep.20141
Drenth, J. P. H., te Morsche, R. H. M., Smink, R., Bonifacino, J. S., and Jansen, J. B. M. J. (2003). Germline Mutations in PRKCSH Are Associated with Autosomal Dominant Polycystic Liver Disease. Nat. Genet. 33, 345–347. doi:10.1038/ng1104
Everson, G. T., and Taylor, M. R. G. (2005). Management of Polycystic Liver Disease. Curr. Gastroenterol. Rep. 7, 19–25. doi:10.1007/s11894-005-0061-6
Gao, H., Wang, Y., Wegierski, T., Skouloudaki, K., Pütz, M., Fu, X., et al. (2010). PRKCSH/80K-H, the Protein Mutated in Polycystic Liver Disease, Protects polycystin-2/TRPP2 against HERP-Mediated Degradation. Hum. Mol. Genet. 19, 16–24. doi:10.1093/hmg/ddp463
Gevers, T. J. G., and Drenth, J. P. H. (2013). Diagnosis and Management of Polycystic Liver Disease. Nat. Rev. Gastroenterol. Hepatol. 10, 101–108. doi:10.1038/nrgastro.2012.254
Gkika, D., Mahieu, F., Nilius, B., Hoenderop, J. G. J., and Bindels, R. J. M. (2004). 80K-H as a New Ca2+ Sensor Regulating the Activity of the Epithelial Ca2+ Channel Transient Receptor Potential Cation Channel V5 (TRPV5). J. Biol. Chem. 279, 26351–26357. doi:10.1074/jbc.m403801200
Höck, M., Wegleiter, K., Ralser, E., Kiechl-Kohlendorfer, U., Scholl-Bürgi, S., Fauth, C., et al. (2015). ALG8-CDG: Novel Patients and Review of the Literature. Orphanet J. Rare Dis. 10, 73. doi:10.1186/s13023-015-0289-7
Hoevenaren, I. A., Wester, R., Schrier, R. W., McFann, K., Doctor, R. B., Drenth, J. P., et al. (2008). Polycystic Liver: Clinical Characteristics of Patients with Isolated Polycystic Liver Disease Compared with Patients with Polycystic Liver and Autosomal Dominant Polycystic Kidney Disease. Liver Int. 28, 264–270. doi:10.1111/j.1478-3231.2007.01595.x
Inoki, K., Ouyang, H., Zhu, T., Lindvall, C., Wang, Y., Zhang, X., et al. (2006). TSC2 Integrates Wnt and Energy Signals via a Coordinated Phosphorylation by AMPK and GSK3 to Regulate Cell Growth. Cell 126, 955–968. doi:10.1016/j.cell.2006.06.055
Janssen, M. J., Salomon, J., Te Morsche, R. H. M., and Drenth, J. P. H. (2012). Loss of Heterozygosity Is Present in SEC63 Germline Carriers with Polycystic Liver Disease. PLoS One 7, e50324. doi:10.1371/journal.pone.0050324
Janssen, M. J., Waanders, E., Te Morsche, R. H. M., Xing, R., Dijkman, H. B. P. M., Woudenberg, J., et al. (2011). Secondary, Somatic Mutations Might Promote Cyst Formation in Patients with Autosomal Dominant Polycystic Liver Disease. Gastroenterology 141, 2056–2063. doi:10.1053/j.gastro.2011.08.004
Johnson, M. L., Harnish, K., Nusse, R., and Van Hul, W. (2004). LRP5 and Wnt Signaling: a union Made for Bone. J. Bone Miner Res. 19, 1749–1757. doi:10.1359/jbmr.040816
Kim, S., Nie, H., Nesin, V., Tran, U., Outeda, P., Bai, C.-X., et al. (2016). The Polycystin Complex Mediates Wnt/Ca2+ Signalling. Nat. Cel Biol 18, 752–764. doi:10.1038/ncb3363
Kleffmann, J., Frank, V., Ferbert, A., and Bergmann, C. (2012). Dosage-sensitive Network in Polycystic Kidney and Liver Disease: Multiple Mutations Cause Severe Hepatic and Neurological Complications. J. Hepatol. 57, 476–477. doi:10.1016/j.jhep.2012.03.001
Kühl, M., Sheldahl, L. C., Park, M., Miller, J. R., and Moon, R. T. (2000). The Wnt/Ca2+ Pathway. Trends Genet. 16, 279–283. doi:10.1016/s0168-9525(00)02028-x
Lang, S., Pfeffer, S., Lee, P.-H., Cavalié, A., Helms, V., Förster, F., et al. (2017). An Update on Sec61 Channel Functions, Mechanisms, and Related Diseases. Front. Physiol. 8, 887. doi:10.3389/fphys.2017.00887
Lee-Law, P. Y., van de Laarschot, L. F. M., Banales, J. M., and Drenth, J. P. H. (2019). Genetics of Polycystic Liver Diseases. Curr. Opin. Gastroenterol. 35, 65–72. doi:10.1097/mog.0000000000000514
