
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 25 July 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.845081
This article is part of the Research Topic Exploiting Genetics and Genomics to Improve the Understanding of Eye Diseases View all 14 articles
 Junkai Tan1†
Junkai Tan1† Liuzhi Zeng2†Yun Wang3†Guo Liu4†Longxiang Huang5
Liuzhi Zeng2†Yun Wang3†Guo Liu4†Longxiang Huang5 Defu Chen6Xizhen Wang3Ning Fan3Yu He2
Defu Chen6Xizhen Wang3Ning Fan3Yu He2 Xuyang Liu1,7*
Xuyang Liu1,7*The molecular mechanisms underlying the pathogenesis of pigment dispersion syndrome and pigmentary glaucoma remain unclear. In pedigree-based studies, familial aggregation and recurrences in relatives suggest a strong genetic basis for pigmentary glaucoma. In this study, we aimed to identify the genetic background of two Chinese pedigrees with pigmentary glaucoma. All members of these two pedigrees who enrolled in the study underwent a comprehensive ophthalmologic examination, and genomic DNA was extracted from peripheral venous blood samples. Whole-exome sequencing and candidate gene verifications were performed to identify the disease-causing variants; in addition, screening of the CPAMD8 gene was performed on 38 patients of sporadic pigmentary glaucoma. Changes in the structure and function of abnormal proteins caused by gene variants were analyzed with a bioinformatics assessment. Pigmentary glaucoma was identified in a total of five patients from the two pedigrees, as were compound heterozygous variants of the CPAMD8 gene. No signs of pigmentary glaucoma were found in carriers of monoallelic CPAMD8 variant/variants. All four variants were inherited in an autosomal recessive mode. In addition to the 38 patients of sporadic pigmentary glaucoma, 13 variants of the CPAMD8 gene were identified in 11 patients. This study reported a possible association between CPAMD8 variants and pigment dispersion syndrome/pigmentary glaucoma.
Pigment dispersion syndrome (PDS) is a relatively rare ocular disorder that affects mostly males, especially in Caucasian countries (Niyadurupola and Broadway, 2008). PDS can lead to pigmentary glaucoma (PG), a type of open-angle glaucoma secondary to excessive deposition of pigment in the trabecular meshwork (TM). The deposited pigment blocks the conventional outflow pathway, leading to an increased resistance to aqueous humor outflow and elevated intraocular pressure (IOP). It is generally believed that the dispersed pigment originates from the shedding of the pigment epithelium located on the posterior surface of the iris (Scuderi et al., 2019). In a prospective study, the criteria for PDS were set as two of three signs: Krukenberg’s spindle, iris transillumination defects, and/or TM pigmentation; in contrast, PG was diagnosed when two of three signs were seen in PDS eyes: IOP greater than 21 mmHg, glaucomatous optic nerve damage, and/or glaucomatous visual field (VF) loss (Tandon et al., 2019). However, iris transillumination defects were not common in East Asian populations probably because these defects cannot be detected in darkly pigmented irides of East Asian patients via slit-lamp examination (Qing et al., 2009). To date, the molecular mechanisms underlying the pathogenesis of PDS and PG remain unclear. In pedigree-based studies, familial aggregation and recurrences in relatives suggest a strong genetic basis for PG (Siddiqui et al., 2003). In addition, variants of the premelanosome (PMEL) gene were associated with heritable patients of PDS/PG (Lahola-Chomiak et al., 2019), and variants of the pigment dispersion–associated genes in animals, such as Dct, Tyrp1, and Lyst, mimicked some clinical features of PDS and PG (Chang et al., 1999; Anderson et al., 2008; Trantow et al., 2009); however, no studies have found a causal relationship between any variants within their human orthologs and PG. Therefore, it is likely that there are other genes involved in the pathogenesis of PDS/PG. Posterior bowing of the iris is frequently present in PDS/PG, causing iridozonular contact, zonular rubbing, and pigment dispersion, which results in excessive pigment deposition in the anterior chamber angle tissues, especially the TM, as aqueous humor and dispersed pigment follow the normal aqueous humor dynamics through the outflow pathway (Lyons and Amram, 2019). Giardina et al. identified the haplotypes of the LOXL1 gene in a cohort of patients with PDS/PG and validated the pathogenic effects of the LOXL1 variant in an animal model. They also speculated that LOXL1 variant–induced defects of the iris stroma are the reason for iris concavity, a phenotype often present in PDS/PG patients (Giardina et al., 2014). However, another study showed that there was no correlation between LOXL1 and PDS/PG (Rao et al., 2008). Hence, the specific pathogenic factor and mechanism of PDS/PG remain poorly understood. Nevertheless, these studies indicate that genetic factors could be associated with an increased risk of developing PDS/PG.
The human CPAMD8 gene (complement 3- and pregnancy zone protein–like, alpha-2-macroglobulin domain–containing 8; OMIM 608841) encodes a protein involved in the immune system (Nagase et al., 1999; Li et al., 2004; Wiggs, 2020). Variants of the CPAMD8 gene have been recently identified as causative factors for anterior segment dysgenesis (ASD) (Cheong et al., 2016; Bonet-Fernandez et al., 2020; Ma et al., 2020), morgagnian cataract (Hollmann et al., 2017), and different subtypes of glaucoma, including POAG, PACG, and congenital glaucoma (Bonet-Fernandez et al., 2020; Siggs et al., 2020; Wiggs, 2020; Li X. et al., 2021b), indicating that CPAMD8 variants may lead to primary and secondary glaucoma; different variants of the CPAMD8 gene may also determine the presentation and severity of ocular phenotypes.
In the current study, compound heterozygous CPAMD8 variants were identified in two pedigrees with PG. Moreover, autosomal recessive inheritance was found in the study of PG pedigrees. Our study reported a possible association between the biallelic CPAMD8 gene variants and PDS/PG.
Two Chinese pedigrees of PG and 38 sporadic PG patients were recruited at Shenzhen Eye Hospital and Chengdu First People’s Hospital, respectively. Members of both pedigrees and sporadic PG patients were systematically interviewed about their medical history and clinically examined by experienced glaucoma specialists; in addition, peripheral blood was collected for DNA analysis. This study was approved by the Medical Ethics Committees of Shenzhen Eye Hospital, Chengdu First People’s Hospital, and Xiamen Eye Center and was conducted according to the principles of the Declaration of Helsinki. Informed written consent was obtained from all participants prior to inclusion in the study.
All members enrolled in the study underwent a comprehensive ophthalmologic examination, which included best corrected visual acuity, IOP measurement, slit-lamp biomicroscopy, gonioscopy, and fundus examination. The ocular biometric parameters of each participant were measured via optical biometry (IOL Master 700, Carl Zeiss, Germany). Glaucomatous optic neuropathy (GON) was evaluated using slit-lamp examination with a concave preset lens (Hruby lens), fundus photography, and spectral domain optical coherence tomography (SD-OCT, Carl Zeiss). VF was examined using the Humphrey VF Analyzer (Carl Zeiss). The anterior chamber angle structure was recorded via ultrasound biomicroscopy (UBM, Tianjin Suowei Electronic Technology Co., Ltd., China) and gonioscopic photography.
