
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 31 March 2022
Sec. Neurogenomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.840577
This article is part of the Research TopicIdentifying the Key Pathogenic Factors of Neurological Disorders by Integrating Multi-omics DataView all 16 articles
 Guanting Lu1†
Guanting Lu1† Liya Ma2†
Liya Ma2† Pei Xu1Binqiang Xian2Lianying Wu1Jianying Ding2Xiaoyan He1Huiyun Xia2
Pei Xu1Binqiang Xian2Lianying Wu1Jianying Ding2Xiaoyan He1Huiyun Xia2 Wuwu Ding1Zhirong Yang1*
Wuwu Ding1Zhirong Yang1* Qiongling Peng2*
Qiongling Peng2*Background: Neurodevelopmental disorder with dysmorphic facies and distal skeletal anomalies (NEDDFSA) is a rare syndromic disorder characterized by global neurodevelopmental delay, early-onset hypotonia, poor overall growth, poor speech/language ability, and additional common phenotypes such as eye anomalies, joint hypermobility, and skeletal anomalies of the hands and feet. NEDDFSA is caused by heterozygous pathogenic variants in the ZMIZ1 gene on chromosome 10q22.3 with autosomal dominant (AD) mode of inheritance. All the 32 reported cases with variants in ZMIZ1 gene had a genetic background in Caucasian, Hispanic, North African, and Southeastern Asian. Until now, there are no reports of Chinese patients with ZMIZ1 pathogenic variants.
Methods: A 5-year-old girl was found to have the characteristic phenotypes of NEDDFSA. Array-Comparative Genomic Hybridization (array-CGH) and whole exome sequencing (WES) were applied for the trio of this female patient. Sanger sequencing was used to verify the selected variants. A comprehensive molecular analysis was carried out by protein structure prediction, evolutionary conservation, motif scanning, tissue-specific expression, and protein interaction network to elucidate pathogenicity of the identified ZMIZ1 variants.
Results: The karyotype was 46, XX with no micro-chromosomal abnormalities identified by array-CGH. There were 20 variants detected in the female patient by WES. A de novo heterozygous missense variant (c.2330G > A, p.Gly777Glu, G777E) was identified in the exon 20 of ZMIZ1. No variants of ZMIZ1 were identified in the non-consanguineous parents and her healthy elder sister. It was predicted that G777E was pathogenic and detrimental to the spatial conformation of the MIZ/SP-RING zinc finger domain of ZMIZ1.
Conclusion: Thus far, only four scientific articles reported deleterious variants in ZMIZ1 and most of the cases were from Western countries. This is the first report about a Chinese patient with ZMIZ1 variant. It will broaden the current knowledge of ZMIZ1 variants and variable clinical presentations for clinicians and genetic counselors.
Neurodevelopmental disorder with dysmorphic facies and distal skeletal anomalies (NEDDFSA; OMIM #618659) is a rare syndromic disorder characterized by global neurodevelopmental delay, hypotonia, poor overall growth, poor speech/language ability, and other common phenotypes such as eye anomalies, joint hypermobility, and distal skeletal anomalies of the hands and feet (Carapito et al., 2019). A balanced translocation t (10; 19) (q22.3; q13.33) was first reported in 2015, involving zinc finger MIZ-type containing 1 (ZMIZ1, OMIM #607159) and proline-rich protein 12 (PRR12, OMIM #616633). It produced two types of fusion genes, ZMIZ1-PRR12 and PRR12-ZMIZ1, which might be related to the occurrence of intellectual disability (ID) and neuropsychiatric alterations (Córdova-Fletes et al., 2015). Later, in 2019, pathogenic variants involving the gene ZMIZ1 were identified in a cohort of 19 NEDDFSA cases from a transatlantic collaborative effort (Carapito et al., 2019). In the same year of 2019, an affected father and his two sons were identified to be suffering from the ZMIZ1-related neurodevelopmental disorder in Florida (Latchman et al., 2020). In 2021, Phetthong et al. reported a 5-year-old Thai girl with developmental delay, facial phenotypes resembling Williams syndrome, and cardiac defects. She carried three types of compound variants, a heterozygous ZMIZ1 variant (c.1497+2T > C), a heterozygous frameshift variant of OTUD6B (OMIM #612021) (c.873delA, p.Lys291AsnfsTer3), and a 0.118 Mb 8q21.3 microdeletion involving OTUD6B (Phetthong et al., 2021).
The gene ZMIZ1 was mapped to chromosome 10q22.3 and it contains 21 exons to produce a 1067-amino acid protein with a calculated molecular mass of 123 kDa (Sharma et al., 2003). According to the Conserved Domain database (CDD) (Lu et al., 2020), ZMIZ1 contains a Zmiz1 N-terminal tetratricopeptide repeat domain (Zmiz1_N, 8–100), Med15 domain (184–557), a nuclear localization signal (NLS, 697–711), a SP-RING zine finger domain (SP-RING_ZMIZ, 739–786), and a transactivation domain (TAD, 837–1067). In 1999, Nagase et al. identified the gene ZMIZ1 (previously called KIAA1224) from a fetal brain cDNA library (Nagase et al., 1999). The encoded protein is a transcriptional co-activator, which belongs to the Protein Inhibitor of Activated STAT (PIAS) family. As a member of the PIAS family, ZMIZ1 has a highly conserved MIZ (Msx-interacting zinc finger) domain which is important for protein-protein interaction and SUMOylation (Sharma et al., 2003; Beliakoff and Sun, 2006). It had been reported that ZMIZ1 could regulate the activity of many transcription factors, such as androgen receptor (AR) (Beliakoff and Sun, 2006), SMAD3 (Li et al., 2006), SMAD4 (Li et al., 2006), and p53 (Lee et al., 2007). As an ortholog of ZMIZ1, tonalli (tna) was identified in Drosophila melanogaster and interacted with the ATP-dependent SWI/SNF complexes, which suggested a potential role in chromatin remodeling (Gutiérrez et al., 2003). Recently, ZMIZ1 was identified to be interacted with BRG1 (SMARCA4) (Li et al., 2011), BAF57 (SMARCE1) (Li et al., 2011), or SATB1 (Pinnell et al., 2015) to regulate the chromatin remodeling in humans. Chromatin remodeling complex could regulate the expression of genes which were essential for the normal dendrite development, synaptic plasticity, and synapse formation (Wu et al., 2007; Vogel-Ciernia et al., 2013; Vogel-Ciernia and Wood, 2014). It has been reported that in utero electroporation of ZMIZ1 pathogenic variants into the progenitor cells in the ventricular zone (VZ) of mice cortices (E14.5) resulted in impaired neuronal positioning with an accumulation in the ventricular and subventricular zones (VZ/SVZ) and intermediate zone (IZ) and a corresponding depletion in the upper cortical plate (CP). Therefore, ZMIZ1 variants were regarded as the causal genetic factors for NEDDFSA (Carapito et al., 2019).
Thus far, no patients with ZMIZ1 variants have been reported in Chinese. In order to decipher the genetic factors for neurodevelopmental disorder or intellectual disability (NEDD/ID) in China, array-CGH and WES were carried out for a cohort of 54 patients with NEDD/ID living in Shenzhen, Guangdong Province, China. After comprehensive bioinformatic analysis, a de novo missense variant (c.2330G > A, p.Gly777Glu, or p.G777E) was identified in the exon 20 of ZMIZ1 in a 5-year-old girl with mild development delay, mild intellectual disability, bilateral hip dysplasia, joint hypermobility, amblyopia in both eyes, strabismus in the right eye, and dysmorphic facial features. According to the criteria proposed by the American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015), this variant was classified as PS2 + PM1 + PM2 + PP2 + PP3 and annotated as “Pathogenic.” After comparing the clinical phenotypes described for NEDDFSA with the clinical phenotypes of our current Chinese patient, this girl was diagnosed as NEDDFSA. This variant is located in the highly conserved zf-MIZ domain and affected the three-dimensional conformation which might be detrimental for the binding of ZMIZ1 to its partners.
To our knowledge, this is the first case of Chinese NEDD/ID caused by a ZIMI1 variant. Due to the huge population, more patients with ZMIZ1-related disorder will be found in the near future.
This study was conducted in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans. This study was approved by the Ethics Committee of the Shenzhen Baoan Women’s and Children’s Hospital. Written informed consent was obtained from each individual.
Peripheral venous blood was collected from the 54 NEDD/ID patients and their parents. Genomic DNA was extracted using the TIANamp Blood DNA Kit (DP348, Tiangen Biotech, Beijing, China) according to the manufacturer’s instructions.
