 Markus Schmid
Markus Schmid Joana Stock
Joana Stock Jörn Bennewitz
Jörn Bennewitz Robin Wellmann
Robin Wellmann
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 24 March 2022
Sec. Livestock Genomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.840815
Numerically small breeds have often been upgraded with mainstream breeds. This historic introgression predisposes the breeds for joint genomic evaluations with mainstream breeds. The linkage disequilibrium structure differs between breeds. The marker effects of a haplotype segment may, therefore, depend on the breed from which the haplotype segment originates. An appropriate method for genomic evaluation would account for this dependency. This study proposes a method for the computation of genomic breeding values for small admixed breeds that incorporate phenotypic and genomic information from large introgressed breeds by considering the breed origin of alleles (BOA) in the evaluation. The proposed BOA model classifies haplotype segments according to their origins and assumes different but correlated SNP effects for the different origins. The BOA model was compared in a simulation study to conventional within-breed genomic best linear unbiased prediction (GBLUP) and conventional multi-breed GBLUP models. The BOA model outperformed within-breed GBLUP as well as multi-breed GBLUP in most cases.
The efficiency of breeding programs for local breeds is often compromised by the limited number of individuals and has resulted in a decreasing competitiveness with high yielding breeds, especially with the advent of genomic selection (GS). In GS, large reference populations are required to accurately predict breeding values of the individuals (Goddard and Hayes 2009) and are therefore difficult to establish in small local breeds. In order to improve the performance of local breeds, sires of closely related high-yielding breeds were frequently used in the past and genetic gain has been generated by introgression. Such strategies increase the genetic relatedness between breeds because a certain number of alleles of the high yielding breed segregate within the target breed after introgression.
Several studies were conducted using different approaches to enable GS in numerically small breeds using the reference population of a second breed (across-breed prediction) or extending the own reference population by adding the reference population of the second breed (multi-breed prediction) as reviewed by Lund et al. (2014, 2016). The major findings were that across-breed prediction is often not suitable to improve the accuracy of prediction and that the benefit of multi-breed reference populations strongly depends on the relatedness between the breeds and density of the SNP panels. A substantial increase in accuracy can only be expected when the breeds are closely related and the number of SNPs is high to capture across-breed linkage disequilibrium (LD) between markers and QTLs. However, variation of LD as well as differences of allelic effects across populations limit the application of such approaches. Different models were proposed accounting for breed-specific effects (e.g., Makgahlela et al., 2012; Thomasen et al., 2013; Hamidi and Rekaya., 2015; van den Berg et al., 2020) and differences in LD (Rahimi et al., 2020) in the field of multi-breed dairy cattle evaluation. One way is to assign the breed origin of alleles (BOA) (Wellmann 2019; Vandenplas et al., 2016) that allows for models assuming SNP effects to be different but correlated across breeds. Such models were applied to simulated and real datasets of crossbred or admixed populations in cattle (Karaman et al., 2021) as well as other livestock (e.g., Duenk et al., 2019) or plant species (Rio et al., 2020) and are reviewed in Stock et al. (2020) and Duenk et al. (2021). The studies have shown that considering BOA has the potential to increase the accuracy of multi-breed GS.
In many numerical small dairy cattle breeds sires from a large and high yielding breed were used in order to speed up genetic gain in the small breed. This resulted in some cases in a substantial amount of introgressed genes and in a mosaic-like haplotype pattern with a mix of native and introgressed haplotypes. For example, in the German Angler breed located in the northern part of Germany, admixture plays a substantial role in the population structure and the proportion of migrant alleles from other breeds is remarkable (Addo et al., 2019, Wang et al., 2017a,b, Schmidtmann et al., 2021). A very close relationship to the Holstein Friesian breed, especially the Red Holstein breed, was observed (Wang et al., 2017b). A similar level of admixture was observed for the German Vorderwald breed, where the genetic progress was mostly driven by the introgressed genes (Hartwig et al., 2014; Hartwig et al., 2015). For these kinds of breeds, a genomic model that considers the mosaic pattern of the haplotype structure would be beneficial in multi-breed genomic evaluations.