The PDS/PG was diagnosed according to the criteria previously reported (Tandon et al., 2019) with minor modifications. In brief, for PDS, subjects should have two signs in at least one eye as follows: Krukenberg’s spindle and heavily pigmented TM (Grade 3 or higher). For PG, subjects should have PDS plus at least two of the three signs in at least one eye as follows: IOP > 21 mmHg, GON (C/D ratio > 0.5), and glaucomatous VF defect. The degree of TM pigmentation was graded from 0 to 4 according to severity (0 = no pigment; 1 = light pigment; 2 = moderate pigment; 3 = heavy and non-confluent pigment; and 4 = heavy and confluent pigment) (Tandon et al., 2019). The severity of Krukenberg’s spindle was also graded from 0 to 4 (0 = none; 1 = few flecks; 2 = subtle spindle; 3 = dense spindle; 4 = diffuse pigment) (Tandon et al., 2019).
Genomic DNA of all individuals was extracted from peripheral venous blood samples using the Qiamp Blood DNA Mini Kit (QIAGEN GmbH, Hilden, Germany) according to the manufacturer’s instructions. DNA integrity was evaluated via 1% agarose gel electrophoresis. Whole-exome sequencing (WES) was performed on three patients in pedigree 1 and the proband in pedigree 2 by Shanghai Genesky Biotechnologies, Inc., and Shanghai WeHealth BioMedical Technology, Co., Ltd, respectively, according to previously described methods (Bao et al., 2019). Exons and adjacent splicing regions (about 20 bp), as well as the full length of the mitochondrial genome, were captured and enriched by SeqCap EZ Exome Probes v3.0 (Roche, Switzerland) hybridization. After the quality control steps, the enriched genes were sequenced using an Illumina HiSeq X Ten platform (San Diego, CA).
The original WES data, excluding unqualified reads according to the quality control standard, were compared with the human reference genome sequence from the University of California Santa Cruz Genome Browser Database using the BWA software (hg19 version). The single-nucleotide variation and insertion and deletion (indel) variants were found using HaplotypeCaller of the genome analysis toolkit. The identified variants were classified in silico and filtered against the genome aggregation database (gnomAD, version 3.1, https://gnomad.broadinstitute.org/). Variants were filtered for minor allele frequency (MAF) ≤ 1% (0.01) in gnomAD. Professional databases and bioinformation prediction software, including SnapGene (version 4.3, www.snapgene.com), Ensembl (https://asia.ensembl.org/index.html), DUET (http://biosig.unimelb.edu.au/duet/stability), and VarSite (https://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/VarSite/GetPage.pl?home=TRUE), were used for further annotation and screening of variants of interest, such as missense, synonymous, and noncanonical splicing variants, according to reference sequences NM_015692.5 and NP_056507.3. SnapGene was used to align the protein sequences among different species. The possible functional impact of an amino acid change was predicted using PolyPhen-2, SIFT, CADD, DUET, and VarSite. The first three prediction data (PolyPhen-2, SIFT, and CADD) were acquired from the Ensembl website for uniforming their versions used in the study. The XHMM and CLAMMS algorithms were employed to analyze the copy number variation of the probe coverage area.
The variant-filtering steps were summarized as follows: according to the autosomal recessive inheritance or compound heterozygous inheritance model, the exome sequencing data of patients from family 1 (II:1, II:4, and II:5) and family 2 (II:3) were screened. Homozygous variants and two or more heterozygous variants of the same gene were obtained after filtration. Genes fully or partially related to clinical phenotype were taken into consideration. Glaucoma-associated genes, including MYOC, CYP1B1, FOXC1, LRP2, PITX2, PAX6, LTBP2, TEK, ANGPT1, and PMEL, were given special attention. Information on candidate genes and variants from both families were obtained through literature and human gene mutation database (HGMD) search using the American College of Medical Genetics and Genomics (ACMG)/AMP (PMID) criteria (Richards et al., 2015).
Direct Sanger sequencing was used to determine the co-segregation of identified variants with the clinical phenotype in pedigrees. Candidate variants identified through the aforementioned variant-filtering steps were validated by Sanger sequencing in all members of the two pedigrees. The primer flanking regions of candidate variants of the CPAMD8 gene (Gene ID 27151) were designed using Primer Premier 5 (Table 1) and synthesized by BGI (Guangzhou, China). Polymerase chain reaction (PCR) was performed using the MyCycler thermal cycler (Bio-Rad, Hercules, CA) in a 25-µL reaction system, which contained 0.1-µg genomic DNA, 40-μmol/L forward and reverse primers, 3-mmol/L magnesium chloride, and 2 × Taq Master Mix (SinoBio, Shanghai, China). The PCR conditions used were as follows: 5 min at 95°C for initial denaturation; 35 cycles of denaturation at 95°C for 30°s for melting; corresponding annealing temperature (TM value) in Table 1 lasting for 30°s; 30°s at 72°C for extension; and a final additional extension step of 10 min at 72°C. Before sequencing, 1% agarose gel electrophoresis was used to purify the target PCR fragments using the QIAquick Gel Extraction Kit (QIAGEN, Shanghai, China). Sanger sequencing was performed on an ABI 377XL automated DNA sequencer (Applied Biosystems, Foster City, CA). Sequence data were compared in a pairwise manner with the related human genome database.

TABLE 1. Primers used in polymerase chain reaction (PCR) for the amplification of the CPAMD8 gene.
The CPAMP8 gene was screened in 38 sporadic PG patients via WES as described above. Variants were filtered with gnomAD (East Asian population); variants with MAF ≤ 0.01 were considered variants or functional SNPs (non-synonymous SNPs).
Five patients from two pedigrees who carried biallelic CPAMD8 compound heterozygous variants presented with typical clinical phenotypes of PDS/PG, including Krukenberg’s spindle on the posterior surface of the cornea, heavily pigmented TM, elevated IOP, and GON. The clinical data of the patients in the two PG pedigrees with biallelic CPAMD8 variants were summarized in Table 2.
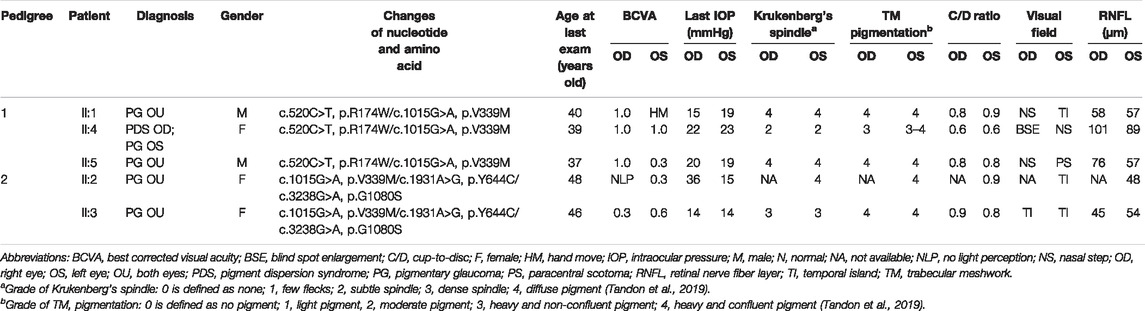
TABLE 2. Clinical data of patients in two pigmentary glaucoma (PG) pedigrees.
