 Luyang Sun1,2
Luyang Sun1,2 Kaixing Qu3Xiaohui Ma2
Kaixing Qu3Xiaohui Ma2 Quratulain Hanif4Jicai Zhang1Jianyong Liu1
Quratulain Hanif4Jicai Zhang1Jianyong Liu1 Ningbo Chen2Quji Suolang5
Ningbo Chen2Quji Suolang5 Chuzhao Lei2*
Chuzhao Lei2* Bizhi Huang1*
Bizhi Huang1*- 1Yunnan Academy of Grassland and Animal Science, Kunming, China
- 2Key Laboratory of Animal Genetics, Breeding and Reproduction of Shaanxi Province, College of Animal Science and Technology, Northwest A&F University, Yangling, China
- 3Academy of Science and Technology, Chuxiong Normal University, Chuxiong, China
- 4National Institute for Biotechnology and Genetic Engineering, Faisalabad, Pakistan
- 5Institute of Animal Science, Tibet Academy of Agricultural and Animal Husbandry Science, Lhasa, China
The location on the Yunnan border with Myanmar and its unique cultural landscape has shaped Lincang humped cattle over time. In the current study, we investigated the genetic characteristics of 22 Lincang humped cattle using whole-genome resequencing data. We found that Lincang humped cattle derived from both Indian indicine and Chinese indicine cattle depicted higher levels of genomic diversity. Based on genome-wide scans, candidate genomic regions were identified that were potentially involved in local thermal and humid environmental adaptions, including genes associated with the body size (TCF12, SENP2, KIF1C, and PFN1), immunity (LIPH, IRAK3, GZMM, and ELANE), and heat tolerance (MED16, DNAJC8, HSPA4, FILIP1L, HELB, BCL2L1, and TPX2). Missense mutations were detected in candidate genes IRAK3, HSPA4, and HELB. Interestingly, eight missense mutations observed in the HELB gene were specific to the indicine cattle pedigree. These mutations may reveal differences between indicine and taurine cattle adapted to variable climatic conditions. Our research provides new insights into the genetic characteristics of Lincang humped cattle representing Lincang and Pu’er areas as an important channel for the migration of Indian indicine from domestication centers toward southwestern China.
Introduction
Domestic cattle comprise two subspecies, humpless taurine (Bos taurus) and humped indicine or zebu (Bos indicus), both of which are derived from extinct wild aurochs (Bos primigenous) (Decker et al., 2014). Existing research has recognized that worldwide cattle can be divided into five continental groups, European taurine, Eurasian taurine, East Asian taurine, Chinese indicine, and Indian indicine, through whole-genome sequencing analysis (Chen et al., 2018). As for current distribution patterns, modern cattle live in different geographical and climatic zones worldwide. Taurine cattle mainly inhabit temperate environments. In contrast, indicine cattle adapt to continuous high and variable temperate climates (Barendse, 2017).
Yunnan province in China is traversed by the Tropic of Cancer, which is mainly a tropical and subtropical climate zone. Recent studies have identified the complex genetic diversity and admixture patterns of cattle breeds in Yunnan province (Chen et al., 2018; R.; Li et al., 2019; Liu et al., 2020). Lincang humped cattle is the more primitive regional livestock breed mainly distributed in the southern part of Lincang and Pu’er cities in Yunnan province bordering Myanmar (Gan, 2011). The exact history of the formation of Lincang humped cattle has not been verified. However, the reason for the formation of this breed is the adaptive selection and breeding of local farmers according to their social needs. Moreover, the local ethnicity (Wa ethnic) regards cattle as a totem, and the birth of the Wa ethnicity culture is closely associated with cattle. Lincang humped cattle displays superior characteristics of heat tolerance and resistance to disease and are one kind of Yunnan high-humped cattle (Y. Zhang, 2011). Long-term strong natural selection and human-mediated selection might have potentially affected the structure of the Lincang humped cattle genome by forming detectable selection signals in functional genes (Andersson and Georges, 2004; Hoffmann, 2010).
The unique adaptive characterization of indigenous African and Asian cattle breeds has become a hot topic based on whole-genome sequencing. (Ben-Jemaa, Mastrangelo, Lee, Lee, and Boussaha, 2020; J.; Kim et al., 2017; Xia et al., 2021). In the current study, the whole genome of 22 Lincang humped cattle was resequenced and compared with the sequence data of 61 cattle from five continental groups. Our analysis reports the genome characterization of Lincang humped cattle using whole-genome resequencing, providing many insights into their candidate signatures of positive selection.