Array-CGH was performed using the Fetal DNA Chip (Version 1.2) designed by The Chinese University of Hong Kong (CUHK) (Leung et al., 2011; Huang et al., 2014). The chip contains a total of 60,000 probes for more than 100 diseases caused by known microduplication/microdeletions. It does not include small-size chromosomal abnormalities, copy number polymorphism, chimerism, or chromosomal rearrangement (Iafrate et al., 2004). The experimental procedures were carried out according to the standard Agilent protocol (Agilent Oligonucleotide Array-Based CGH for Genomic DNA Analysis, version 3.5). Hybridized slides were scanned with SureScan High-Resolution Microarray Scanner (G2505B, Agilent Technologies, Santa Clara, CA), and the image data were extracted and converted to text files using Agilent Feature Extraction software (Version 10.5.1.1). The data were graphed and analyzed using Agilent CGH Analytics software.
Only gains or losses that were encompassed by at least three consecutive oligomers on the array were considered. Then, the clinical relevance of observed chromosomal aberrations was estimated according to data found in the scientific literature and databases for each of the regions and genes involved, using the DECIPHER database (Swaminathan et al., 2012) for known microdeletion and microduplication syndromes and the Online Mendelian Inheritance in Man (OMIM) (Sayers et al., 2021) for known disease-causing genes, gene functions, and inheritance patterns. Copy number variations were considered as “likely pathogenic/pathogenic” when they involved regions known to be associated with microdeletion or microduplication syndromes.
WES was performed for family trios (trio-WES) without chromosomal abnormalities at MyGenostics or BerryGenomics Co. LTD. Briefly, the fragmented genomic DNAs were ligated with the 3ʹ end of the Illumina adapters and amplified by polymerase chain reaction (PCR). The amplified DNA was captured with Gencap Human whole Exon Kit (52M) at MyGenostics or with xGen Exome Research Panel v2.0 (Integrated DNA Technologies, Coralville, IA) at BerryGenomics. The capture procedure was performed in accordance with the manufacturer’s protocol. Finally, the generated libraries were sequenced on Illumina HiSeq 2500 platform for paired-end sequencing.
The sequencing depth of each sample was about 100. Sequencing reads were aligned with the human reference genome (UCSC hg19). The workflow of the screening for causal variants was depicted in Figure 1. Briefly, clean reads were obtained after removal of adaptors and low-quality reads. GATK (Genome Analysis Toolkit) was used to trim the variant calling in the trimmed WES clean data (https://gatk.broadinstitute.org/hc/en-us). ANNOVAR was applied to annotate the generated VCF file (Wang et al., 2010). Deleted variants with a minor allele frequency (MAF) > 5% in the 1000 Genome Project, MAF >2% in in-house data, or synonymous single nucleotide variants (SNVs) were removed. SNVs that caused splicing, frameshift, stop gain, or stop loss were retained for subsequent analysis. A position was called as heterozygous if 25% or more of the reads identify the minor allele.
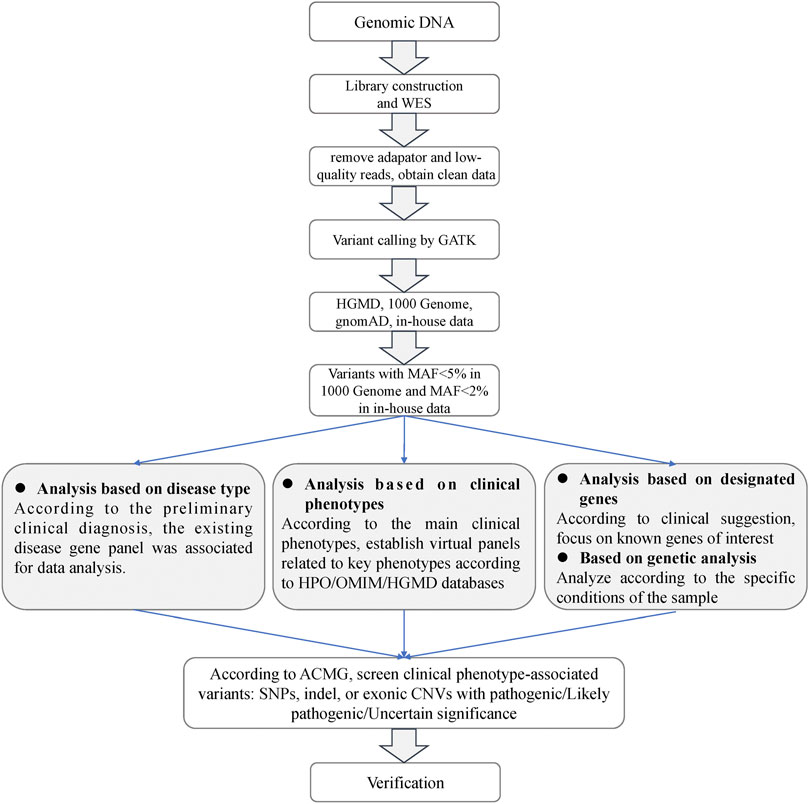
FIGURE 1. Analysis flowchart of the whole exome sequencing data.
The chromosomal location and type of the identified variants were retrieved in UCSC Genome Browser (Navarro Gonzalez et al., 2021) and NCBI dbSNP (Bhagwat, 2010). The MAFs of the variants were screened in several public databases with a large number of human samples, such as 1000 Genome Project (n = 2504) (Siva, 2008), NHLBI Exome Sequencing Project (GO-ESP) (n = 6503) (Amendola et al., 2015), The Exome Aggregation Consortium (ExAC) (n = 60,706) (Lek et al., 2016), gnomAD (n = 15,708) (Scheps et al., 2020), and NHLBI Trans-Omics for Precision Medicine (TOPMED) (n = 60,000) (Taliun et al., 2021). The function prediction of these variants was carried out by online software, PolyPhen-2 (Adzhubei et al., 2010), and PROVEAN (Choi et al., 2012). Pathogenicity of the variants was evaluated according to the American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015). The selected variants were verified by Sanger sequencing using the ABI 3500 Genetic Analyzer (Applied Biosystems, Foster City, CA).
Protein sequences of ZMIZ1 in 34 species were downloaded from NCBI GenBank, including five primates (Homo sapiens, Pan troglodytes, Gorilla Gorilla gorilla, Hylobates moloch, and Macaca fascicularis), one cattle (Bos taurus), one horse (Equus caballus), one dog (Canis lupus familiar), three carnivores (Neogale vison, Panthera tigris, and Halichoerus grypus), three rodents (Eptesicus fuscus, Mus musculus, and Rattus norvegicus), five reptiles (Crotalus tigris, Varanus komodoensis, Dermochelys coriacea, Chelonoidis abingdonii, and Mauremys mutica), two birds (Falco rusticolus, Gallus gallus), three amphibians (Bufo bufo, Xenopus tropicalis, and Oryzias latipes), three fish (Takifugu rubripes, Hippocampus comes, and Danio rerio), three arthropods (Limulus polyphemus, Penaeus monodon, and Ceratitis capitata), and four mollusks (Acropora millepora, Crassostrea gigas, Octopus sinensis, and Exaiptasia diaphana). The protein sequences were aligned by the ClustalW alignment algorithms of MEGA X (Kumar et al., 2018) (gap opening penalty and gap extension penalty for pairwise alignment and multiple alignment were set as 10.00, 0.10 and 10.00, 0.20, respectively; the delay divergent cutoff was 30%).
The intrinsically disordered regions of ZMIZ1 protein (NP_065071) were analyzed using the online web server IUPred2A (https://iupred2a.elte.hu/) with long disorder setting to identify probable disordered regions using the IUPred2 model and disordered binding regions using the ANCHOR2 model (Erdos and Dosztanyi, 2020). The distinct motifs of ZMIZ1 were analyzed using the online software Motif Scan (https://myhits.sib.swiss/cgi-bin/motif_scan) under default settings to search all known motifs in HAMAP (Pedruzzi et al., 2015), PROSITE (Hulo et al., 2006), Pfam (Mistry et al., 2021), and InterPro databases (Mitchell et al., 2019). The possible phosphorylation sites of ZMIZ1 were predicted by Disorder Enhanced Phosphorylation Predictor (DEEP, http://www.pondr.com/cgi-bin/depp.cgi) using 0.50 as the cutoff value (Iakoucheva et al., 2004).