This study proposes a method for the computation of genomic breeding values for small admixed breeds that incorporates phenotypic and genomic information from large introgressed breeds. The start and the end of the introgression events are considered to be in the past, which is applicable to many small local admixed breeds. A multi-breed BOA model is derived for multi-breed genomic selection that is suitable for application when the individuals have fragmented genomes. It classifies haplotype segments according to their origins and assumes different SNP effects for the different origins.
For validation, it was compared with models that did not consider the breed-origin of QTL alleles. All models were applied to simulated datasets. In the simulation, the genotypes of the small admixed breed were derived from German Angler cattle, while the genotypes of the introgressed breed were derived from German Holstein cattle. Different scenarios were investigated in which the number of genotyped animals of the target breed, i.e., the numerically small Angler breed, varied, while the number of genotyped animals of the large introgressed breed, i.e., the German Holstein, remained constant.
The data basis for the simulation study were 50k SNP-chip (Illumina BovineSNP50 BeadChip, Illumina Inc., San Diego, CA) genotypes of Angler (AN) (Wang et al., 2017a) and Holstein (HF) (Streit et al., 2013) individuals from the German population. Starting with the base generation, one further generation was simulated for each breed according to the simulation protocol of Stock et al. (2021) with R-package x-breed (Esfandyari and Sørensen 2017). The resulting simulated HF dataset (simHF) consisted of 6,000 individuals and the simulated Angler data set contained 3,000 individuals.
Several subsets of the total Angler data set were sampled to mimic different population sizes for the small breed. Subset simAN1 consisted of 750 individuals, simAN2 consisted of 1,500 individuals and simAN3 contained all 3,000 simulated Angler individuals. The different simAN populations are referred to as breed size scenarios. The sample sizes represent 12.5, 25 and 50% of the number of simHF individuals. In each of the subsets, all sires had the same number of offspring.
From the 23,448 SNPs that segregated in both breeds, 1,000 SNPs were randomly selected as QTLs, while the remaining SNPs were used as markers for genomic prediction.
The QTL effects for the simAN datasets and the simHF dataset were correlated. The additive effects
Hence, the correlation of QTL effects between the two simulated breeds was 0.95. Dominance was not modelled. The additive effects were scaled to represent a trait with an additive variance of
The true breeding values (TBV) were calculated as
where
The BOA model was compared with two conventional methods for the prediction of genomic breeding values, which are within-breed prediction with GBLUP for the simulated Angler cattle, and a multi-breed prediction with GBLUP.
It is assumed that genotypes and phenotypes from several breeds or crosses are available, which includes the target breed. The number of SNP is denoted as
and
The model equation for the phenotypic value of individual
where
where
with
Alternative representations of the model and the mixed model equations are given in the Supplementary Appendix.
The BOA model requires the breed origins
The covariance matrix
For within-breed genomic prediction we used the model
where the
The SNP markers for genomic prediction were chosen as follows. From the 22,448 SNPs that were not chosen as QTLs, all SNPs that segregated with a minor allele frequency (MAF) <0.03 within one of the simulated breeds and SNPs that did not segregate in both breeds were omitted. Across all replicates, on average 21,670 SNPs remained and were used for genomic prediction.
The genomic predictions were done separately for each breed-size scenario and each replicate. The accuracies of prediction were assessed by a 5-fold cross validation. The individuals of the respective simAN dataset were assigned to five different classes such that individuals from different classes had no sires in common. Hence, each class included the offspring of 10 sires. In each cross-validation cycle, one class was used as the validation set, and the four remaining classes were used as the reference population.
For multi-breed GBLUP and for the BOA model, the respective simAN reference set was joined with the simHF individuals. Consequently, the number of individuals from the simAN population in the reference population varied, while the number of simHF individuals was constant.
An overview on the sample sizes is given in Table 1. The reference populations for within-breed prediction consisted of 600, 1,200, and 2,400 simAN individuals, respectively. The reference populations for multi-breed prediction were enlarged by the 6,000 simHF individuals. The proportions of simAN individuals in the multi-breed reference population were thus 9, 17 and 29% for the simAN1, simAN2 and simAN3 scenario, respectively.