Methods
Samples and Resequencing
A total of 22 domestic Lincang humped cattle (NCBI: PRJNA781760) from Cangyuan Wa Ethnicity Autonomous County, Lincang City, Yunnan Province, China, were sequenced. The purity of that breed was ensured throughout the sampling. However, one cattle might fall under hybrid cattle. Genomic DNA was extracted from the ear tissue samples. Twenty-two paired-end DNA libraries were constructed for the 22 pieces (500 bp insert size) and subjected to Illumina NovaSeq sequencing at the Novogene Bioinformatics Institute, Beijing, China. The genome sequence data of 61 cattle from five continental groups including European cattle breeds [Angus and Simmental (n = 17)], Chinese native breeds (Leiqiong, Guangfeng, Jian, Jingjiang, Wannan, and Wenshan n = 24), Indian cattle breeds (Sahiwal, Hariana, Tharparkar, Nelore, Gir, and Brahman, n = 10), and Korean native breed (Hanwoo, n = 10) were used for the combined analysis (Supplementary Table S1).
Reads Mapping and SNP Calling
The clean reads were mapped to the latest Bos taurus reference genome (ARS-UCD1.2_Btau5.0.1Y.fa) using BWA-MEM (0.7.13-r1126) (H. Li & Durbin, 2009). The average mapping rate of the reads was 99.72%, and the sequencing coverage was approximately 9.75 × (ranging from 8.94 to 11.78) per individual. Duplicate reads were removed using Picard Tools (http://broadinstitute.github.io/picard). The genome analysis toolkit (GATK, version 3.8) (Using the HaplotypeCaller, GenotypeGVCFs, and Select Variants module) was used to detect SNPs. Previous studies have referred to the SNP calling parameters (Chen et al., 2018). Moreover, ANNOVAR (Using the table_annovar.pl module) (K. Wang, Li, & Hakonarson, 2010) was used to annotate the functions of the SNPs.
Population Genetic Analysis
VCFtools (Danecek et al., 2011) was used to estimate the nucleotide diversity of each breed in window sizes of 50 kb with 20 kb increment. Furthermore, the linkage disequilibrium (LD) decay between pairwise SNPs was calculated by PopLDdecay software (C. Zhang, Dong, Xu, He, & Yang, 2019). PLINK was used to detect the runs of homozygosity (ROH) in each cattle population. The number and length of ROH for each population were estimated and classified into three categories: 0.5–1 Mb, 1–2 Mb, and 2–4 Mb (Sun et al., 2021). PLINK (version 1.9) (Purcell et al., 2007) was again used to remove the linkage sites in genomic data (--indep-pair-wise 50 5 0.2) to perform principal component analysis (PCA) and ADMIXTURE analysis. To accurately identify the components of Lincang humped cattle, ADMIXTURE software (Alexander & Lange, 2011) was used to analyze the population structure with a kinship (K) set from 2 to 5. The aforementioned results were visualized via RStudio software (Loraine et al., 2015). A phylogenetic tree was constructed using the neighbor-joining (NJ) method by PLINK with the matrix of pairwise genetic distances and visualized in MEGA7 (Kumar, Stecher, & Tamura, 2016) and FigTree v1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/).
Selective Sweep Identification
To identify selective sweep regions in Lincang humped cattle, three methods were used (I) The fixation index (FST) values (Weir & Cockerham, 1984; Porto-Neto et al., 2013), (II) Cross-population extended haplotype homozygosity (XP-EHH) (Sabeti et al., 2007), and (III) The composite likelihood ratio (CLR) (Nielsen et al., 2005). FST and XP-EHH were calculated with a 50-kb sliding window and 20-kb steps along the autosomes using VCFtools and in-house scripts between Lincang humped cattle and the reference group. The CLR test was calculated for sites in non-overlapping 50-kb windows using “SweepFinder”. Tajima’s D statistics and nucleotide diversity were calculated for each candidate gene using VCFtools. Furthermore, functional annotation (GO analysis) and KEGG pathway enrichment were performed by DAVID 6.8 (Huang da, Sherman, & Lempicki, 2009), and FDR <0.05 was used as a threshold to detect significantly enriched genes and pathways. The LD heatmap was visualized for candidate genes using VCFtools based on LDBlockShow (Dong et al., 2021).