A relatively mild phenotype was observed in patient II:4 from pedigree 1, including subtle Krukenberg’s spindle and moderate pigmentation of the TM in both eyes, with elevated IOP, VF damage, and inferotemporal retinal nerve fiber layer (RNFL) defect in the left eye (Table 2). The patient’s elder and younger brothers, the other two patients in pedigree 1 (II:1 and II:5), presented with more significant glaucomatous manifestations, including increased IOP and GON. Diffuse Krukenberg’s spindle and grade 4 TM pigmentation were observed in both eyes. An enlarged C/D ratio (0.8–0.9), VF defects, and thinning RNFL confirmed the diagnosis of GON. UBM photographs of patient II:1 and II:5 (pedigree 1) revealed that the iris was bowed backward bilaterally, creating a reverse pupillary block and leading to a possible contact between the posterior surface of the iris and the zonules or anterior lens capsule (Figure 1 and Table 2).
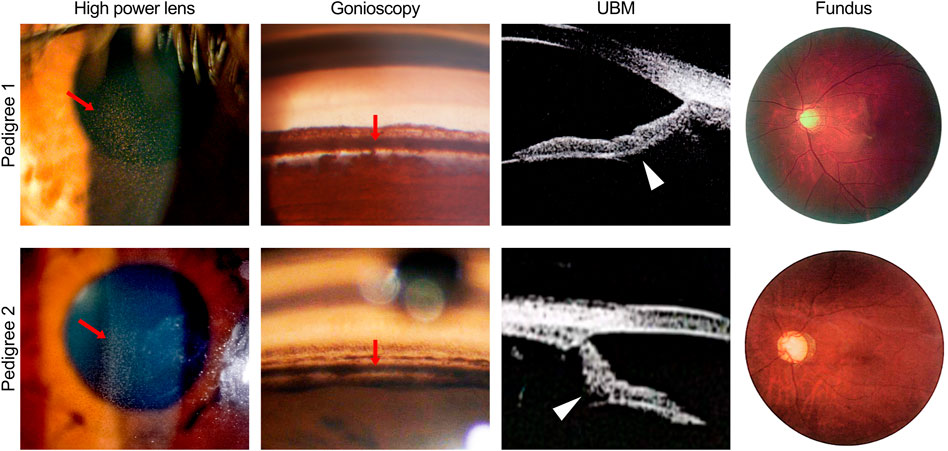
FIGURE 1. Ocular phenotypes of patients in two pigmentary glaucoma (PG) pedigrees associated with CPAMD8 variants. Clinical photography including slit-lamp biomicroscopy, gonioscopy, ultrasound biomicroscopy, and fundus photography. Representative signs of PG, such as Krukenberg’s spindle, TM pigmentation, and reverse pupillary block, were indicated by red arrows and white arrowheads, respectively. In pedigree 1, representative images were acquired from the right eye of patient II:5. In pedigree 2, representative images were acquired from the left eye of patient II:2 (slit-lamp biomicroscopy and fundus photography) and the right eye of patient II:3 (gonioscopy and ultrasound biomicroscopy).
The proband of pedigree 2 (II:3), a 46-year-old woman, underwent trabeculectomy in the right eye 2 years ago and presented with high myopia, poor corrected visual acuity, and controlled IOP with IOP-lowering medications in both eyes. Krukenberg’s spindle, lens opacity, and heavily pigmented TM were seen in both eyes. Bilateral typical GON with C/D ratio close to 1.0 was also observed (Figure 1 and Table 2).
The proband’s sister (II:2) from pedigree 2 had undergone glaucoma surgeries three or four times and presented with poor visual acuity and elevated IOP in both eyes. Due to the uncontrolled IOP and corneal endothelial decompensation in the right eye, the KP, TM, and optic disc could not be observed in detail. Krukenberg’s spindle, homogeneously and densely pigmented TM, and GON were detected in the left eye. UBM examination revealed a concave iris in both eyes of II:2 and II:3 in pedigree 2 (Figure 1 and Table 2).
The IOP presented in Table 2 were measured at the last follow-up visit of the patients, prior to which both II:1, II:5 in Family 1 and II:2, II:3 in Family 2 had undergone a single or multiple anti-glaucoma surgeries; five patients are currently treated with at least one anti-glaucoma drug. All the affected individuals, but not healthy individuals, in this study presented with elevated IOP before and during the treatments.
Elevated IOP, Krukenberg’s spindle, homogeneously and densely pigmented TM, and GON were also seen in 38 sporadic PG patients (data not shown). As expected, iris transillumination was not observed in patients enrolled in this study.
Carriers of the monoallelic CPAMD8 variant/variants were asymptomatic (Supplementary Table S1), except for the mother of two affected daughters in pedigree 2, who had a long history of inflammatory keratitis and presented with leucoma and normal IOP in both eyes. She denied a history of elevated IOP.
Common genetic characteristics were found in both three-generation pedigrees as follows: no history of consanguineous marriage; all patients were first filial generation; parents and second filial generation were not diagnosed with either PDS or PG; and segregation was consistent with autosomal recessive inheritance. WES and Sanger sequencing were used to investigate the underlying genetic alterations in the two pedigrees as described below, using pedigree 2 as an example (Figure 2A). The original sequencing data was 11,820.95 MB, and the average sequencing depth of the target region was 142.71X, with a 98.15% coverage of average sequencing depth of up to ×30. No pathogenic or possible pathogenic variation of mitochondrial genome and no copy number variation fully or partially related to the clinical phenotype of the patients were detected. After performing the variant-filtering steps according to the autosomal recessive inheritance or compound heterozygous inheritance model, a total of 176 homozygous variants of 67 genes (Supplementary Table S2) or two or more heterozygous variants of the same genes were shared by all affected individuals (II:1, II:4, and II 5) of family 1; on the contrary, a total of 230 variants of 100 genes (Supplementary Table S3) using similar filtering strategy were identified in the proband from family 2 (II:3). Information of all candidate genes in both families was obtained through literature and HGMD search, in which only CPAMD8 was previously reported in relation to glaucoma (Bonet-Fernandez et al., 2020; Siggs et al., 2020; Wiggs, 2020; Li X. et al., 2021b). Furthermore, Sanger sequencing verification of CPAMD8 was carried out with all family members in both pedigrees. It was confirmed that compound heterozygous variants (c.520C>T, p.R174W and c.1015G>A, p.V339M in pedigree 1; c.1015G>A, p.V339M; c.1931A>G, p.Y644C and c.3238G>A, p.G1080S in pedigree 2) in CPAMD8 were co-segregated in two pedigrees with PDS/PG patients (patient II:1, II:4, and II:5 from pedigree 1; patient II:2 and II:3 from pedigree 2; Figure 2A). All variants in this study were rare, with one (p.G1080S) unavailable in gnomAD (East Asian), at a frequency consistent with that of rare recessive disease (Table 3).