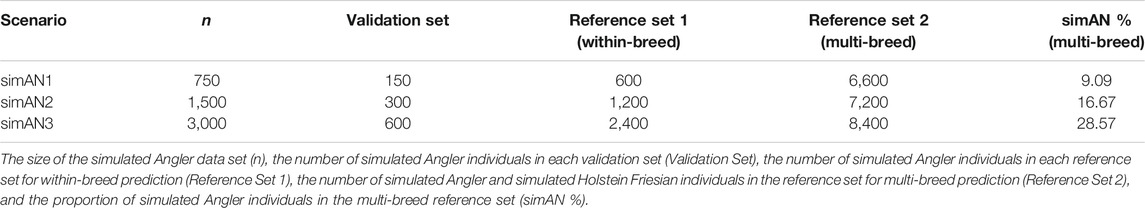
TABLE 1. Numbers of genotyped animals in reference and validation sets for the three investigated scenarios.
The marker effects were estimated with all three models for each cross-validation cycle.
The genomic breeding values of the individuals in the validation set were computed for the BOA model as
where
where
For each method, the prediction accuracy was calculated as the correlation between the GEBVs and the TBVs of the validation individuals. The accuracies presented in the results are the averages, taken over all cross-validation cycles and replicates.
The mean proportion of SNPs with Holstein origin across all replicates was 0.157 ± 0.007. Table 2 shows the results of the model comparison for all investigated breed-size scenarios. In general, the prediction accuracies increased with increasing size of the reference population. The BOA model provided the highest accuracies for simAN1 and simAN2, whereas it showed the same mean accuracy as within-breed GBLUP for simAN3. Multi-breed GBLUP was inferior to the other models in simAN1 and simAN3. Within-breed GBLUP resulted in the lowest accuracies in the medium-sized reference population scenario simAN2. The standard deviations (SD) of the accuracies were highest using the small reference set, while it showed the smallest SD values in the medium-sized reference set. The standard errors of the accuracies were relatively small (0.011–0.017).
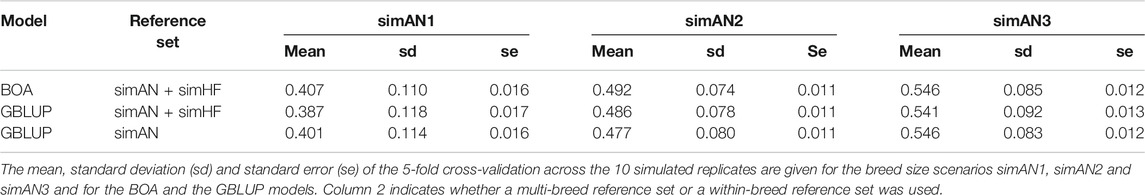
TABLE 2. Mean accuracies of genomic prediction in the simulated breed size scenarios.
It was shown in this study that a multi-breed genomic prediction with the proposed BOA model can increase the accuracies of the GEBVs for numerically small admixed populations over multi-breed and within-breed genomic predictions with GBLUP under certain conditions. The model considers the breed origin of each haplotype in individuals with admixed genomes consisting of native and introgressed haplotype segments. This makes it especially interesting for numerical small breeds with historic introgression from high yielding breeds, as it was observed e.g., in the German Angler or Vorderwald breed (Addo et al., 2019; Wang et al., 2017a; Wang et al., 2017b; Schmidtmann et al., 2021; Hartwig et al., 2014, 2015).
The simulated QTL positions were the same in both breeds. Recent mutations that could have created new QTLs were ignored. The QTL effects of both genetic groups were assumed to be highly correlated with a correlation of 0.95. The QTL positions were chosen from the 50k chip, so the simulated QTLs are common variants whereas a large fraction of the additive variance is expected to come from rare QTL variants (e.g., Kemper and Goddard 2012; Visscher et al., 2017). This can compromise a direct transition of the simulation results to real data.
The LD structures in the simulation are expected to be similar to those investigated in real populations (Qanbari et al., 2010; Addo et al., 2019; Schmidtmann et al., 2021) because only one generation was simulated, so recombination could occur only within one meiotic division. Although the QTL effects were highly correlated, the correlation of the marker effects was only 0.75. The reason for the relatively low correlation of marker effects is that the QTLs were excluded from the marker set. Therefore, the effect of a single QTL is captured by several markers and the LD between markers and QTLs is different in both genetic groups. The shorter ranges of LD in admixed populations like the German Angler compared with other breeds (Addo et al., 2019; Schmidtmann et al., 2021) contributes to the observed low correlation. A higher correlation between marker effects might be observed when more dense marker panels would be used and a heavy-tailed distribution of marker effects would be assumed.