Results
Resequencing, Identification, and Diversity of Single Nucleotide Polymorphisms
The Lincang humped cattle (n = 22) were selected for genome resequencing (Supplementary Figure S1A). The data were combined with the available dataset (n = 61) from 15 breeds, giving a total of 83 individuals (Supplementary Table S1). Nelore, Gir, and Brahman were also used to represent Indian indicine cattle in this study. In total, 2.58 billion clean reads were generated and aligned to the reference genome ARS-UCD1.2_Btau5.0.1Y.fa with an average alignment rate of 99.72% and an average depth of 11.87 × in Lincang humped cattle.
Furthermore, 34,636,187 SNPs were detected in mapped reads across 22 Lincang humped cattle. Approximately, 0.7% of SNPs with 98,226 nonsynonymous and 154,537 synonymous SNPs were detected in exonic regions. Then, 37.9% of SNPs were found in intronic regions, 59.1% were found in intergenic regions, 1% of SNPs were observed in untranslated regions (UTR), and 1.2% of SNPs were found in upstream and downstream of genes (Figure 1A). The highest number of SNPs was observed in the Chinese indicine, while the number was lower in taurine cattle than in indicine cattle (Supplementary Table S2). Moreover, the highest and second highest numbers of specific SNPs were found in Chinese indicine and Lincang humped cattle, respectively (Supplementary Figure S1B). The difference in the number of SNPs and specific SNPs might indicate the differences in the cattle numbers and different populations.
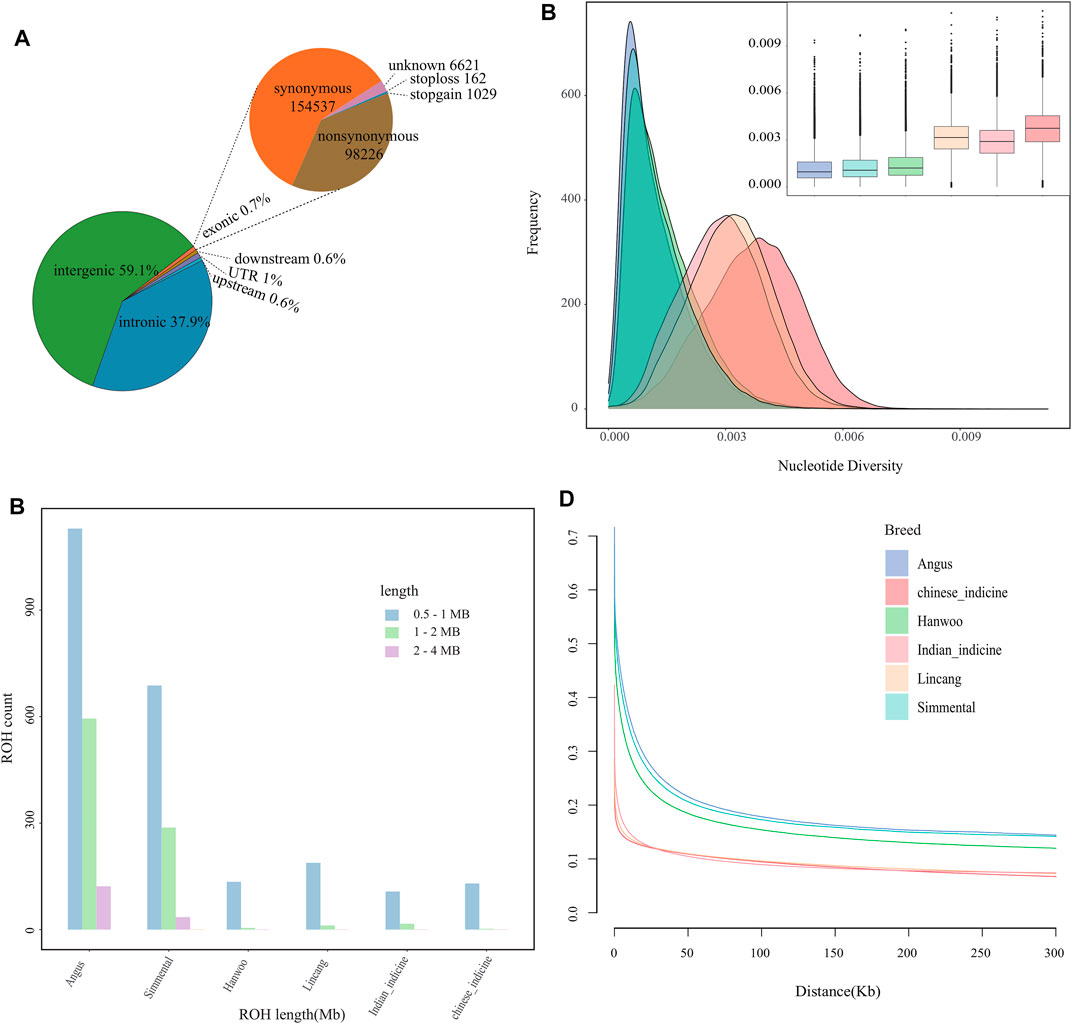
FIGURE 1. Genomic characteristics of cattle population. (A) Functional classification of the detected SNPs in Lincang cattle. (B) The estimation of number and length of ROH for each group. (C) Estimated nucleotide diversity for each group. (D) Linkage disequilibrium (r2) decay on cattle autosomes estimated from each group.