The effect of G777E on the structural change was predicted by the online protein structure and function prediction tool, I-TASSER (Iterative Threading ASSEmbly Refinement) under default parameters (Yang and Zhang, 2015) for the whole second globular region (aa575-820) and visualized using Mol* 3D Viewer (Sehnal et al., 2021). The gene expression data of ZMIZ1 were evaluated according to the normalized signal intensity of probe 212124 at which were extracted from a gene atlas of human protein-encoding transcriptomes for 79 human tissues (NCBI GEO #GSE1133) (Su et al., 2004). The protein interaction network with ZMIZ1 (PPI enrichment p value = 1.51E-03) was generated by STRING (version 11.5, https://string-db.org/) under default settings. Gene Ontology (GO) analysis was performed on the nine members of the network in the GO knowledgebase (http://geneontology.org/) under default parameters.
There were 54 cases in our current NEDD/ID cohort collected from southern China. There were 48.15% (26/54) women and 51.85% (28/54) men. The mean age of women and men patients was 2.45 ± 1.15 and 2.67 ± 1.71, respectively. In these 54 samples, pathogenic variants were found in 33 patients, 5 with microdeletions and 28 with variants in protein-coding genes (Supplementary Table S1). The positive rate was 61.11% (33/54). In one patient, ZMIZ1 was detected to have a pathogenic missense variant (c.2330G > A, p.G777E). This patient was a 5-year-old girl who was referred to our department because of psychomotor developmental delay. She was the second child of a non-consanguineous couple (Figure 2A). The proband was delivered at term to a 36-year-old mother by Cesarean section due to breech position at 2016-10. Her birth weight was 3000 g, and there was no history of asphyxia at birth. At the sixth month after birth, asymmetric dermatoglyphs were found on both of her lower limbs after a physical examination and later diagnosed as dysplasia of bilateral hip joints. At 1 year old, the patient had chronic constipation. Since 2017, she has been sent to the ophthalmology department several times due to binocular weak eyesight and strabismus in the right eye. After 1 year old, she was still unable to speak and walk without support and was sent to the rehabilitation center for special training. Until 2 years old, she was able to speak simple words and walk, and was finally diagnosed as “developmental delay”.
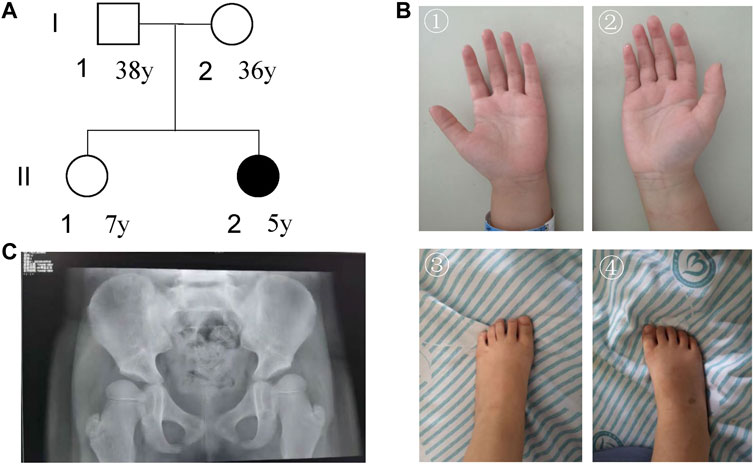
FIGURE 2. Characterization of the patient’s information. (A) Pedigree; (B) pictures of hands and foot; (C) DR X-ray film for bilateral hip joints.
For facial features, the patient had epicanthus, ptosis, up-slanting eyelid fissure, wide eye distance, wide nose bridge, Cupid lip arch and low-set ear. Regarding skeletal abnormalities, she had short fingers and toes, conical fingers (Figure 2B), and excessive joint mobility. As for the gross motor, the patient could walk alone. She could not stand on one foot for more than 3 s and jump on one foot. The trotting posture was slightly abnormal, and easy to fall. In terms of fine motor, she could draw a straight line, pull a zipper, unbutton buckles, cut paper inflexibly with scissors, eat with spoons and chopsticks, but couldn’t draw circles, squares, and triangles. In terms of the language, she could speak simple and long sentences with clear pronunciation, understand a few Chinese characters, recite numbers from 1 to 20, answer simple questions, but sometimes with confused word order and logic. For the social adaptive capacity, she could wear and take off clothes, shoes, and socks, could go to the bathroom, and eat by herself. She had the initiative to share and express her needs but without the initiative to say hello and goodbye. Besides, she had poor name calling response, poor sitting quietly ability, and hyperactive behavior.
The visually evoked potential (VEP) test showed that after blink flash stimulation for both eyes, N75, P100, and N145 waves could be induced with good repeatability. However, the latency of the P100 and N145 waves on both sides was prolonged, which was slightly abnormal. The evaluations for audiology system, heart, and urinary system were normal. DR X ray film for hip joint anteroposterior projection at 4-years-7-months old showed that the left and right acetabular angles were about 22 and 25°, respectively (Figure 2C). She was diagnosed with congenital dysplasia of the hip by an orthopedic surgeon at a tertiary children’s hospital. She received 2 brain MRI scans (March 28, 2019and October 26, 2021) and 3 electroencephalogram (EEG) examinations (March 15, 2019, August 20, 2020, and June 23, 2021). No obvious irregularities were identified.
Neuropsychological development assessment was performed for the patient at 5-years-1-month old using the Wechsler Preschool and Primary Scale of Intelligence Fourth Edition (WPPSI-IV) and the parent-rated Adaptive Behavior Assessment System II (ABAS-II) infant version. Her score on the full-scale intelligence quotient of WPPSI-IV was 75 (95% CI: 70-82, P5). The verbal comprehension index, visual spatial index, perceptual reasoning index, working memory index, and processing speed index of WPPSI-IV were 77 (95% CI: 71-86, P6), 83 (95% CI 76-94, P13), 79 (95% CI 73-87, P8), 76 (95% CI 76-94, P5), and 71 (95% CI 66-85, P3), respectively. The overall adaptive function score of ABAS-II was 77 (95% CI: 73-81, P6). The scores of social skills, conceptual skills, and practical skills in the three composite areas of adaptive function were 71 (95% CI 64-78, P3), 84 (95% CI 77-91, P14), and 80 (95% CI 74-86, P9), respectively. According to the clinical evaluation, she was at the edge level of intellectual development.
Whole exome sequencing was performed for the trio to identify possible genetic factors of the proband. After removal of adaptors and low-quality reads, the obtained total clean data obtained for the trio were 11,959.04 (Mb) for the proband, 15,681.80 (Mb) for the father, and 12,202.51 (Mb) for the mother (Table 1). The target coverage was at least or more than 98%. The average depth of target region was more than 100X. The on-target ratio was more than 35%. In these samples, the total numbers of identified SNVs were 175,809 for the proband, 197,560 for the father, and 179,722 for the mother, respectively. The percentages of pathogenic variants were around 4%. The total number of small insertions and deletions were 36,452, 42,698, and 36,816 for the proband, father, and mother, respectively.
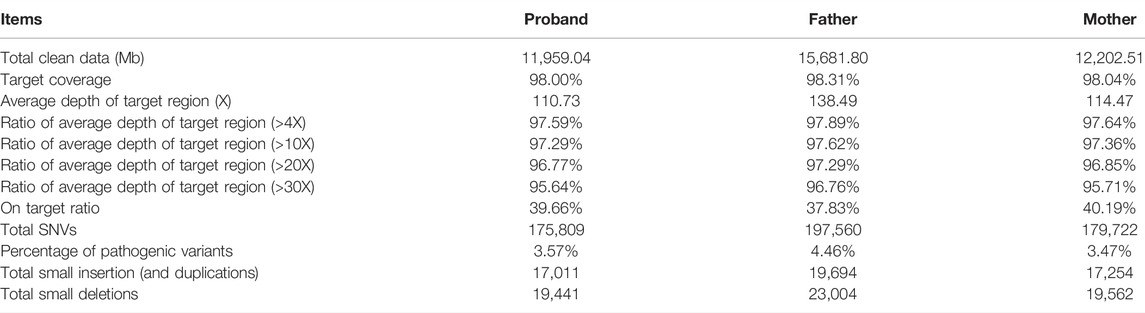
TABLE 1. Characterization of the whole exome sequencing for the trio.