This paper focused on methods to improve GS in small admixed populations. It compared the prediction accuracies of various methods in a simulated population that had a similar LD structure as the target breeds. A detailed quantification of the impact of influencing factors (e.g., LD and its consistency across populations, or the relatedness and genetic correlation between the populations) on the accuracies was beyond the scope of the study. The study explicitly focused on admixed populations where the events of introgression were in the past and breeding programs aim at reducing migrant contributions from other (high yielding) breeds or at least keep the amount of migrant contributions constant (e.g., Wang et al., 2017b). Therefore, the impact of the approach on crossbred individuals was not determined. But still, models that include BOA information have been shown to be beneficial for crosses (e.g., Duenk et al., 2019).
In this study, the multi-breed BOA model was compared with conventional multi-breed GBLUP and within-breed GBLUP. The multi-breed BOA approach led to an increase in the accuracy of the genomic breeding values when the number of genotyped AN individuals was small and medium, and showed similar results as the within-breed GBLUP method for the large reference sets. The difference between the prediction accuracies of the models, however, tend to decrease with an increasing number of genotyped AN individuals. The multi-breed prediction with GBLUP was not superior to within-breed prediction when the number of genotyped AN individuals was large. The reason is possibly that the multi-breed GBLUP model assumes a perfect correlation between the marker effects of both breeds. This assumption was certainly violated in the simulation. The BOA model, which accounts for the correlation between marker effects, could improve upon single-breed evaluations and outperformed multi-breed GBLUP in all cases.
For the prediction of genomic breeding values of Angler in practice, to date, a joint reference population of several Scandinavian red dairy breeds (i.e., Danish Red, Norwegian Red, Swedish Red, and Finnish Ayrshire) is used. To increase the accuracies of the GEBVs for Angler, about 170 genotyped and progeny-tested German Angler bulls have been included to this reference set as well (private communication RSHeG, 2021). Hence, the findings of the study in scenario simAN1 might be most relevant for the current Angler cattle breeding program.
In the past decades, the Angler breed has been upgraded with other breeds, such as Red Holstein and Holstein Friesian to increase its economic value. This has led to relatively high kinships between them (Wang et al., 2017b). However, in this study only Holstein Friesian genotypes were available and considered, and thus the total amount of introgression was probably not detected completely. In addition, the available Holstein Friesian genotypes originated from the current population, which might have also biased the categorization of the native parts of the genome. The use of the most closely related introgressed breed is expected to bear the greatest potential in multi-breed predictions in the target breed when applying BOA models and should therefore preferably be used if applicable. This was not shown here as such datasets were not available. At an animal level, the proposed BOA model considers the genetic connectedness of individuals from both breeds. The closer individuals of the high yielding breed are related with the individuals of the target breed, the more informative they are for multi-breed prediction and thus contribute more to the accuracy of breeding value estimation. Generally, multi-breed prediction is increasingly beneficial when applied to high density marker information or whole-genome sequence data (Lund et al., 2014), however, such datasets are mostly not available in cost-efficient breeding programs of small local cattle populations.
A multi-breed genomic prediction with the proposed BOA model increased the accuracies of the estimated genomic breeding values for numerically small admixed populations over multi-breed and within-breed genomic predictions with GBLUP. The BOA model assumes that the additive effect of an allele depends on the genetic group from which the respective haplotype segment originates. It is of special interest for multi-breed genomic predictions for numerically small breeds with past introgression from high yielding breeds.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
RW, JB, and MS designed the study. RW developed the proposed BOA model. JS and MS simulated the data. MS did the statistical analyses. All authors drafted and approved the manuscript.