In the absence of pedigree records, ROH may help to infer the level of inbreeding. At the ROH threshold of >2 Mb, Lincang humped cattle showed low levels of genomic inbreeding. Cattle of European origin appeared to be more inbred than other groups (Figure 1B). The nucleotide diversity was the highest in Chinese indicine cattle, followed by Lincang and Indian indicine cattle (Figure 1C). The lowest nucleotide diversity was found in taurine cattle. The genome-wide LD was lower for the indicine cattle than for the taurine cattle, which might indicate faster LD decay in indicine cattle than in taurine cattle (Figure 1D).
Phylogenetic Relationship, Principal Component Analysis, and Population Structure
The phylogenetic relationship among 83 cattle samples was explored based on the autosomal SNPs. The NJ tree separated taurine and indicine in its clade. The taurine clade clustered Angus, Simmental, and Hanwoo, whereas Chinese indicine, Indian indicine, and Lincang humped cattle were clustered into the indicine clade (Figure 2A). An individual of Lincang humped cattle appeared alone between the taurine and the indicine clade, indicating it as a hybrid. Principal component analysis (PCA) demonstrated a clear genetic structure. PC1 explained 7.73% of the total variation and was driven by the difference between taurine and indicine cattle. Within indicine, a separation was found between Chinese indicine and Indian indicine along PC2. The Lincang humped cattle were found at an intermediate position between Chinese indicine and Indian indicine (Figure 2B). The admixture estimated from K = 2 to K = 5 showed gradual separation of Lincang humped cattle. When K = 2, the CV error value was the lowest, which means