In this proband, 20 specific variants were selected. Twelve of them were heterozygous in 10 genes (ANKRD36C, MUC2, MUC4, HRCT1, KLHL29, MYO15B, PER3, RPTN, TWIST1, and ZMIZ1) and 8 homozygous in 8 genes (AGAP3, CCDC177, CGN, DSPP, ESX1, FOXN4, MUC4, POTEB3, and SLC35E2A) (Table 2). According to the criteria of ACMG guidelines, 10 heterozygous and 8 homozygous variants were annotated as variants of uncertain significance (VUS). Most of these variants were predicted to be “neutral” by Provean or “benign” by Polyphen. PER3 (OMIM #603427), TWIST1 (OMIM #601622), and DSPP (OMIM #125485) were also recorded in the OMIM database as disease-causing genes. However, the phenotypes caused by these genes were not in line with our female proband. Besides, two rare heterozygous variants (c.148C > T, p.R50W in KLHL29 and c.2330G > A, p.G777E in ZMIZ1) were annotated as “likely pathogenic” (PS2 + PM2 + PP2) and “pathogenic” (PS2 + PM1 + PM2 + PP2 + PP3), respectively.
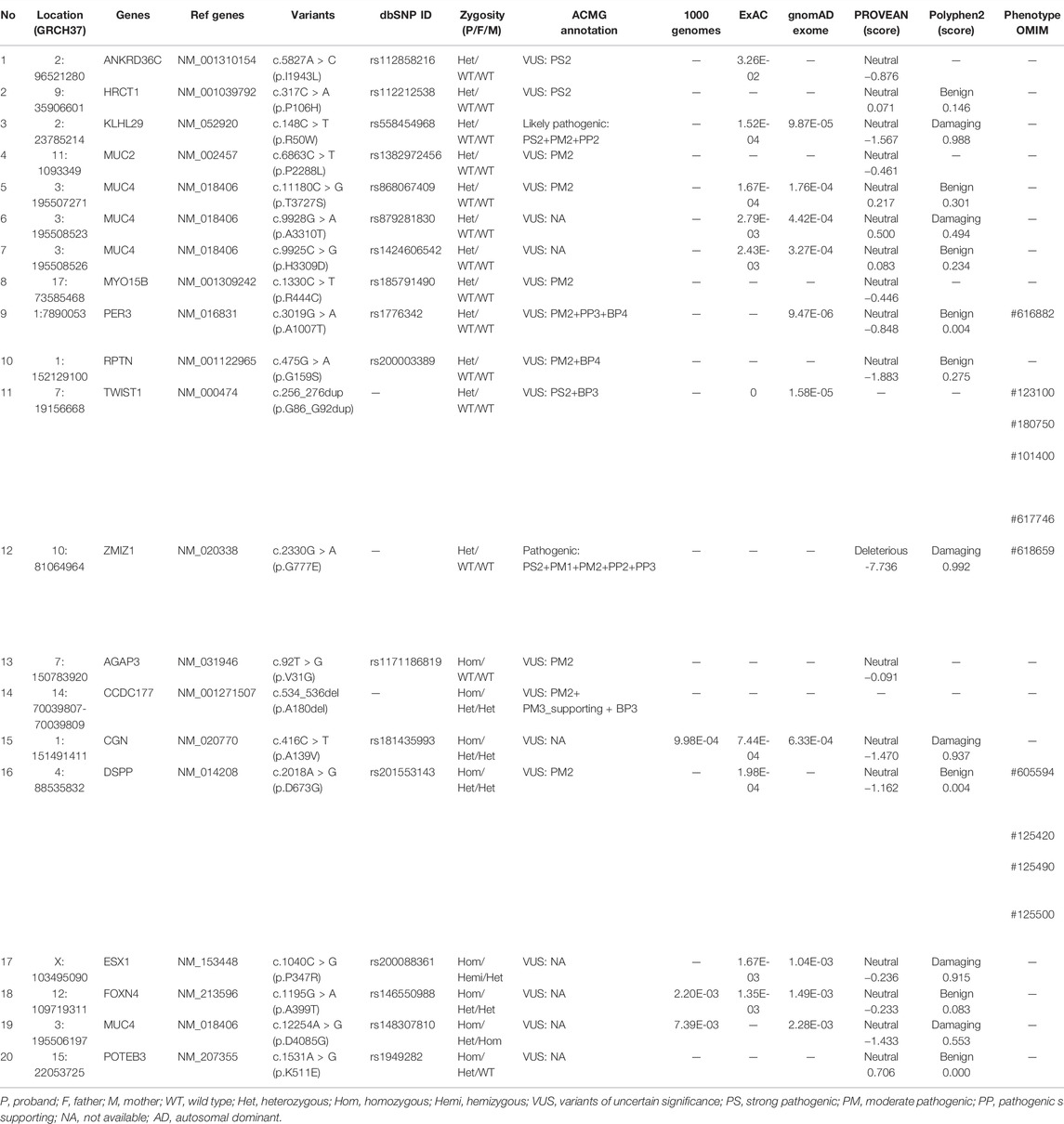
TABLE 2. Identified variants in the five-year-old proband.
Since there was no experimental evidence for KLHL29 leading to neurodevelopmental disorder, ZMIZ1 was considered as the most potential disease-causing gene. The c.2330G > A (p.G777E) was a heterozygous SNV in the exon 20 of ZMIZ1 gene (NM_020338) (Figures 3A,B) and confirmed by Sanger sequencing (Figure 3C) only in the patient, but not in her healthy parents or her elder sister. Thus, it was a de novo variant. The protein sequences of ZMIZ1 from more than 34 species (Mollusca, crabs, fish, amphibians, insects, reptiles, rodents, dogs, cats, cattle, and primates) were downloaded from NCBI GenBank and aligned by the ClustalW alignment algorithms of MEGA 11, the G777 was highly conserved in the animals during evolution (Figure 3D). G777E was localized in the functional MSX-interacting zinc finger (zf-MIZ) domain (Figure 3E) and predicted to be “deleterious” with a score of 0.536, “deleterious” with a score of -7.736 (Provean) and “probably damaging” with a score of 0.992 (PolyPhen-2). In addition, this SNV has not been detected in multiple public genome databases, such as 1000 Genome Project (n = 2504), NHLBI Exome Sequencing Project (GO-ESP) (n = 6503), the Exome Aggregation Consortium (ExAC) (n = 60,706), Genome Aggregation database (gnomAD) (n = 15,708), and NHLBI Trans-Omics for Precision Medicine (TOPMED) (n = 60,000). Since this amino acid changing variant was only identified in the patient, and not in her parents, it was regarded as “spontaneous.” According to the Probability of Loss-of-function Intolerance (pLI) analysis, the pLI value of ZMIZ1 was 1.000, which indicated ZMIZ1 being a haploinsufficient gene. It has been reported that ZMIZ1 could cause the occurrence of a rare neurodevelopmental disorder, neurodevelopmental disorder with dysmorphic facies and distal skeletal anomalies (NEDDFSA). Based on the recorded clinical phenotypes (Supplementary Table S2), this patient was finally diagnosed as NEDDFSA. Besides, four rare variants in ZMIZ1 were also detected in another four NEDD patients, namely, c.3096 + 15C > T, c.1024A > G (p.M342V), c.540 + 20T > C and c.679G > A (p.A227T) (Supplementary Table S3).