Li, A., Davila, S., Furu, L., Qian, Q., Tian, X., Kamath, P. S., et al. (2003). Mutations in PRKCSH Cause Isolated Autosomal Dominant Polycystic Liver Disease. Am. J. Hum. Genet. 72, 691–703. doi:10.1086/368295
Mao, J., Wang, J., Liu, B., Pan, W., Farr, G. H., Flynn, C., et al. (2001). Low-density Lipoprotein Receptor-Related Protein-5 Binds to Axin and Regulates the Canonical Wnt Signaling Pathway. Mol. Cel 7, 801–809. doi:10.1016/s1097-2765(01)00224-6
Masyuk, T. V., Masyuk, A. I., and LaRusso, N. F. (2018). Polycystic Liver Disease: The Interplay of Genes Causative for Hepatic and Renal Cystogenesis. Hepatology 67, 2462–2464. doi:10.1002/hep.29708
Misselwitz, B., Staeck, O., Matlack, K. E. S., and Rapoport, T. A. (1999). Interaction of BiP with the J-Domain of the Sec63p Component of the Endoplasmic Reticulum Protein Translocation Complex. J. Biol. Chem. 274, 20110–20115. doi:10.1074/jbc.274.29.20110
Nusse, R., and Clevers, H. (2017). Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 169, 985–999. doi:10.1016/j.cell.2017.05.016
Oriol, R., Martinez-Duncker, I., Chantret, I., Mollicone, R., and Codogno, P. (2002). Common Origin and Evolution of Glycosyltransferases Using Dol-P-Monosaccharides as Donor Substrate. Mol. Biol. Evol. 19, 1451–1463. doi:10.1093/oxfordjournals.molbev.a004208
Peces, R., Drenth, J. P., Te Morsche, R. H., González, P., and Peces, C. (2005). Autosomal Dominant Polycystic Liver Disease in a Family without Polycystic Kidney Disease Associated with a Novel Missense Protein Kinase C Substrate 80K-H Mutation. Wjg 11, 7690–7693. doi:10.3748/wjg.v11.i48.7690
Pinson, K. I., Brennan, J., Monkley, S., Avery, B. J., and Skarnes, W. C. (2000). An LDL-Receptor-Related Protein Mediates Wnt Signalling in Mice. Nature 407, 535–538. doi:10.1038/35035124
Porath, B., Gainullin, V. G., Cornec-Le Gall, E., Dillinger, E. K., Heyer, C. M., Hopp, K., et al. (2016). Mutations in GANAB , Encoding the Glucosidase IIα Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. Am. J. Hum. Genet. 98, 1193–1207. doi:10.1016/j.ajhg.2016.05.004
Qian, Q., Du, H., King, B. F., Kumar, S., Dean, P. G., Cosio, F. G., et al. (2008). Sirolimus Reduces Polycystic Liver Volume in ADPKD Patients. Jasn 19, 631–638. doi:10.1681/asn.2007050626
Qian, Q. (2010). Isolated Polycystic Liver Disease. Adv. Chronic Kidney Dis. 17, 181–189. doi:10.1053/j.ackd.2009.12.005
Ren, Q., Chen, J., and Liu, Y. (2021). LRP5 and LRP6 in Wnt Signaling: Similarity and Divergence. Front. Cel Dev. Biol. 9, 670960. doi:10.3389/fcell.2021.670960
Reynolds, D. M., Falk, C. T., Li, A., King, B. F., Kamath, P. S., Huston, J., et al. (2000). Identification of a Locus for Autosomal Dominant Polycystic Liver Disease, on Chromosome 19p13.2-13.1. Am. J. Hum. Genet. 67, 1598–1604. doi:10.1086/316904
Russell, R. T., and Pinson, C. W. (2007). Surgical Management of Polycystic Liver Disease. Wjg 13, 5052–5059. doi:10.3748/wjg.v13.i38.5052
Sawakami, K., Robling, A. G., Ai, M., Pitner, N. D., Liu, D., Warden, S. J., et al. (2006). The Wnt Co-receptor LRP5 Is Essential for Skeletal Mechanotransduction but Not for the Anabolic Bone Response to Parathyroid Hormone Treatment. J. Biol. Chem. 281, 23698–23711. doi:10.1074/jbc.m601000200
Schnell, D. J., and Hebert, D. N. (2003). Protein Translocons. Cell 112, 491–505. doi:10.1016/s0092-8674(03)00110-7
Serra, A. L., Poster, D., Kistler, A. D., Krauer, F., Raina, S., Young, J., et al. (2010). Sirolimus and Kidney Growth in Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 363, 820–829. doi:10.1056/nejmoa0907419