the most reasonable biological explanation was obtained. Lincang humped cattle belonged to Bos indicus, composed of crosses with Indian–Chinese indicine genotypes (Figure 2C). In particular, hybrid cattle between taurine and indicine cattle appeared in Lincang humped cattle, which might represent the recent introduction of Simmental cattle. To ensure the accuracy of studying Lincang humped cattle, this particular sample was removed in the follow-up analysis.
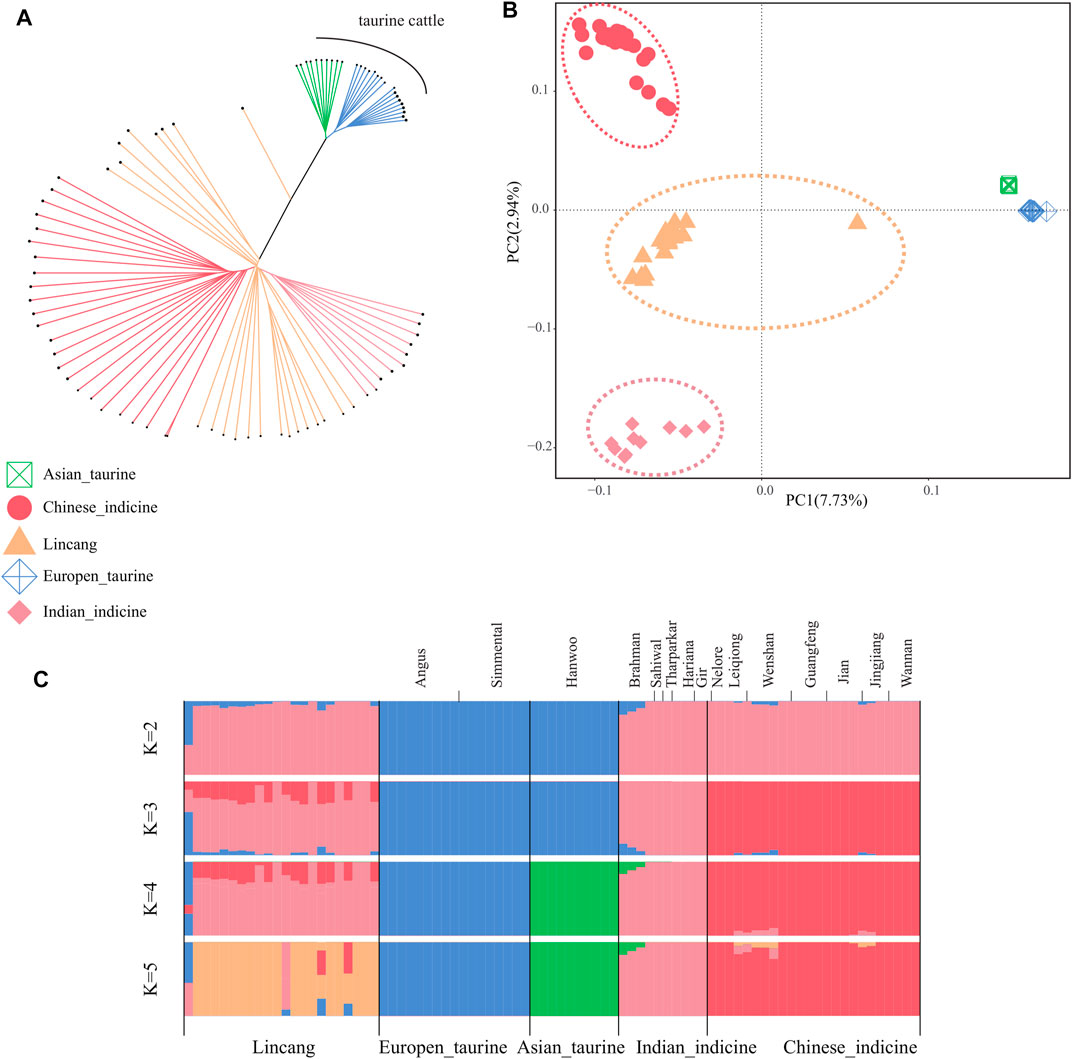
FIGURE 2. Population structure and relationships of Lincang cattle. (A) Neighbor-joining tree of the relationships between the cattle group. (B) Principal component analysis of the cattle group. (C) Model-based clustering of the cattle group using ADMIXTURE (with K = 2 to K = 5). Breeds are colored by geographic regions and labeled with breed names.
Candidate Regions and Genes Under Positive Selection
The composite likelihood method (CLR) was applied to detect the selection signals in Lincang humped cattle (Figure 3A). The top 1% signal window was selected as candidate regions, while 618 genes were annotated with selection characteristics (Supplementary Table S3). KEGG pathway and gene ontology (GO) analyses were used to perform functional enrichment analysis. However, no significant enrichment pathway was found. Surprisingly, the TCF12 gene was annotated in the top 10 signal windows of CLR. The primary biological process of TCF12 is to orchestrate the activity of myogenic factors through myogenic differentiation (Parker, Perry, Fauteux, Berkes, & Rudnicki, 2006). Throughout the entire TCF12 region, Lincang humped cattle showed low nucleotide diversity and constant haplotype diversity patterns (Figure 3B).