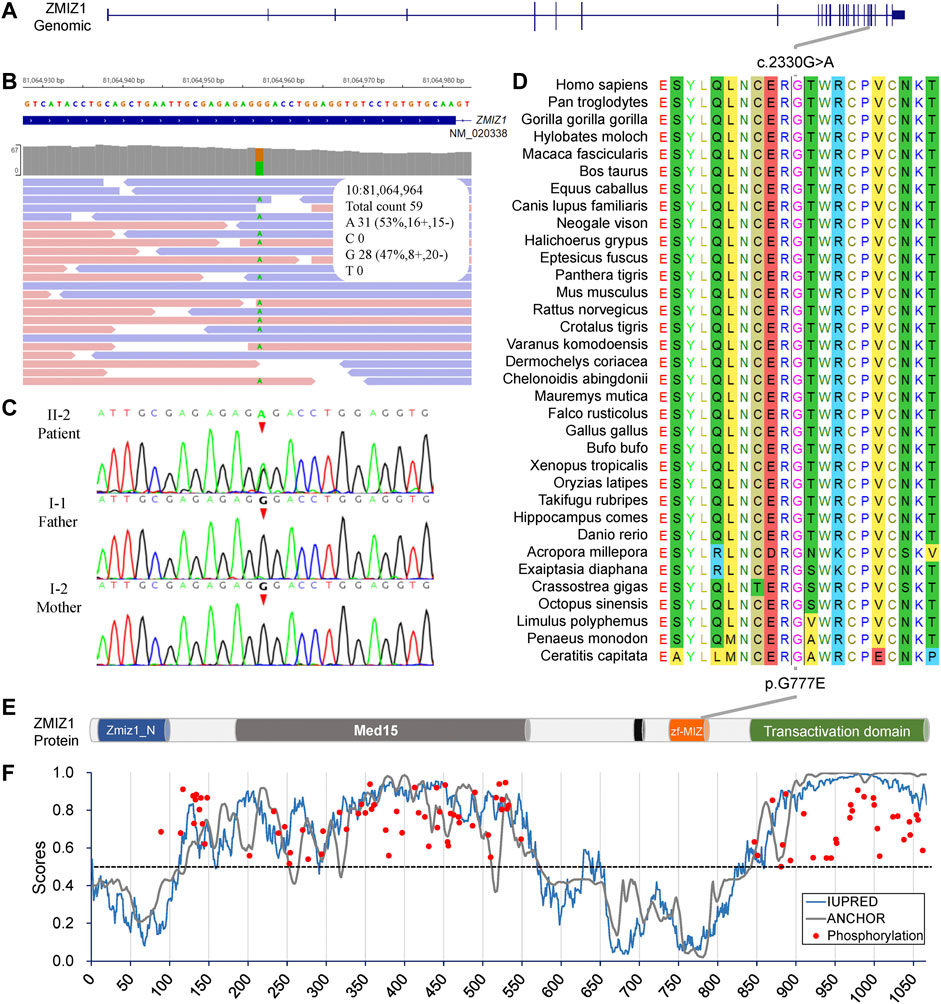
FIGURE 3. Molecular analysis of the c.2330G > A (p.G777E) in ZMIZ1 gene. (A) Gene structure of ZMIZ1; (B) IGV view of the c.2330G > A identified by WES; (C) Sanger sequencing of the c.2330G > A variant; (D) evolutionary conservation analysis; (E) protein structure of ZMIZ1; (F) analysis for intrinsically disordered regions and phosphorylation sites.
The intrinsically disordered regions of ZMIZ1 protein (NP_065071) were analyzed using the online web server IUPred2A. Two functional globular regions were identified at two portions (aa2-110 and 575-820), which overlapped with the two important functional domains, Zmiz1 N-terminal tetratricopeptide repeat domain (Zmiz1_N, aa8-100) and MIZ/SP-RING zinc finger (zf-MIZ, aa739-786), respectively. The predicted two globular regions and two intrinsically disordered regions are displayed in Figure 3E. The possible phosphorylation sites of ZMIZ1 were predicted using DEEP under default parameters. Interestingly, most of the phosphorylation sites were located in the two long disordered regions (Figure 3F). As for G777E, it was localized in the second globular regions containing zf-MIZ domain and might affect the probable tertiary structures as predicted by I-TASSER (Supplementary Figure S1).
The distinct regions of ZMIZ1 were analyzed using the online software Motif Scan under default settings. Seven distinct regions were identified, one alanine-rich region (aa280-305, E-score = 2.1E-06), two proline-rich regions (aa334-555, E-score = 3.9E-16; aa867-1002, E-score = 3.8E-07), one bipartite nuclear localization signal (NLS, aa697-711, E-score = 2.1E+04), MIZ/SP-RING zinc finger (aa738-787, E-score = 1.1E-33), and one copper binding octapeptide (aa947-954, E-value = 1.5). All variants of ZMIZ1 were also recruited from the DECIPHER database (Swaminathan et al., 2012) and the four published articles (Córdova-Fletes et al., 2015; Carapito et al., 2019; Latchman et al., 2020; Phetthong et al., 2021). A total of 33 patients with ZMIZ1 pathogenic variants were collected, 1 from our current cohort (Figures 4A,B), 8 from the DECIPHER database (Figure 4C), and 24 from published articles (Figure 4D). Except for K91R, H581R, and H683Y, other variants were localized in the low-complexity regions, such as the alanine-rich region and the proline-rich region of the Med15 (mediator complex subunit 15) domain, and the proline-rich region in the C-terminal transactivation domain (TAD). There were nine amino acid-changing variants, which were strongly conserved during evolution (Figure 4E). Six of them were in the alanine-rich region, accounting for 66.67% (6/9). From the phosphorylation prediction by DEEP, except for T300M, other variants could distinctly change the phosphorylation pattern of the alanine-rich region (Figure 4F).
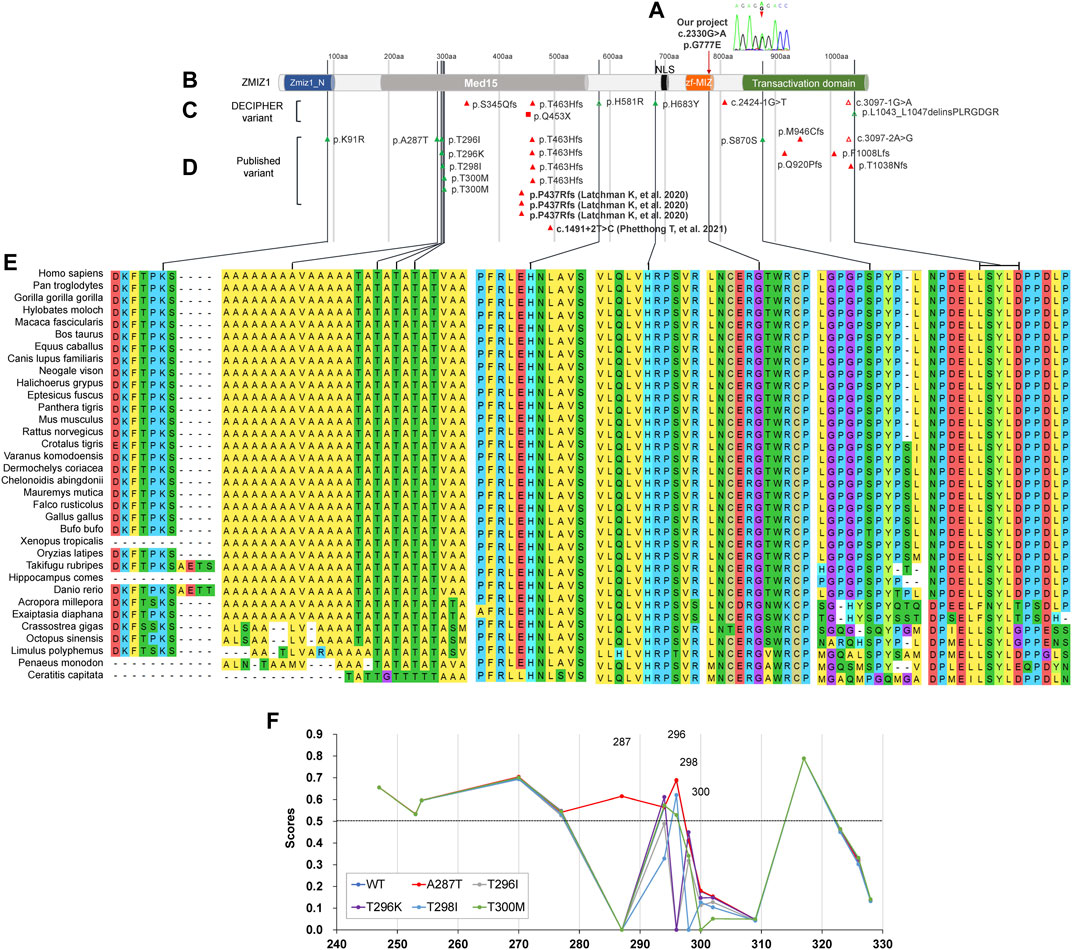
FIGURE 4. Molecular analysis of variants of ZMIZ1. (A) Variant in our cohort; (B) diagram of ZMIZ1 protein; (C) variants in DECIPHER; (D) variants in reported articles; (E) evolutionary conservation; (F) phosphorylation analysis in the alanine-rich region.
The gene expression data of 79 human tissues showed that ZMIZ1 was expressed in the heart, thyroid, immune cells, ovary, retina, and brain, with the highest in the pineal (Figure 5A). The protein interaction network with ZMIZ1 indicated that ZMIZ1 could interact with SMAD3, SMAD4, MYC, NOTCH1, RBPJ, SMARCA4, ETS1, and UBE2I (Figure 5B). According to the GO analysis for the 9 members (Figure 5C), the network was involved significantly in mesenchyme morphogenesis, hypoxia, tube morphogenesis, regulation of transcription, response to stimulus, endocardium development, epithelial to mesenchymal transition, and cardiac left ventricle morphogenesis in GO term “biological process.” In “molecular function,” transcription, SMAD binding, and SUMOylation were significantly enriched. As for “cellular component” and “subcellular localization,” members of this network were localized in nuclear to form multiple protein complexes, mainly MAML1-RBP-Jκ-ICN1 (Intracellular Notch1) complex and SMAD protein complex to regulate the expression of target genes.