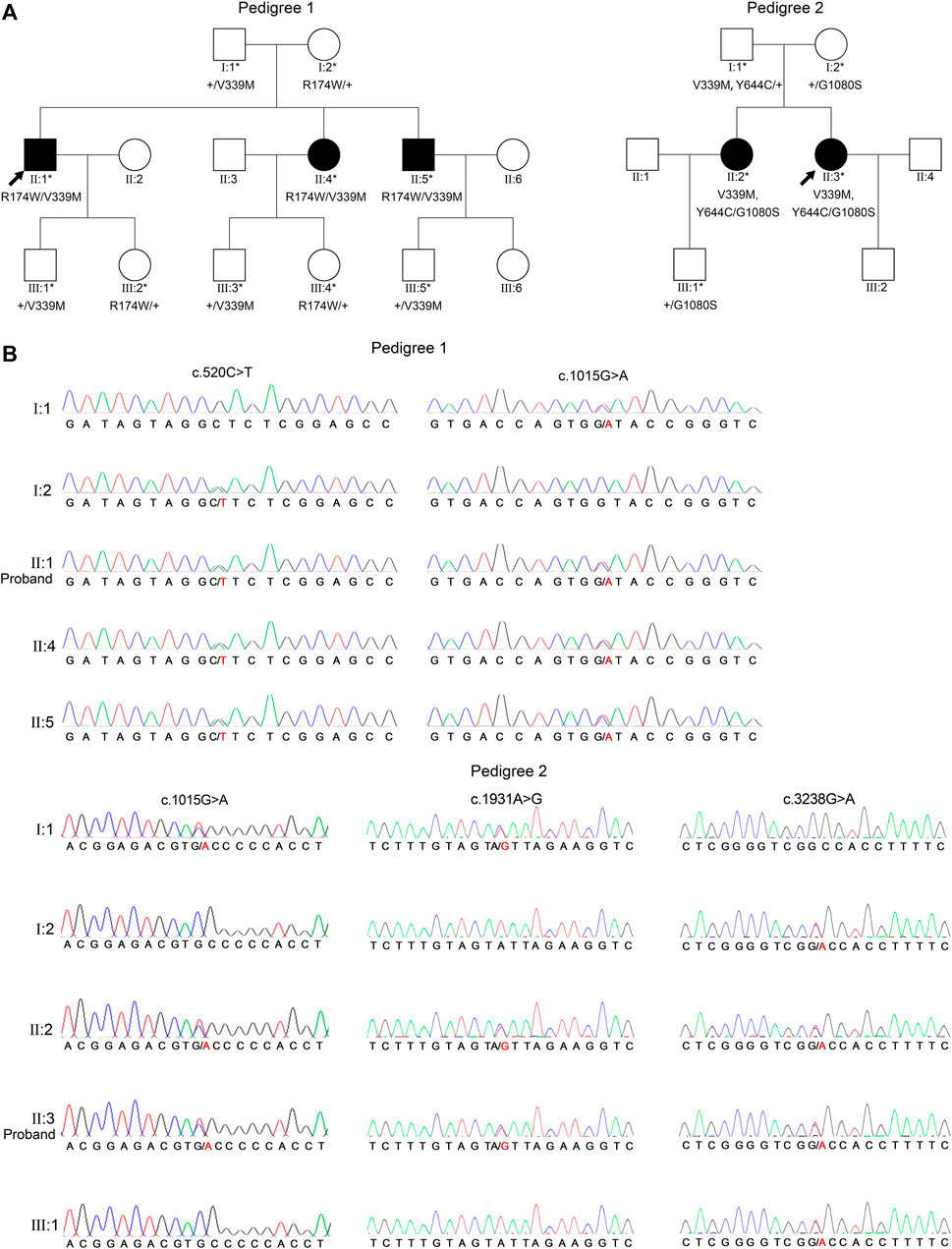
FIGURE 2. Two PG pedigrees with CPAMD8 variants and Sanger sequencing results of the CPAMD8 gene. (A) The two PG pedigrees described from our research. Round symbols indicate female individuals; square symbols indicate male individuals; black symbols indicate PG patients; arrow marks indicate the proband; asterisks represent subjects involved in genetic research. The plus signs denote reference allele. (B) Sanger sequencing verification and segregation analysis of biallelic CPAMD8 variants in two PG pedigrees. Variants are denoted in red font. Two different compound heterozygous variants of CPAMD8 were identified in two PG pedigrees. Abbreviations: PG, pigmentary glaucoma.
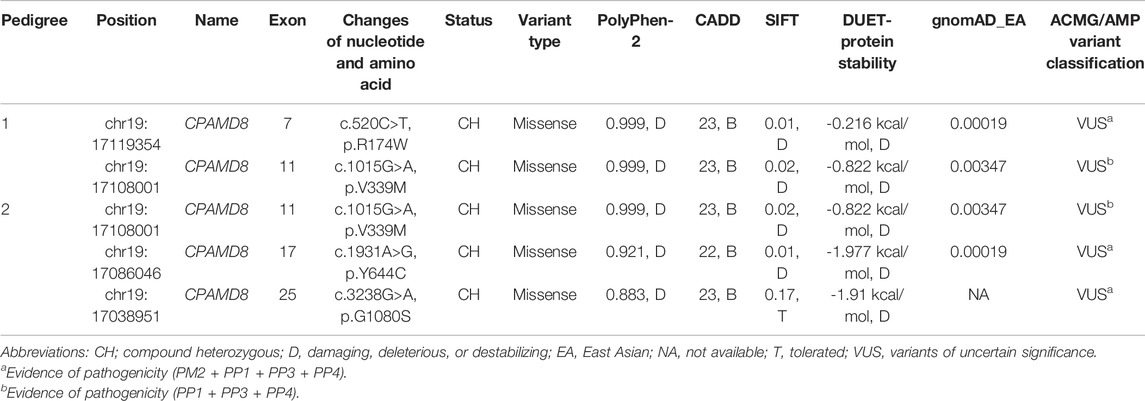
TABLE 3. Summary of PG pedigrees with CPAMD8 Variants.
In pedigree 1, compound heterozygous variants p.R174W (rs201333165) and p.V339M (rs369985652) in the CPAMD8 gene were identified via WES. The genotype-phenotype co-segregation was verified via Sanger sequencing in the whole pedigree. Variant p.R174W of CPAMD8 was inherited from the mother, whereas the other variant, p.V339M, was inherited from the father. The mode of inheritance was in accordance with compound heterozygous variation under a recessive inheritance pattern (Figure 2).
Two missense variants found in the CPAMD8 gene of the subject were classified as “variants of uncertain significance” according to the ACMG guidelines. Three online predictive software, PolyPhen-2, SIFT, and DUET, classified these two variants as damaging, destabilizing, or deleterious (Table 3). The macroglobulin domain 2 (MG2) (Figure 3A) is a domain of α-2-macroglobulin (A2M) in eukaryotes (Janssen et al., 2005). A2Ms, among the core domains of CPAMD8, are plasma proteins that trap and inhibit a broad range of proteases and are major components of the eukaryotic innate immune system (Wong and Dessen, 2014). The arginine (Arg) residue at position 174 is highly conserved (Figure 3B; conservation = 0.9 from 37 aligned protein sequences from different organisms). The change from an Arg to a tryptophan side chain was highly unfavored in terms of conserved amino acid properties, which might result in a change in protein function.