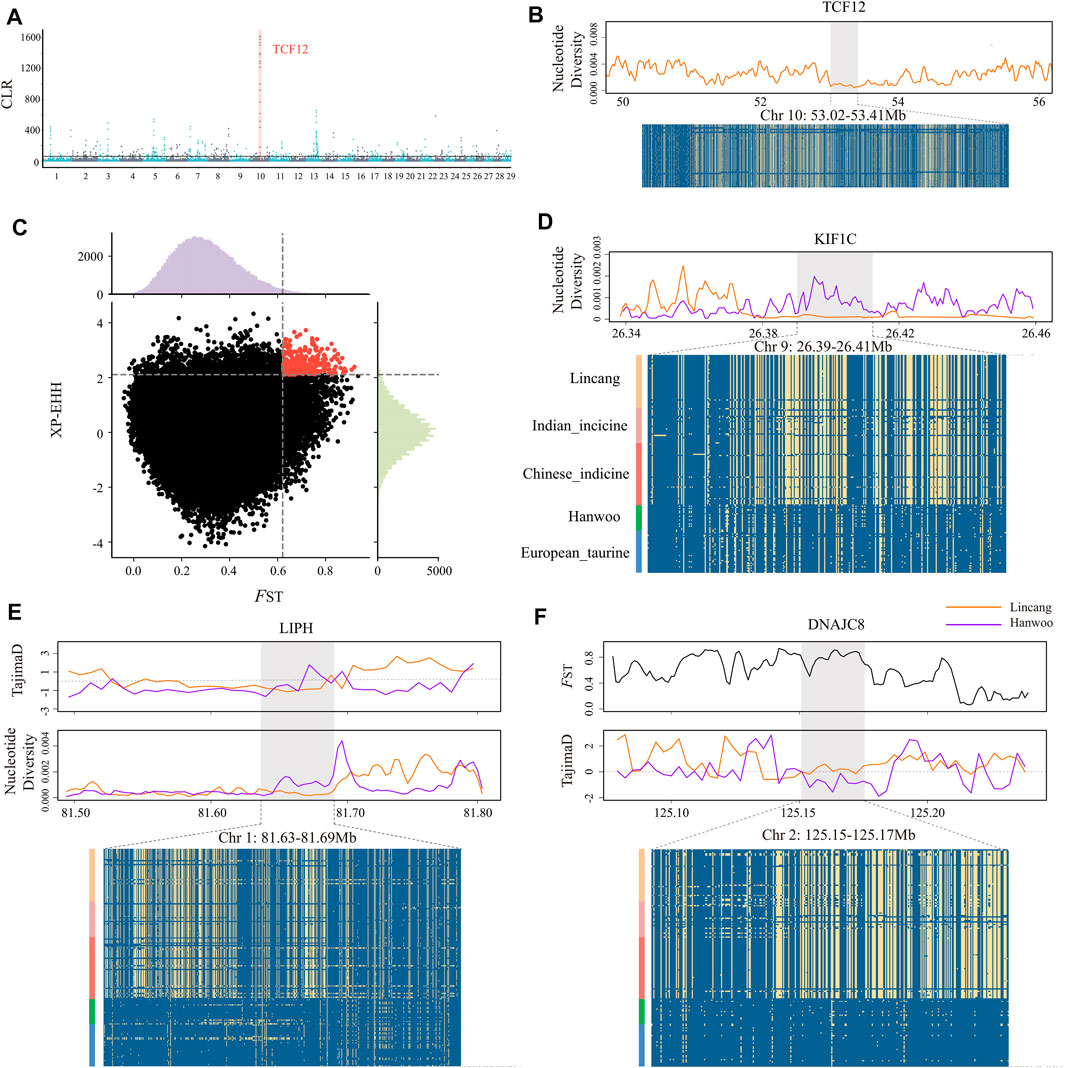
FIGURE 3. Selective signals in Lincang cattle. (A) Manhattan plot of selective sweeps by the CLR method. The horizontal lines indicate the significance threshold (where CLR >61) used for extracting outliers. (B) Nucleotide diversity plots and haplotype pattern heatmaps of the TCF12 gene region. (C) Distribution of the FST (x axis) and XP-EHH (y axis) between Lincang and Hanwoo cattle. The dashed vertical and horizontal lines indicate the significance threshold (where FST > 0.62 and XP-EHH > 2.1) used for extracting outliers. (D) Nucleotide diversity and degree of haplotype sharing across populations at the KIF1C gene region. (E) TajimaD, nucleotide diversity, and degree of haplotype sharing across populations at the LIPH gene region. (F) FST, TajimaD, and degree of haplotype sharing across populations at the DNAJC8 gene region. The major allele at each SNP position is colored in yellow.
The fixation index (FST) test was performed on various groups (I) Lincang humped cattle and Indian indicine; (II) Lincang humped cattle and Chinese indicine; (III) Lincang humped cattle and Hanwoo cattle; and (IV) Lincang humped cattle and European taurine, averaging 0.047, 0.048, 0.32, and 0.34, respectively. Moreover, the FST and XP-EHH methods were performed on Lincang and Hanwoo cattle to detect the significant positive selection signatures, owing to their vast genetic differences between both breeds pertaining to the ecological adaption (Figure 3C). The outlier regions were screened in the top 1% of the empirical distribution of FST and XP-EHH statistics (Supplementary Table S4) and annotated (316 genes). The pathway “negative regulation of protein kinase activity” (FDR = 0.03) was significantly enriched in the KEGG pathway, which might play an important role in the adaptation of Lincang humped cattle to stressful environments.