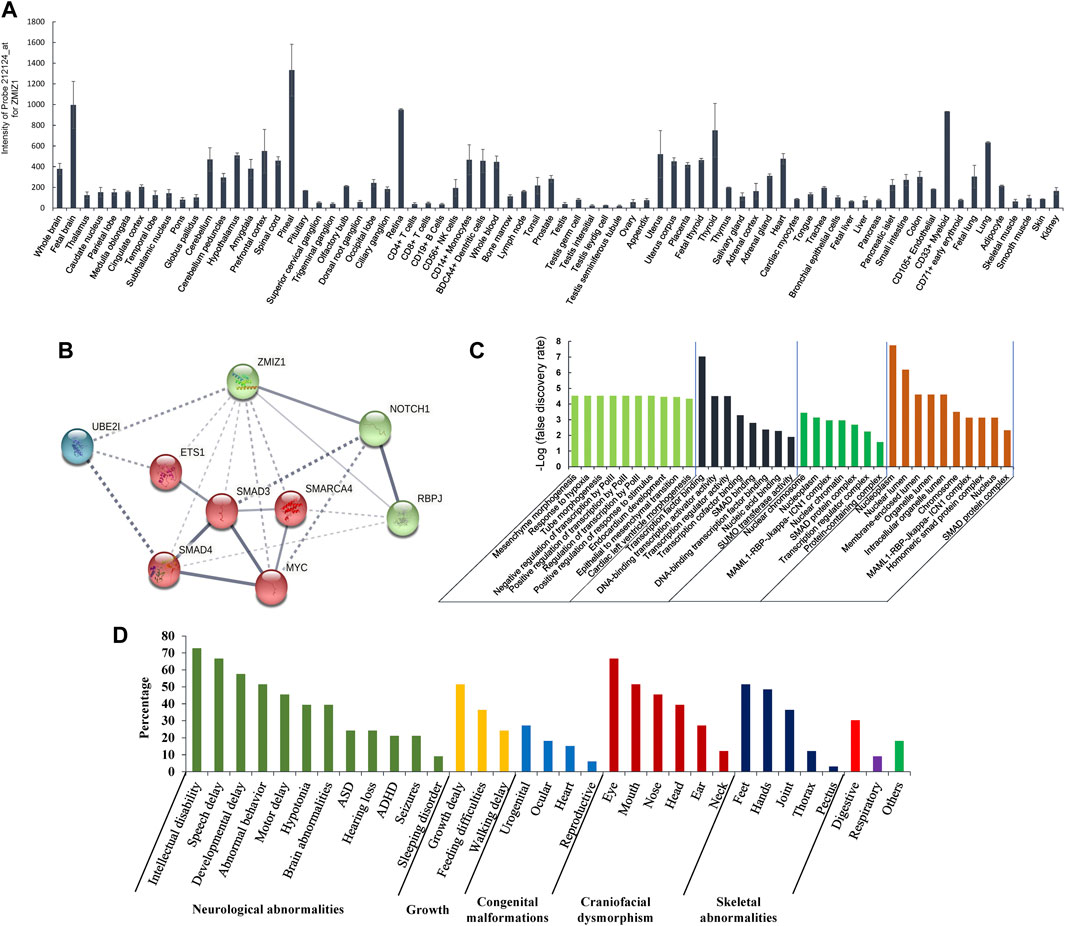
FIGURE 5. Expression of ZMIZ1 in different human tissues, and network analysis. (A) Tissue-specific expression of ZMIZ1; (B) protein interaction network produced by STRING; (C) GO analysis for the 9 members of the network; (D) phenotypes of the patients carrying ZMIZ1pathogenic variants.
Pathogenic variants of the zinc finger MIZ-type containing 1 (ZMIZ1, OMIM#607159) could cause the occurrence of a rare syndromic disease, neurodevelopmental disorder with dysmorphic facies and distal skeletal anomalies (NEDDFSA) with an autosomal dominant (AD) mode of inheritance. Currently, 32 patients with neurodevelopmental disorders have reported carrying pathogenic variants in the protein coding sequences of ZMIZ1 (n = 29) and chromosomal translocations involving ZMIZ1 (n = 3) (Supplementary File S2). Among these patients, except for c.1491+2T > C identified in a Thai female, the remaining 31 variants were detected in patients with Caucasian origin in Western countries. In our small cohort of NEDD/ID cases in China, a de novo missense pathogenic variant c.2330G > A (p.G777E) was detected in a 5-year-old girl. This patient presented the characteristic clinical phenotypes of NEDDFSA, such as neurodevelopmental delay, mild intellectual disability, hypotonia, language delay, dysmorphic facial features, joint hypermobility, and hand and foot anomalies, which were the common features of NEDDFSA (Figure 5D, Supplementary Table S2). As far as we know, this was the first report of ZMIZ1 variant in Chinese. Besides, we also identified four other rare variants in the ZMIZ1 gene (c.540 + 20T > C, c.679G > A, c.1024A > G, and c.3096 + 15C > T) (Supplementary Table S3). Although predicted as “neutral” or “benign” to the function of ZMIZ1, it still could not rule out their pathogenicity. Cellular and animal experiments should be taken to verify the function of these variants, including the c.2330G > A (p.G777E).
ZMIZ1 was previously known as ZIMP10, RAI17, or KIAA1224. In 1999, Nagase et al. identified the gene ZMIZ1 (previously called KIAA1224) from a fetal brain cDNA library (Nagase et al., 1999). According to the human tissue-specific transcriptomes, it was expressed in the heart, thyroid, immune cells, ovary, retina, and brain, with the highest in the pineal gland (Su et al., 2004). The encoded protein is a transcriptional co-activator, which belongs to the Protein Inhibitor of Activated STAT (PIAS) family. As a member of the PIAS family, ZMIZ1 has a highly conserved MIZ (Msx-interacting zinc finger) domain which is important for protein-protein interaction and SUMOylation (Sharma et al., 2003; Beliakoff and Sun, 2006). It had been reported that ZMIZ1 could regulate the activity of many transcription factors, such as androgen receptor (AR) (Beliakoff and Sun, 2006), SMAD3 (Li et al., 2006), SMAD4 (Li et al., 2006), and p53 (Lee et al., 2007). As an ortholog of ZMIZ1, tonalli (tna) was identified in Drosophila melanogaster and interacted with the ATP-dependent SWI/SNF complexes, which suggested a potential role in chromatin remodeling (Gutiérrez et al., 2003). Recently, ZMIZ1 was identified to be interacted with BRG1 (SMARCA4) (Li et al., 2011), BAF57 (SMARCE1) (Li et al., 2011), or SATB1 (Pinnell et al., 2015) to regulate the chromatin remodeling in humans. The protein-protein interaction network showed that ZMIZ1 could interact with SMAD3, SMAD4, MYC, NOTCH1, RBPJ, SMARCA4, ETS1, and UBE2I. According to the GO analysis for the 9 members of the protein network containing ZMIZ1, the network was significantly involved in mesenchyme morphogenesis, hypoxia, tube morphogenesis, regulation of transcription, response to stimulus, endocardium development, epithelial to mesenchymal transition, and cardiac left ventricle morphogenesis. This explained why ZMIZ1 pathogenic variant could affect the normal development of multiple systems, such as nerve, heart, and bones. GO analysis also showed that members of this network were localized in the nucleus to form two multiple protein complexes, mainly MAML1-RBP-Jκ-ICN1 complex and SMAD protein complex, to regulate the expression of target genes. The proper expression of ZMIZ1 was essential for the standard embryonic development. It has been revealed in mice embryos at different stages that ZMIZ1 was expressed dynamically in the neural tissues, craniofacial tissues, mandibular, foregut, limb buds, optic vesicle and otic pit, and somite (Beliakoff et al., 2008; Rodriguez-Magadán et al., 2008). This was consistent with the above-mentioned clinical features produced by the mutant ZMIZ1.