JS was partly supported by the H. Wilhelm Schaumann Foundation, Hamburg, Germany, which is gratefully acknowledged.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.840815/full#supplementary-material
Addo, S., Klingel, S., Hinrichs, D., and Thaller, G. (2019). Runs of Homozygosity and NetView Analyses Provide New Insight into the Genome-wide Diversity and Admixture of Three German Cattle Breeds. PLoS ONE 14 (12), e0225847–20. doi:10.1371/journal.pone.0225847
Duenk, P., Bijma, P., Wientjes, Y. C. J., and Calus, M. P. L. (2021). Review: Optimizing Genomic Selection for Crossbred Performance by Model Improvement and Data Collection. J. Anim. Sci. 99 (8), 1–24. doi:10.1093/jas/skab205
Duenk, P., Calus, M. P. L., Wientjes, Y. C. J., Breen, V. P., Henshall, J. M., Hawken, R., et al. (2019). Validation of Genomic Predictions for Body Weight in Broilers Using Crossbred Information and Considering Breed-Of-Origin of Alleles. Genet. Sel Evol. 51 (1), 1–12. doi:10.1186/s12711-019-0481-7
Esfandyari, H., and Sørensen, A. C. (2017). “Xbreed: An R Package for Genomic Simulation of Purebreds and Crossbreds,” in 68th Annual Meeting of the European Federation of Animal Science, Tallinn, Estonia, 28 Aug - 1 Sep 2017.
Falconer, D. S., and Mackay, T. F. C. (1996). Introduction to Quantitative Genetics. 4th ed. London: Longman Group Ltd.
Goddard, M. E., and Hayes, B. J. (2009). Mapping Genes for Complex Traits in Domestic Animals and Their Use in Breeding Programmes. Nat. Rev. Genet. 10 (6), 381–391. doi:10.1038/nrg2575
Hartwig, S., Wellmann, R., Emmerling, R., Hamann, H., and Bennewitz, J. (2015). Short Communication: Importance of Introgression for Milk Traits in the German Vorderwald and Hinterwald Cattle. J. Dairy Sci. 98 (3), 2033–2038. doi:10.3168/jds.2014-8571
Hartwig, S., Wellmann, R., Hamann, H., and Bennewitz, J. (2014). The Contribution of Migrant Breeds to the Genetic Gain of Beef Traits of German Vorderwald and Hinterwald Cattle. J. Anim. Breed. Genet. 131, 496–503. doi:10.1111/jbg.12099
Hay, E. H., and Rekaya, R. (2015). A Multi-Compartment Model for Genomic Selection in Multi-Breed Populations. Livestock Sci. 177, 1–7. doi:10.1016/j.livsci.2015.03.027
Karaman, E., Su, G., Croue, I., and Lund, M. S. (2021). Genomic Prediction Using a Reference Population of Multiple Pure Breeds and Admixed Individuals. Genet. Sel Evol. 53 (1), 1–15. doi:10.1186/s12711-021-00637-y
Kemper, K. E., and Goddard, M. E. (2012). Understanding and Predicting Complex Traits: Knowledge from Cattle. Hum. Mol. Genet. 21 (R1), R45–R51. doi:10.1093/hmg/dds332
Lund, M. S., Su, G., Janss, L., Guldbrandtsen, B., and Brøndum, R. F. (2014). Genomic Evaluation of Cattle in a Multi-Breed Context. Livestock Sci. 166 (1), 101–110. doi:10.1016/j.livsci.2014.05.008
Lund, M. S., van den Berg, I., Ma, P., Brøndum, R. F., and Su, G. (2016). Review: How to Improve Genomic Predictions in Small Dairy Cattle Populations. Animal 10 (6), 1042–1049. doi:10.1017/S1751731115003031
Makgahlela, M. L., Mäntysaari, E. A., Strandén, I., Koivula, M., Nielsen, U. S., Sillanpää, M. J., et al. (2012). Across Breed Multi-Trait Random Regression Genomic Predictions in the Nordic Red Dairy Cattle. J. Anim. Breed. Genet. 130 (1), 10–19. doi:10.1111/j.1439-0388.2012.01017.x
Mohammad Rahimi, S., Rashidi, A., and Esfandyari, H. (2020). Accounting for Differences in Linkage Disequilibrium in Multi-Breed Genomic Prediction. Livestock Sci. 240 (June), 104165. doi:10.1016/j.livsci.2020.104165
Qanbari, S., Pimentel, E. C. G., Tetens, J., Thaller, G., Lichtner, P., Sharifi, A. R., et al. (2009). The Pattern of Linkage Disequilibrium in German Holstein Cattle. Anim. Genet. 41 (4), 346–356. doi:10.1111/j.1365-2052.2009.02011.x
Rio, S., Moreau, L., Charcosset, A., and Mary-Huard, T. (2020). Accounting for Group-specific Allele Effects and Admixture in Genomic Predictions: Theory and Experimental Evaluation in Maize. Genetics 216 (1), 27–41. doi:10.1534/genetics.120.303278
RSHeG (2021). Personal Communication. Washington, D.C., United States: American Psychological Association.