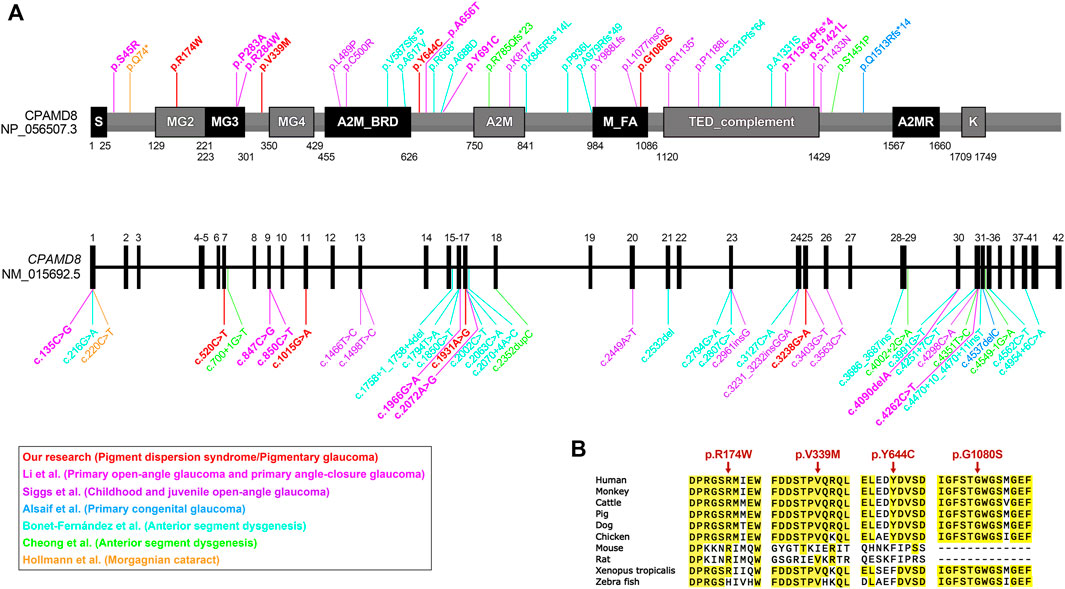
FIGURE 3. Bioinformatics analyses of CPAMD8 variants in two PG pedigrees. (A) Schematic representations showing variant sites on the sequence of the CPAMD8 gene and CPAMD8 protein identified in our study and other studies of the association between CPAMD8 gene and inherited ocular disorders (Cheong et al., 2016; Hollmann et al., 2017; Alsaif et al., 2019; Bonet-Fernandez et al., 2020; Siggs et al., 2020; Li X. et al., 2021b). Each study was plotted in a distinct color. Domains were indicated according to the Pfam database (https://pfam.xfam.org/). Abbreviations: S, signal peptide; A2M, α-2-macroglobulin domain; A2M_BRD, α-2-macroglobulin bait region domain; A2MR, α-macroglobulin receptor binding domain; MG, macroglobulin domain of α-2-macroglobulin; M_FA, farnesoic acid O-methyl transferase domain; TED, α-macroglobulin thioester domain; K, kazal-2 serine protease inhibitor domain. (B) Sequence alignments of the CPAMD8 protein among different species. Four missense variants were highly conserved in various species. The positions of the variant amino acids are indicated by red arrows.
In contrast to Arg at position 174, the valine residue at position 339 is relatively less conserved (Figure 3B; conservation = 0.7 from 43 aligned protein sequences from different organisms). This may not result in an obvious change in protein function.
In pedigree 2, three variants, p.V339M, p.Y644C, and p.G1080S, in the CPAMD8 gene were identified via WES. Two variants, p.V339M and p.Y644C, were inherited from their father, whereas another variant, p.G1080S, was inherited from their mother. These variants were co-segregating with the clinical phenotype verified via Sanger sequencing and segregation analysis and were accordant with compound heterozygous variation under a recessive inheritance pattern (Figure 2).
Three missense variants found in the CPAMD8 gene of the subject were classified as “variants of uncertain significance” according to the ACMG/AMP guidelines for variant classification. Variant p.V339M was presented in both pedigrees. Another two variants (p.Y644C and p.G1080S) were identified as highly conservative in different species (Figure 3B) and might result in instability of the CPAMD8 protein, as predicted by DUET (Table 3). The p.Y644C and p.G1080S variants were predicted to be damaging or deleterious by PolyPhen-2. SIFT predicted that the p.Y644C variant was deleterious and the p.G1080S variant was tolerable (Table 3). The heterozygous variant c.1931A>G of exon 17 in CPAMD8 would result in the substitution of tyrosine (Tyr), a neutral amino acid at position 644, by hydrophobic cysteine. The Tyr residue at position 644 is very highly conserved (Figure 3B; conservation = 0.9 from 43 aligned protein sequences from different organisms), rendering it significant for protein function.
The heterozygous variant c.3238G>A of exon 25 in CPAMD8 would result in the substitution of glycine (Gly) at position 1080 by serine, which is located at the end of the methyltransf-FA (farnesoic acid O-methyl transferase) domain (Figure 3A), a well-known key protein domain of the enzyme that catalyzes the formation of methyl farnesoate from farnesoic acid in the biosynthetic pathway of juvenile hormone (Li J. et al., 2021a). The Gly residue at position 1080 is fairly well conserved (Figure 3B; conservation = 0.8 from 47 aligned protein sequences), which means it is significant for protein function.
By screening the CPAMD8 gene on 38 sporadic PG patients (Table 4), 13 variants (c.1015G>A, c.503T>C, c.5242C>G, c.1173_1174del, c.92+1G>T, c.3786+1G>A, c.2117G>A, c.215C>T, c.959C>T, c.979G>T, c.2009G>A c.5342C>G, and c.4685+1G>A) of the CPAMD8 gene with MAF ≤0.01 were identified in 11 sporadic PG patients. Among them, one variant of CPAMD8 (c.1015G>A, p.V339M) was identified in pedigree 1 and pedigree 2 as well as one sporadic patient (patient 1) of PG, which was not reported previously. In addition, one sporadic PG patient (patient 2) carried three heterozygous variants of CPAMD8.
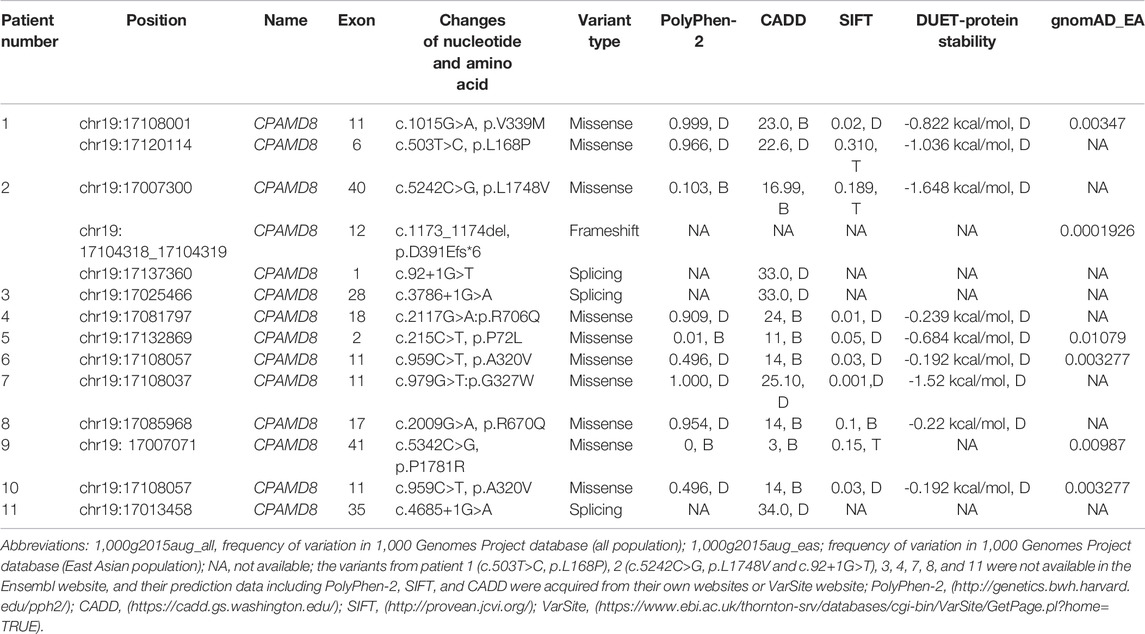
TABLE 4. Summary of sporadic PG patients with CPAMD8 variants.