After compiling all the ZMIZ1 variants in the DECIPHER database, published articles, and our cohort (Supplementary Table S2), 12 patients were found to carry amino-acid changing variants, and half of them (6/12) had variants in the alanine-rich sequence. The alanine-rich low-complexity region (LCR) was localized in the N-terminal intrinsic disordered region of ZMIZ1. The alanine-rich sequences were extremely conserved in different species during evolution, suggesting its importance for the proper function of ZMIZ1. According to the reports, many transcription factors or transcription mediators, such as FUS (FUS RNA binding protein), EWSR1 (EWS RNA binding protein 1), TAF15 (TATA-box binding protein associated factor 15), Sp1 (Sp1 transcription factor), and AR could interact with ZMIZ1 at the transcriptional start sites via their extremely low-complexity regions (LCRs) to form local phase-separated condensates (or called droplets) to stabilize DNA binding, recruit RNA polymerase II (RNA Pol II), and activate transcription (Chong et al., 2018; Zamudio et al., 2019). These special condensates were a trade-off between proper functionality and risk of abnormal aggregation. The aberrant phase transitions within liquid-like droplets lie at the heart of many kinds of diseases, such as TATA box-binding protein (TBP, OMIM#600075) for spinocerebellar ataxia 17 (SCA17, OMIM#607136) (Friedman et al., 2007), FUS (OMIM#137070) for amyotrophic lateral sclerosis 6 (ALS6, OMIM#608030) (Patel et al., 2015), and androgen receptor (AR, OMIM#313700) for spinal and bulbar muscular atrophy (SBMA, OMIM#313200). As predicted by IUPred2A, ZMIZ1 contained three low-complexity regions (one alanine-rich and two proline-rich regions). It is reasonable that the alanine-rich region might be indispensable for the phase separation of ZMIZ1 to carry out the transcription mediator function. As predicted, the variants could change the phosphorylation pattern in the alanine-rich region, which might affect the local conformation. This might be the underlying molecular mechanism for the alanine-rich region being the variation hotspot of ZMIZ1. However, this has not yet been experimentally verified.
In conclusion, a de novo missense variant was first discovered in a Chinese female with a rare heterozygous syndromic disease, neurodevelopmental disorder with dysmorphic facies, and distal skeletal anomalies (NEDDFSA). Currently, a total of 32 patients with 27 types of variants of ZMIZ1 (24 in protein-coding sequences and 3 translocations) have been identified globally. However, the underlying molecular mechanism of these variants has not been elucidated. Further experimental studies should be carried out to clarify these unknown fields to determine potential drug targets for the treatment of NEDDFSA.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding authors.
This study was conducted in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans. This study was approved by the Ethical Committee of the Shenzhen Baoan Women’s and Children’s Hospital. We obtained written informed consent from all individual members of the study. For minors, written informed consent has been obtained from their parents or legal guardians.
GL, LM, ZY, and QP conceived of the project and wrote the manuscript. GL analyzed the data. PX, BX, LW, JD, HX, and WD collected samples, consulted with patients, and extracted genomic DNAs. QP conducted genetic counseling, analyzed the clinical data, and revised the manuscript.
This work was funded by grants from the Shenzhen Science and Technology Innovation Commission (JCYJ20180305164359668 to QP), Shenzhen Municipal Health Commission (SZBC2018020 to QP), Project of Sichuan Provincial Department of Science and Technology (2019YFS0443), Key Research and Development Project of Deyang City’s Science and Technology Bureau (2021SZ003 to GL), and Special Fund for Incubation Projects of Deyang People’s Hospital (FHG202004 to GL).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank the patient and parents for their cooperation and consent to this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.840577/full#supplementary-material
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 7, 248–249. doi:10.1038/nmeth0410-248
Amendola, L. M., Dorschner, M. O., Robertson, P. D., Salama, J. S., Hart, R., Shirts, B. H., et al. (2015). Actionable Exomic Incidental Findings in 6503 Participants: Challenges of Variant Classification. Genome Res. 25, 305–315. doi:10.1101/gr.183483.114
Beliakoff, J., and Sun, Z. (2006). Zimp7 and Zimp10, Two Novel PIAS-like Proteins, Function as Androgen Receptor Coregulators. Nucl. Recept Signal. 4, e017. doi:10.1621/nrs.04017
Beliakoff, J., Lee, J., Ueno, H., Aiyer, A., Weissman, I. L., Barsh, G. S., et al. (2008). The PIAS-like Protein Zimp10 Is Essential for Embryonic Viability and Proper Vascular Development. Mol. Cel Biol 28, 282–292. doi:10.1128/mcb.00771-07
Bhagwat, M. (2010). Searching NCBI's dbSNP Database. Curr. Protoc. Bioinformatics Chapter 1, Unit 1–19. doi:10.1002/0471250953.bi0119s32
Carapito, R., Ivanova, E. L., Morlon, A., Meng, L., Molitor, A., Erdmann, E., et al. (2019). ZMIZ1 Variants Cause a Syndromic Neurodevelopmental Disorder. Am. J. Hum. Genet. 104, 319–330. doi:10.1016/j.ajhg.2018.12.007
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R., and Chan, A. P. (2012). Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS One 7, e46688. doi:10.1371/journal.pone.0046688
Chong, S., Dugast-Darzacq, C., Liu, Z., Dong, P., Dailey, G. M., Cattoglio, C., et al. (2018). Imaging Dynamic and Selective Low-Complexity Domain Interactions that Control Gene Transcription. Science 361, eaar2555. doi:10.1126/science.aar2555
Córdova-Fletes, C., Domínguez, M. G., Delint-Ramirez, I., Martínez-Rodríguez, H. G., Rivas-Estilla, A. M., Barros-Núñez, P., et al. (2015). A De Novo t(10;19)(q22.3;q13.33) Leads to ZMIZ1/PRR12 Reciprocal Fusion Transcripts in a Girl with Intellectual Disability and Neuropsychiatric Alterations. Neurogenetics 16, 287–298. doi:10.1007/s10048-015-0452-2
Erdos, G., and Dosztanyi, Z. (2020). Analyzing Protein Disorder with IUPred2A. Curr. Protoc. Bioinformatics 70, e99. doi:10.1002/cpbi.99
Friedman, M. J., Shah, A. G., Fang, Z.-H., Ward, E. G., Warren, S. T., Li, S., et al. (2007). Polyglutamine Domain Modulates the TBP-TFIIB Interaction: Implications for its normal Function and Neurodegeneration. Nat. Neurosci. 10, 1519–1528. doi:10.1038/nn2011
Gutiérrez, L., Zurita, M., Kennison, J. A., and Vázquez, M. (2003). The Drosophila Trithorax Group Gene Tonalli (Tna) Interacts Genetically with the Brahma Remodeling Complex and Encodes an SP-RING finger Protein. Development 130, 343–354. doi:10.1242/dev.00222
Huang, J., Poon, L. C., Akolekar, R., Choy, K. W., Leung, T. Y., and Nicolaides, K. H. (2014). Is High Fetal Nuchal Translucency Associated with Submicroscopic Chromosomal Abnormalities on Array CGH. Ultrasound Obstet. Gynecol. 43, 620–624. doi:10.1002/uog.13384
Hulo, N., Bairoch, A., Bulliard, V., Cerutti, L., De Castro, E., Langendijk-Genevaux, P. S., et al. (2006). The PROSITE Database. Nucleic Acids Res. 34, D227–D230. doi:10.1093/nar/gkj063
Iafrate, A. J., Feuk, L., Rivera, M. N., Listewnik, M. L., Donahoe, P. K., Qi, Y., et al. (2004). Detection of Large-Scale Variation in the Human Genome. Nat. Genet. 36, 949–951. doi:10.1038/ng1416
Iakoucheva, L. M., Radivojac, P., Brown, C. J., O'Connor, T. R., Sikes, J. G., Obradovic, Z., et al. (2004). The Importance of Intrinsic Disorder for Protein Phosphorylation. Nucleic Acids Res. 32, 1037–1049. doi:10.1093/nar/gkh253