Sherstha, R., McKinley, C., Russ, P., Scherzinger, A., Bronner, T., Showalter, R., et al. (1997). Postmenopausal Estrogen Therapy Selectively Stimulates Hepatic Enlargement in Women with Autosomal Dominant Polycystic Kidney Disease. Hepatology 26, 1282–1286. doi:10.1002/hep.510260528
Spirli, C., Mariotti, V., Villani, A., Fabris, L., Fiorotto, R., and Strazzabosco, M. (2017). Adenylyl Cyclase 5 Links Changes in Calcium Homeostasis to cAMP-dependent Cyst Growth in Polycystic Liver Disease. J. Hepatol. 66, 571–580. doi:10.1016/j.jhep.2016.10.032
Sun, L., Eklund, E. A., Van Hove, J. L. K., Freeze, H. H., and Thomas, J. A. (2005). Clinical and Molecular Characterization of the First Adult Congenital Disorder of Glycosylation (CDG) Type Ic Patient. Am. J. Med. Genet. 137A, 22–26. doi:10.1002/ajmg.a.30831
Tu, X., Joeng, K. S., Nakayama, K. I., Nakayama, K., Rajagopal, J., Carroll, T. J., et al. (2007). Noncanonical Wnt Signaling through G Protein-Linked PKCδ Activation Promotes Bone Formation. Develop. Cel 12, 113–127. doi:10.1016/j.devcel.2006.11.003
Ushioda, R., Hoseki, J., and Nagata, K. (2013). Glycosylation-independent ERAD Pathway Serves as a Backup System under ER Stress. MBoC 24, 3155–3163. doi:10.1091/mbc.e13-03-0138
van Aerts, R. M. M., van de Laarschot, L. F. M., Banales, J. M., and Drenth, J. P. H. (2018). Clinical Management of Polycystic Liver Disease. J. Hepatol. 68, 827–837. doi:10.1016/j.jhep.2017.11.024
van de Laarschot, L. F. M., and Drenth, J. P. H. (2018). Genetics and Mechanisms of Hepatic Cystogenesis. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis. 1864, 1491–1497. doi:10.1016/j.bbadis.2017.08.003
van de Laarschot, L. F. M., Te Morsche, R. H. M., Hoischen, A., Venselaar, H., Roelofs, H. M., Cnossen, W. R., et al. (2020). Novel GANAB Variants Associated with Polycystic Liver Disease. Orphanet J. Rare Dis. 15, 302. doi:10.1186/s13023-020-01585-4
Van Keimpema, L., De Koning, D. B., Van Hoek, B., Van Den Berg, A. P., Van Oijen, M. G. H., De Man, R. A., et al. (2011). Patients with Isolated Polycystic Liver Disease Referred to Liver Centres: Clinical Characterization of 137 Cases. Liver Int. 31, 92–98. doi:10.1111/j.1478-3231.2010.02247.x
Waanders, E., te Morsche, R. H. M., de Man, R. A., Jansen, J. B. M. J., and Drenth, J. P. H. (2006). Extensive Mutational Analysis ofPRKCSH andSEC63 Broadens the Spectrum of Polycystic Liver Disease. Hum. Mutat. 27, 830. doi:10.1002/humu.9441
Waanders, E., Venselaar, H., te Morsche, R., de Koning, D., Kamath, P., Torres, V., et al. (2010). Secondary and Tertiary Structure Modeling Reveals Effects of Novel Mutations in Polycystic Liver Disease Genes PRKCSH and SEC63. Clin. Genet. 78, 47–56. doi:10.1111/j.1399-0004.2009.01353.x
Walz, G., Budde, K., Mannaa, M., Nürnberger, J., Wanner, C., Sommerer, C., et al. (2010). Everolimus in Patients with Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 363, 830–840. doi:10.1056/nejmoa1003491
Keywords: polycystic liver disease, PLD, PCLD, PRKCSH, Sec63, LRP5, alg8
Citation: Yu Z, Shen X, Hu C, Zeng J, Wang A and Chen J (2022) Molecular Mechanisms of Isolated Polycystic Liver Diseases. Front. Genet. 13:846877. doi: 10.3389/fgene.2022.846877
Received: 31 December 2021; Accepted: 23 March 2022;
Published: 26 April 2022.
Edited by:
Olga Zolotareva, Technical University of Munich, GermanyReviewed by:
Neera Dahl, Yale Medicine, United StatesEvgenia Zotova, FSBI “National Medical Research Center of Endocrinology” of the Ministry of Health of Russia, Russia
Copyright © 2022 Yu, Shen, Hu, Zeng, Wang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianyong Chen, Y2p5YWN5NjlAMTYzLmNvbQ==
†These authors have contributed equally to this work