In this study, compound heterozygous variants of the CPAMD8 gene were identified in five patients from two unrelated Chinese PG pedigrees via filtering of WES data and Sanger sequencing verification. These compound heterozygous variants of the CPAMD8 gene have not been previously reported in literatures. All variants appear to be damaging, destabilizing, or deleterious based on bioinformatics analysis. Through WES and filtering against several publicly accessible variation databases and eliminating all previously reported variants, other glaucoma-associated genes, including MYOC, CYP1B1, FOXC1, LRP2, PITX2, PAX6, LTBP2, TEK, ANGPT1, and PMEL, were excluded in these two pedigrees. Of the 13 variants of CPAMD8 identified in 38 sporadic PG patients@ with MAF ≤ 0.01, compound heterozygous variants were likely present in two of them (Table 4), and their parents were asymptomatic. These results indicate that the CPAMD8 gene variant is potentially associated with PDS/PG.
In previous studies, PDS was thought to be an autosomal dominant inherited disease (Scheie and Cameron, 1981; Mandelkorn et al., 1983; Tandon et al., 2019); meanwhile, there was only one autosomal recessive inheritance pattern for PG in a four-generation family reported (Stankovic., 1961; Lascaratos et al., 2013). Andersen et al. (Andersen et al., 1997) used microsatellite repeat markers distributed throughout the human genome to perform a genome screen of subjects from four pedigrees with PDS. They found that the responsible gene is located in a 10-centimorgan interval between markers D7S2462 and D7S2423 of chromosome 7. The two pedigrees in this study presented an autosomal recessive inheritance pattern. No PDS/PG phenotypes were found in the carriers of monoallelic CPAMD8 variant/variants from these two pedigrees. For pedigree 1, we have followed up the pedigree for 10 years and never found other pedigree members presenting with any phenotype of PDS/PG or glaucoma. In pedigree 2, patient I:1 with monoallelic CPAMD8 variants (c.1015G>A, p.V339M; c.1931A>G, p.Y644C) had no previous history of ocular abnormalities, and examinations on his eyes were unremarkable. Therefore, our data meet the necessary criteria indicating that CPAMD8 is involved in the inheritance of PG in a rare autosomal recessive pattern.
It was recently suggested that CPAMD8 variants may be involved in the development of ASD (Cheong et al., 2016; Bonet-Fernandez et al., 2020; Ma et al., 2020), morgagnian cataract (Hollmann et al., 2017), and different subtypes of glaucoma, including POAG, PACG, and congenital glaucoma (Bonet-Fernandez et al., 2020; Siggs et al., 2020; Li X. et al., 2021b). However, neither ASD nor morgagnian cataract was present in our study of PG patients with CPAMD8 missense variants, although these ocular disorders share similar background of changed CPAMD8. Li et al. performed a cohort study of the association between CPAMD8 variants and POAG/PACG and showed that biallelic truncation variants were more frequently associated with ASD, which usually presented with more severe phenotype, including iridocorneal adhesion and iris and ciliary body hypoplasia, whereas biallelic missense variants were more common in POAG/PACG (Li X. et al., 2021b), suggesting that the functional damages of CPAMD8 protein could be different for different variants/alterations in the CPAMD8 gene. The type of variants/alterations may determine the presentation and severity of the ocular phenotypes. In agreement with the previous report, all alterations identified in our study of PDS/PG with milder phenotypes than ASD were missense variants. We also found patient II:4 (female) of pedigree 1 who had later-onset and milder PG phenotypes (PDS in the right eye and PG in the left eye) than her elder and younger affected brothers, indicating that the clinical phenotypes of genetic disorders were possibly influenced by other factors, such as gender, epigenetic changes, and environmental exposure (Yan et al., 2010; Sarkar et al., 2014; Choi et al., 2020).
Variants of pigment dispersion–associated genes in animals, such as Dct, Tyrp1, and Lyst, mimicked some clinical features of PDS and PG (Chang et al., 1999; Anderson et al., 2008; Trantow et al., 2009); however, no studies have found a causal relationship between any variants within their human orthologs and PG. It is likely that there are non-pigment–related genes involved in the pathogenesis of PDS/PG. Iris concavity is often present in PDS/PG patients and leads to iridozonular chafing and pigment release (Lyons and Amram, 2019). Potential mechanisms for iridozonular chafing remain unclear. In a cohort study of PDS/PG patients, the LOXL1 variant was identified and further validated to be pathogenic in an animal model. It was speculated that this variant was responsible for iris stromal defects in PDS/PG patients (Giardina et al., 2014). This study provided a possible interpretation for close contact of iris epithelium and zonule, that stromal defects appear to be present in abnormal iris. However, another study showed that there was no correlation between LOXL1 and PDS/PG (Rao et al., 2008). An animal study examined the eyes of old DBA/2J mice (2.5–18 months), characterized its ocular pigment abnormalities, and speculated that the loss of pigment cells in the anterior chamber was at least in part caused by iris stromal atrophy with aging (Schraermeyer et al., 2009). This finding indicates that normal iris stroma may be necessary for the stability of pigment epithelium. In the anterior segment of the eye, CPAMD8 has been demonstrated to be expressed in neural crest–derived tissues, including the cornea, iris, ciliary body, and lens (Wiggs 2020). Siggs et al. found iris stromal hypoplasia in childhood and juvenile POAG patients with biallelic CPAMD8 variants (Siggs et al., 2020), which implied a possible role of CPAMD8 in iris stromal defects. Furthermore, CPAMD8 belongs to the α-2-macroglobulin/complement 3 protein family, whose members are involved in innate and acquired immune responses (Wiggs 2020). Wong et al. found that in the iris of patients with POAG or PCAG, the expression of IL-2 and IFN-γ (Th1 cytokine) increased, IL-6 (Th2 cytokine) decreased, and TGF-β (Th3 cytokine) increased. They also suggested the presence of an immune imbalance in glaucomatous eyes (Wong et al., 2015). In sum, whether CPAMD8 is involved in the immunological abnormalities of iris stroma in some subtypes of glaucoma is unknown. In the current study, we confirmed that CPAMD8 variants co-segregated with PDS/PG in two pedigrees. Further studies are needed to clarify whether CPAMD8 plays a role in the pathogenesis of iris stromal abnormalities (such as immunological abnormalities), iris epithelial cell loss, and pigment dispersion. In addition, the CPAMD8 protein was mainly detected in the nonpigmented epithelial cells of iris tissues (Siggs et al., 2020; Li X. et al., 2021b), which also implied that the effect of CPAMD8 variants on the loss of iris pigment epithelium may be indirect.