Schmidtmann, C., Schönherz, A., Guldbrandtsen, B., Marjanovic, J., Calus, M., Hinrichs, D., et al. (2021). Assessing the Genetic Background and Genomic Relatedness of Red Cattle Populations Originating from Northern Europe. Genet. Sel Evol. 53. doi:10.1186/s12711-021-00613-6
Stock, J., Bennewitz, J., Hinrichs, D., and Wellmann, R. (2020). A Review of Genomic Models for the Analysis of Livestock Crossbred Data. Front. Genet. 11 (June), 1–10. doi:10.3389/fgene.2020.00568
Stock, J., Esfandyari, H., Hinrichs, D., and Bennewitz, J. (2021). Implementing a Genomic Rotational Crossbreeding Scheme to Promote Local Dairy Cattle Breeds-A Simulation Study. J. Dairy Sci. 104, 6873–6884. doi:10.3168/jds.2020-19927
Streit, M., Wellmann, R., Reinhardt, F., Thaller, G., Piepho, H.-P., and Bennewitz, J. (2013). Using Genome-wide Association Analysis to Characterize Environmental Sensitivity of Milk Traits in Dairy Cattle. G3 (Bethesda, Md. 3, 1085–1093. doi:10.1534/g3.113.006536
Thomasen, J. R., Sørensen, A. C., Su, G., Madsen, P., Lund, M. S., and Guldbrandtsen, B. (2013). The Admixed Population Structure in Danish Jersey Dairy Cattle Challenges Accurate Genomic predictions1The Admixed Population Structure in Danish Jersey Dairy Cattle Challenges Accurate Genomic Predictions. J. Anim. Sci. 91 (7), 3105–3112. doi:10.2527/jas.2012-5490
van den Berg, I., MacLeod, I. M., Reich, C. M., Breen, E. J., Breen, E. J., and Pryce, J. E. (2020). Optimizing Genomic Prediction for Australian Red Dairy Cattle. J. Dairy Sci. 103 (7), 6276–6298. doi:10.3168/jds.2019-17914
Vandenplas, J., Calus, M. P. L., Sevillano, C. A., Windig, J. J., and Bastiaansen, J. W. M. (2016). Assigning Breed Origin to Alleles in Crossbred Animals. Genet. Sel Evol. 48 (1), 1–22. doi:10.1186/s12711-016-0240-y
Visscher, P. M., Wray, N. R., Zhang, Q., Sklar, P., McCarthy, M. I., Brown, M. A., et al. (2017). 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 101 (1), 5–22. doi:10.1016/j.ajhg.2017.06.005
Wang, Y., Bennewitz, J., and Wellmann, R. (2017a). Novel Optimum Contribution Selection Methods Accounting for Conflicting Objectives in Breeding Programs for Livestock Breeds with Historical Migration. Genet. Sel Evol. 49 (1), 1–12. doi:10.1186/s12711-017-0320-7
Wang, Y., Segelke, D., Emmerling, R., Bennewitz, J., and Wellmann, R. (2017b). Long-Term Impact of Optimum Contribution Selection Strategies on Local Livestock Breeds with Historical Introgression Using the Example of German Angler Cattle. G3: Genes, Genomes, Genet. 7, 4009–4018. doi:10.1534/g3.117.300272
Keywords: admixed population, multi-breed genomic prediction, BOA model, cattle, allele origin
Citation: Schmid M, Stock J, Bennewitz J and Wellmann R (2022) Improving the Accuracy of Multi-Breed Prediction in Admixed Populations by Accounting for the Breed Origin of Haplotype Segments. Front. Genet. 13:840815. doi: 10.3389/fgene.2022.840815
Received: 21 December 2021; Accepted: 04 March 2022;
Published: 24 March 2022.
Edited by:
Fernando Baldi, São Paulo State University, BrazilReviewed by:
George R. Wiggans, Council on Dairy Cattle Breeding, United StatesCopyright © 2022 Schmid, Stock, Bennewitz and Wellmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus Schmid, TWFya3VzX3NjaG1pZEB1bmktaG9oZW5oZWltLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.