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K., and Mega, X. (2018). MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 35, 1547–1549. doi:10.1093/molbev/msy096
Latchman, K., Calder, M., Morel, D., Rhodes, L., Juusola, J., and Tekin, M. (2020). Autosomal Dominant Inheritance in a Recently Described ZMIZ1‐ Related Neurodevelopmental Disorder: Case Report of Siblings and an Affected Parent. Am. J. Med. Genet. 182, 548–552. doi:10.1002/ajmg.a.61446
Lee, J., Beliakoff, J., and Sun, Z. (2007). The Novel PIAS-like Protein hZimp10 Is a Transcriptional Co-activator of the P53 Tumor Suppressor. Nucleic Acids Res. 35, 4523–4534. doi:10.1093/nar/gkm476
Lek, M., Karczewski, K. J., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., et al. (2016). Analysis of Protein-Coding Genetic Variation in 60,706 Humans. Nature 536, 285–291. doi:10.1038/nature19057
Leung, T. Y., Vogel, I., Lau, T. K., Chong, W., Hyett, J. A., Petersen, O. B., et al. (2011). Identification of Submicroscopic Chromosomal Aberrations in Fetuses with Increased Nuchal Translucency and Apparently normal Karyotype. Ultrasound Obstet. Gynecol. 38, 314–319. doi:10.1002/uog.8988
Li, X., Thyssen, G., Beliakoff, J., and Sun, Z. (2006). The Novel PIAS-like Protein hZimp10 Enhances Smad Transcriptional Activity. J. Biol. Chem. 281, 23748–23756. doi:10.1074/jbc.m508365200
Li, X., Zhu, C., Tu, W. H., Yang, N., Qin, H., and Sun, Z. (2011). ZMIZ1 Preferably Enhances the Transcriptional Activity of Androgen Receptor with Short Polyglutamine Tract. PLoS One 6, e25040. doi:10.1371/journal.pone.0025040
Lu, S., Wang, J., Chitsaz, F., Derbyshire, M. K., Geer, R. C., Gonzales, N. R., et al. (2020). CDD/SPARCLE: the Conserved Domain Database in 2020. Nucleic Acids Res. 48, D265–D268. doi:10.1093/nar/gkz991
Mistry, J., Chuguransky, S., Williams, L., Qureshi, M., Salazar, G. A., Sonnhammer, E. L. L., et al. (2021). Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 49, D412–D419. doi:10.1093/nar/gkaa913
Mitchell, A. L., Attwood, T. K., Babbitt, P. C., Blum, M., Bork, P., Bridge, A., et al. (2019). InterPro in 2019: Improving Coverage, Classification and Access to Protein Sequence Annotations. Nucleic Acids Res. 47, D351–D360. doi:10.1093/nar/gky1100
Nagase, T., Ishikawa, K., Kikuno, R., Hirosawa, M., Nomura, N., and Ohara, O. (1999). Prediction of the Coding Sequences of Unidentified Human Genes.XV. The Complete Sequences of 100 New cDNA Clones from Brain Which Code for Large Proteins In Vitro. DNA Res. 6, 337–345. doi:10.1093/dnares/6.5.337
Navarro Gonzalez, J., Zweig, A. S., Speir, M. L., Schmelter, D., Rosenbloom, K. R., Raney, B. J., et al. (2021). The UCSC Genome Browser Database: 2021 Update. Nucleic Acids Res. 49, D1046–D1057. doi:10.1093/nar/gkaa1070
Patel, A., Lee, H. O., Jawerth, L., Maharana, S., Jahnel, M., Hein, M. Y., et al. (2015). A Liquid-To-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 162, 1066–1077. doi:10.1016/j.cell.2015.07.047
Pedruzzi, I., Rivoire, C., Auchincloss, A. H., Coudert, E., Keller, G., de Castro, E., et al. (2015). HAMAP in 2015: Updates to the Protein Family Classification and Annotation System. Nucleic Acids Res. 43, D1064–D1070. doi:10.1093/nar/gku1002
Phetthong, T., Khongkrapan, A., Jinawath, N., Seo, G. H., and Wattanasirichaigoon, D. (2021). Compound Heterozygote of Point Variant and Chromosomal Microdeletion Involving OTUD6B Coinciding with ZMIZ1 Variant in Syndromic Intellectual Disability. Genes (Basel) 12, 1583. doi:10.3390/genes12101583
Pinnell, N., Yan, R., Cho, H. J., Keeley, T., Murai, M. J., Liu, Y., et al. (2015). The PIAS-like Coactivator Zmiz1 Is a Direct and Selective Cofactor of Notch1 in T Cell Development and Leukemia. Immunity 43, 870–883. doi:10.1016/j.immuni.2015.10.007
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Rodriguez-Magadán, H., Merino, E., Schnabel, D., Ramírez, L., and Lomelí, H. (2008). Spatial and Temporal Expression of Zimp7 and Zimp10 PIAS-like Proteins in the Developing Mouse Embryo. Gene Expr. Patterns 8, 206–213. doi:10.1016/j.modgep.2007.10.005
Sayers, E. W., Beck, J., Bolton, E. E., Bourexis, D., Brister, J. R., Canese, K., et al. (2021). Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 49, D10–D17. doi:10.1093/nar/gkaa892
Scheps, K. G., Hasenahuer, M. A., Parisi, G., Targovnik, H. M., and Fornasari, M. S. (2020). Curating the gnomAD Database: Report of Novel Variants in the Globin‐coding Genes and Bioinformatics Analysis. Hum. Mutat. 41, 81–102. doi:10.1002/humu.23925
Sehnal, D., Bittrich, S., Deshpande, M., Svobodová, R., Berka, K., Bazgier, V., et al. (2021). Mol* Viewer: Modern Web App for 3D Visualization and Analysis of Large Biomolecular Structures. Nucleic Acids Res. 49, W431–W437. doi:10.1093/nar/gkab314
Sharma, M., Li, X., Wang, Y., Zarnegar, M., Huang, C. Y., Palvimo, J. J., et al. (2003). hZimp10 Is an Androgen Receptor Co-activator and Forms a Complex with SUMO-1 at Replication Foci. EMBO J. 22, 6101–6114. doi:10.1093/emboj/cdg585
Su, A. I., Wiltshire, T., Batalov, S., Lapp, H., Ching, K. A., Block, D., et al. (2004). A Gene Atlas of the Mouse and Human Protein-Encoding Transcriptomes. Proc. Natl. Acad. Sci. 101, 6062–6067. doi:10.1073/pnas.0400782101
Swaminathan, G. J., Bragin, E., Chatzimichali, E. A., Corpas, M., Bevan, A. P., Wright, C. F., et al. (2012). DECIPHER: Web-Based, Community Resource for Clinical Interpretation of Rare Variants in Developmental Disorders. Hum. Mol. Genet. 21, R37–R44. doi:10.1093/hmg/dds362
Taliun, D., Harris, D. N., Harris, D. N., Kessler, M. D., Carlson, J., Szpiech, Z. A., et al. (2021). Sequencing of 53,831 Diverse Genomes from the NHLBI TOPMed Program. Nature 590, 290–299. doi:10.1038/s41586-021-03205-y
Vogel-Ciernia, A., Matheos, D. P., Barrett, R. M., Kramár, E. A., Azzawi, S., Chen, Y., et al. (2013). The Neuron-specific Chromatin Regulatory Subunit BAF53b Is Necessary for Synaptic Plasticity and Memory. Nat. Neurosci. 16, 552–561. doi:10.1038/nn.3359
Vogel-Ciernia, A., and Wood, M. A. (2014). Neuron-specific Chromatin Remodeling: a Missing Link in Epigenetic Mechanisms Underlying Synaptic Plasticity, Memory, and Intellectual Disability Disorders. Neuropharmacology 80, 18–27. doi:10.1016/j.neuropharm.2013.10.002
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 38, e164. doi:10.1093/nar/gkq603
Wu, J. I., Lessard, J., Olave, I. A., Qiu, Z., Ghosh, A., Graef, I. A., et al. (2007). Regulation of Dendritic Development by Neuron-specific Chromatin Remodeling Complexes. Neuron 56, 94–108. doi:10.1016/j.neuron.2007.08.021
Yang, J., and Zhang, Y. (2015). I-TASSER Server: New Development for Protein Structure and Function Predictions. Nucleic Acids Res. 43, W174–W181. doi:10.1093/nar/gkv342
Keywords: Zmiz1, NEDDFSA, Chinese, low-complexity region, whole-exome sequencing
Citation: Lu G, Ma L, Xu P, Xian B, Wu L, Ding J, He X, Xia H, Ding W, Yang Z and Peng Q (2022) A de Novo ZMIZ1 Pathogenic Variant for Neurodevelopmental Disorder With Dysmorphic Facies and Distal Skeletal Anomalies. Front. Genet. 13:840577. doi: 10.3389/fgene.2022.840577
Received: 21 December 2021; Accepted: 22 February 2022;
Published: 31 March 2022.
Edited by:
Shicheng Guo, University of Wisconsin-Madison, United StatesReviewed by:
Xianli Shen, Dana-Farber Cancer Institute and Harvard Medical School, United StatesCopyright © 2022 Lu, Ma, Xu, Xian, Wu, Ding, He, Xia, Ding, Yang and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiongling Peng, MzE0NjMzNjIxQHFxLmNvbQ==; Zhirong Yang, MTMyNDk4NjgxN0BxcS5jb20=
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.