It is worth mentioning that iris transillumination defects, which are a feature of PDS and PG, were not observed in any patients in the two pedigrees or in sporadic PG patients, even though they presented with other typical clinical phenotypes of PDS/PG, including Krukenberg’s spindle and heavily and homogenously pigmented TM. One of the reasons is probably the fact that iris transillumination defects cannot be detected in darkly pigmented irides of East Asian patients via traditional slit-lamp examination (Qing et al., 2009). However, infrared imaging techniques may help demonstrate iris transillumination defects in these patients who show other clinical signs of PDS (Roberts and Wernick, 2007).
In conclusion, our study identified compound heterozygous variants of the CPAMD8 gene in these two Chinese pedigrees with PG inherited in an autosomal recessive manner. This study reported a possible association between the biallelic CPAMD8 variants and PDS/PG. Because the CPAMD8 ortholog is conspicuously absent in rodent genomes (Siggs et al., 2020), animal studies of the pathogenesis of PDS/PG with CPAMD8 variants require special considerations.
The data analyzed in this study is subject to the following licenses/restrictions: The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access these datasets should be directed to XL, eGxpdTEyMTNAMTI2LmNvbQ==.
The studies involving human participants were reviewed and approved by the Medical Ethics Committees of Shenzhen Eye Hospital (Number: 20180717-10) and Chengdu First People’s Hospital (Number: 2020-KT-011). The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
XL, JT, LZ, YW, and GL designed and performed most of the studies, analyzed the data, and wrote the manuscript; NF, LH, XW, YH, and D-FC helped conduct this study and write the manuscript. XL coordinated the study, conceived and designed the experimental plan, and finalized the manuscript. All authors reviewed the manuscript.
This study was supported by grants from the National Natural Science Foundation of China (NSFC grants 82070963, 81770924, and 81900829), the Science and Technology Project in Medical and Health of Xiamen (Grants Nos. 3502Z20194066 and 3502Z20214ZD1214), and the Science and Technology Innovation Committee of Shenzhen (Grant No. JCYJ20210324125614039).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank Zhilan Yuan, Yuanbo Liang, Liang Liang, Feng Wang, and Sujie Fan for their help and suggestions.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.845081/full#supplementary-material
Alsaif, H. S., Khan, A. O., Patel, N., Alkuraya, H., Hashem, M., Abdulwahab, F., et al. (2019). Congenital Glaucoma and CYP1B1: an Old Story Revisited. Hum. Genet. 138 (8-9), 1043–1049. doi:10.1007/s00439-018-1878-z
Andersen, J. S., Pralea, A. M., DelBono, E. A., Haines, J. L., Gorin, M. B., Schuman, J. S., et al. (1997). A Gene Responsible for the Pigment Dispersion Syndrome Maps to Chromosome 7q35-Q36. Arch. Ophthalmol. 115 (3), 384–388. doi:10.1001/archopht.1997.01100150386012
Anderson, M. G., Hawes, N. L., Trantow, C. M., Chang, B., and John, S. W. M. (2008). Iris Phenotypes and Pigment Dispersion Caused by Genes Influencing Pigmentation. Pigment. Cell Melanoma Res. 21 (5), 565–578. doi:10.1111/j.1755-148x.2008.00482.x
Bao, Y., Yang, J., Chen, L., Chen, M., Zhao, P., Qiu, S., et al. (2019). A Novel Mutation in the NDP Gene Is Associated with Familial Exudative Vitreoretinopathy in a Southern Chinese Family. Genet. Test. Mol. Biomarkers 23 (12), 850–856. doi:10.1089/gtmb.2019.0099
Bonet-Fernández, J.-M., Aroca-Aguilar, J.-D., Corton, M., Ramírez, A.-I., Alexandre-Moreno, S., García-Antón, M.-T., et al. (2020). CPAMD8 Loss-Of-Function Underlies Non-dominant Congenital Glaucoma with Variable Anterior Segment Dysgenesis and Abnormal Extracellular Matrix. Hum. Genet. 139 (10), 1209–1231. doi:10.1007/s00439-020-02164-0
Chang, B., Smith, R. S., Hawes, N. L., Anderson, M. G., Zabaleta, A., Savinova, O., et al. (1999). Interacting Loci Cause Severe Iris Atrophy and Glaucoma in DBA/2J Mice. Nat. Genet. 21 (4), 405–409. doi:10.1038/7741
Cheong, S.-S., Hentschel, L., Davidson, A. E., Gerrelli, D., Davie, R., Rizzo, R., et al. (2016). Mutations in CPAMD8 Cause a Unique Form of Autosomal-Recessive Anterior Segment Dysgenesis. Am. J. Hum. Genet. 99 (6), 1338–1352. doi:10.1016/j.ajhg.2016.09.022
Choi, H. R., Choi, S. H., Choi, S. M., Kim, J. K., Lee, C. R., Kang, S.-W., et al. (2020). Benefit of Diverse Surgical Approach on Short-Term Outcomes of MEN1-Related Hyperparathyroidism. Sci. Rep. 10, 10634. doi:10.1038/s41598-020-67424-5
Giardina, E., Oddone, F., Lepre, T., Centofanti, M., Peconi, C., Tanga, L., et al. (2014). Common Sequence Variants in the LOXL1gene in Pigment Dispersion Syndrome and Pigmentary Glaucoma. BMC Ophthalmol. 14, 52. doi:10.1186/1471-2415-14-52
Hollmann, A. K., Dammann, I., Wemheuer, W. M., Wemheuer, W. E., Chilla, A., Tipold, A., et al. (2017). Morgagnian Cataract Resulting from a Naturally Occurring Nonsense Mutation Elucidates a Role of CPAMD8 in Mammalian Lens Development. PLoS One 12 (7), e0180665. doi:10.1371/journal.pone.0180665
Janssen, B. J. C., Huizinga, E. G., Raaijmakers, H. C. A., Roos, A., Daha, M. R., Nilsson-Ekdahl, K., et al. (2005). Structures of Complement Component C3 Provide Insights into the Function and Evolution of Immunity. Nature 437, 505–511. doi:10.1038/nature04005
Lahola-Chomiak, A. A., Footz, T., Nguyen-Phuoc, K., Neil, G. J., Fan, B., Allen, K. F., et al. (2019). Non-Synonymous Variants in Premelanosome Protein (PMEL) Cause Ocular Pigment Dispersion and Pigmentary Glaucoma. Hum. Mol. Genet. 28 (8), 1298–1311. doi:10.1093/hmg/ddy429
Lascaratos, G., Shah, A., and Garway-Heath, D. F. (2013). The Genetics of Pigment Dispersion Syndrome and Pigmentary Glaucoma. Surv. Ophthalmol. 58 (2), 164–175. doi:10.1016/j.survophthal.2012.08.002
Li, J., Wang, W., Zhao, Q., Fan, S., Li, Y., Yuan, P., et al. (2021a). A Haemocyte-Expressed Methyltransf_FA Domain Containing Protein (MFCP) Exhibiting Microbe Binding Activity in Oyster Crassostrea gigas. Dev. Comp. Immunol. 122, 104137. doi:10.1016/j.dci.2021.104137
Li, X., Sun, W., Xiao, X., Fang, L., Li, S., Liu, X., et al. (2021b). Biallelic Variants in CPAMD8 Are Associated with Primary Open-Angle Glaucoma and Primary Angle-Closure Glaucoma. Br. J. Ophthalmol. Published Online First: 21 June 2021. doi:10.1136/bjophthalmol-2020-318668
Li, Z.-F., Wu, X.-h., and Engvall, E. (2004). Identification and Characterization of CPAMD8, a Novel Member of the Complement 3/α 2 -macroglobulin Family with a C-Terminal Kazal Domain. Genomics 83 (6), 1083–1093. doi:10.1016/j.ygeno.2003.12.005
Lyons, L., and Amram, A. (2019). Iris Transillumination Defects in Pigment Dispersion Syndrome. N. Engl. J. Med. 381 (20), 1950. doi:10.1056/nejmicm1903842
Ma, A., Yousoof, S., Grigg, J. R., Flaherty, M., Minoche, A. E., Cowley, M. J., et al. (2020). Revealing Hidden Genetic Diagnoses in the Ocular Anterior Segment Disorders. Genet. Med. 22 (10), 1623–1632. doi:10.1038/s41436-020-0854-x
Mandelkorn, R. M., Hoffman, M. E., Olander, K. W., Zimmerman, T., and Harsha, D. (1983). Inheritance and the Pigmentary Dispersion Syndrome. Ann. Ophthalmol. 15 (6), 577–582.
Nagase, T., Ishikawa, K., Kikuno, R., Hirosawa, M., Nomura, N., and Ohara, O. (1999). Prediction of the Coding Sequences of Unidentified Human Genes.XV. The Complete Sequences of 100 New cDNA Clones from Brain Which Code for Large Proteins In Vitro. DNA Res. 6 (5), 337–345. doi:10.1093/dnares/6.5.337
Niyadurupola, N., and Broadway, D. C. (2008). Pigment dispersion syndrome and pigmentary glaucoma - a major review. Clin. Exp. Ophthalmol. 36 (9), 868–882. doi:10.1111/j.1442-9071.2009.01920.x
Qing, G., Wang, N., Tang, X., Zhang, S., and Chen, H. (2009). Clinical characteristics of pigment dispersion syndrome in Chinese patients. Eye 23 (8), 1641–1646. doi:10.1038/eye.2008.328
Rao, K. N., Ritch, R., Dorairaj, S. K., Kaur, I., Liebmann, J. M., Thomas, R., et al. (2008). Exfoliation syndrome and exfoliation glaucoma-associated LOXL1 variations are Not involved in pigment dispersion syndrome and pigmentary glaucoma. Mol. Vis. 14, 1254–1262.
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Roberts, D. K., and Wernick, M. N. (2007). Infrared imaging technique may help demonstrate iris transillumination defects in blacks who show other pigment dispersion syndrome clinical signs. J. Glaucoma 16 (5), 440–447. doi:10.1097/ijg.0b013e3181405e72
Sarkar, K., Sadhukhan, S., Han, S.-S., and Vyas, Y. M. (2014). Disruption of hSWI/SNF complexes in T cells by WAS mutations distinguishes X-linked thrombocytopenia from Wiskott-Aldrich syndrome. Blood 124 (23), 3409–3419. doi:10.1182/blood-2014-07-587642
Scheie, H. G., and Cameron, J. D. (1981). Pigment dispersion syndrome: a clinical study. Br. J. Ophthalmol. 65 (4), 264–269. doi:10.1136/bjo.65.4.264
Schraermeyer, M., Schnichels, S., Julien, S., Heiduschka, P., Bartz-Schmidt, K.-U., and Schraermeyer, U. (2009). Ultrastructural analysis of the pigment dispersion syndrome in DBA/2J mice. Graefes Arch. Clin. Exp. Ophthalmol. 247, 1493–1504. doi:10.1007/s00417-009-1146-y
Scuderi, G., Contestabile, M. T., Scuderi, L., Librando, A., Fenicia, V., and Rahimi, S. (2019). Pigment dispersion syndrome and pigmentary glaucoma: a review and update. Int. Ophthalmol. 39 (7), 1651–1662. doi:10.1007/s10792-018-0938-7
Siddiqui, Y., Ten Hulzen, R. D., Cameron, J. D., Hodge, D. O., and Johnson, D. H. (2003). What is the risk of developing pigmentary glaucoma from pigment dispersion syndrome? Am. J. Ophthalmol. 135 (6), 592794–592799. doi:10.1016/s0002-9394(02)02289-4
Siggs, O. M., Souzeau, E., Taranath, D. A., Dubowsky, A., Chappell, A., Zhou, T., et al. (2020). Biallelic CPAMD8 variants are a frequent cause of childhood and juvenile open-angle glaucoma. Ophthalmology 127 (6), 758–766. doi:10.1016/j.ophtha.2019.12.024
Stankovic, J. (1961). Ein Beitrag zur Kenntnis der Vererbung des Pigmentglaucom. Klin. Monatsbl Augenheilkd 139, 165–175.
Tandon, A., Zhang, Z., Fingert, J. H., Kwon, Y. H., Wang, K., and Alward, W. L. M. (2019). The heritability of pigment dispersion syndrome and pigmentary glaucoma. Am. J. Ophthalmol. 202, 55–61. doi:10.1016/j.ajo.2019.02.017
Trantow, C. M., Mao, M., Petersen, G. E., Alward, E. M., Alward, W. L. M., Fingert, J. H., et al. (2009). LystMutation in Mice Recapitulates Iris Defects of Human Exfoliation Syndrome. Invest. Ophthalmol. Vis. Sci. 50 (3), 1205–1214. doi:10.1167/iovs.08-2791
Wiggs, J. L. (2020). CPAMD8, a new gene for anterior segment dysgenesis and childhood glaucoma. Ophthalmology 127 (6), 767–768. doi:10.1016/j.ophtha.2020.02.035
Wong, M., Huang, P., Li, W., Li, Y., Zhang, S. S., and Zhang, C. (2015). T-helper1/T-helper2 cytokine imbalance in the iris of patients with glaucoma. PLoS One 10 (3), e0122184. doi:10.1371/journal.pone.0122184
Wong, S. G., and Dessen, A. (2014). Structure of a bacterial α2-macroglobulin reveals mimicry of eukaryotic innate immunity. Nat. Commun. 5, 4917. doi:10.1038/ncomms5917
Keywords: CPAMD8, pigmentary glaucoma, autosomal recessive inheritance, pedigree, compound heterozygous variant
Citation: Tan J, Zeng L, Wang Y, Liu G, Huang L, Chen D, Wang X, Fan N, He Y and Liu X (2022) Compound Heterozygous Variants of the CPAMD8 Gene Co-Segregating in Two Chinese Pedigrees With Pigment Dispersion Syndrome/Pigmentary Glaucoma. Front. Genet. 13:845081. doi: 10.3389/fgene.2022.845081
Received: 29 December 2021; Accepted: 20 June 2022;
Published: 25 July 2022.
Edited by:
Yang Sun, Stanford University, United StatesReviewed by:
Bo Lei, Henan Provincial People’s Hospital, ChinaCopyright © 2022 Tan, Zeng, Wang, Liu, Huang, Chen, Wang, Fan, He and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuyang Liu, eGxpdTEyMTNAMTI2LmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.