Moreover, it is noticeable that the regions scanned by FST on BTA1(81.60–81.65 MB), BTA5 (47.58–47.65 MB), BTA7 (43.16–43.27, 43.40–43.45 MB), BTA16 (50.56–50.61 MB), and BTA19 (26.38–26.45 MB) showed a strong positive selection signal, while using XP-EHH, BTA2 (125.14–125.19 MB), BTA7 (44.54–44.63 MB), BTA11 (55.84–55.89 MB), and BTA12 (24.12–24.19, 76.56–76.65 MB) regions showed strong positive selection signals (Table 1). Overall, genes related to the body size (SENP2, KIF1C, and PFN1) (Figure 3D and Supplementary Figure 2A), immunity (LIPH, IRAK3, GZMM, and ELANE) (Figure 3E and Supplementary Figure 2B), and heat resistance (MED16, DNAJC8, and HSPA4) (Figure 3F and Supplementary Figure 2C,D) were identified in the 11 candidate genomic regions. Most target genes exhibited lower nucleotide diversity, higher FST, and differential Tajima’s D values than Hanwoo genomic regions, indicating strong selective sweeps. Furthermore, a missense mutation (rs521365524) was found in the IRAK3 gene, an immune-related gene. This mutation presented a predominant divergence between Lincang humped cattle (allele G frequency = 0.9) and Hanwoo cattle (allele T frequency = 1). Another missense mutation (rs210913195) was detected in the heat-related gene HSPA4, which showed a widespread pattern in Lincang humped cattle (frequency 0.98) and the opposite pattern in Hanwoo cattle (frequency 0.2).
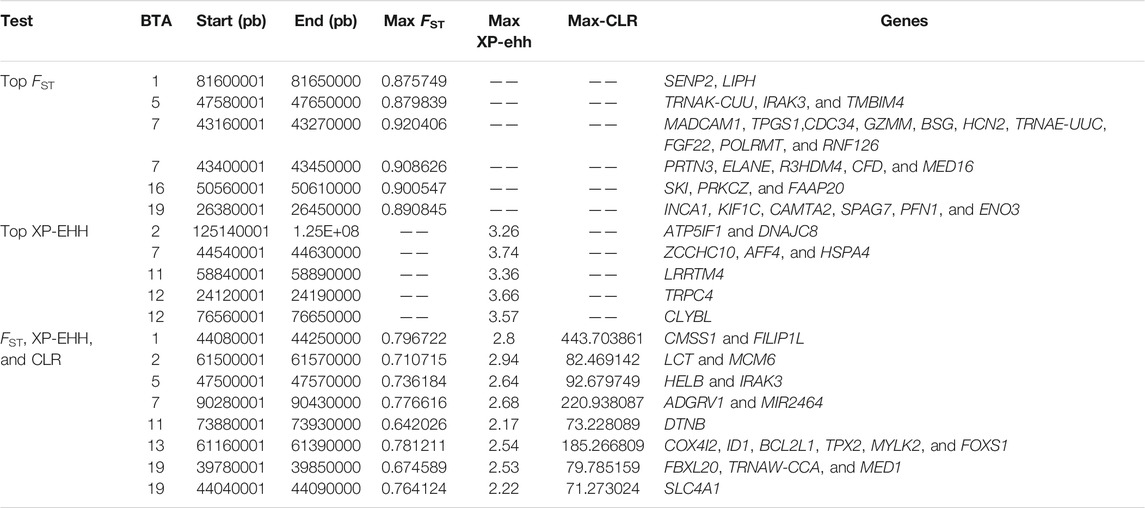
TABLE 1. Genomic regions and associated genes putatively under selection identified using FST, XP-EHH, and CLR statistics.
It is worth noting that eight overlapped genomic regions and 19 genes were detected among the three mentioned selection methods (Table 1), indicating that these were strongly selected in Lincang humped cattle. Among them, FILIP1L, HELB, BCL2L1, and TPX2 genes were all associated with heat stress. Candidate genes showed a stable haplotype diversity pattern in Lincang humped cattle or discrepant Tajima’s D and nucleotide diversity (Figure 4A and Supplementary Figure S2E,F).
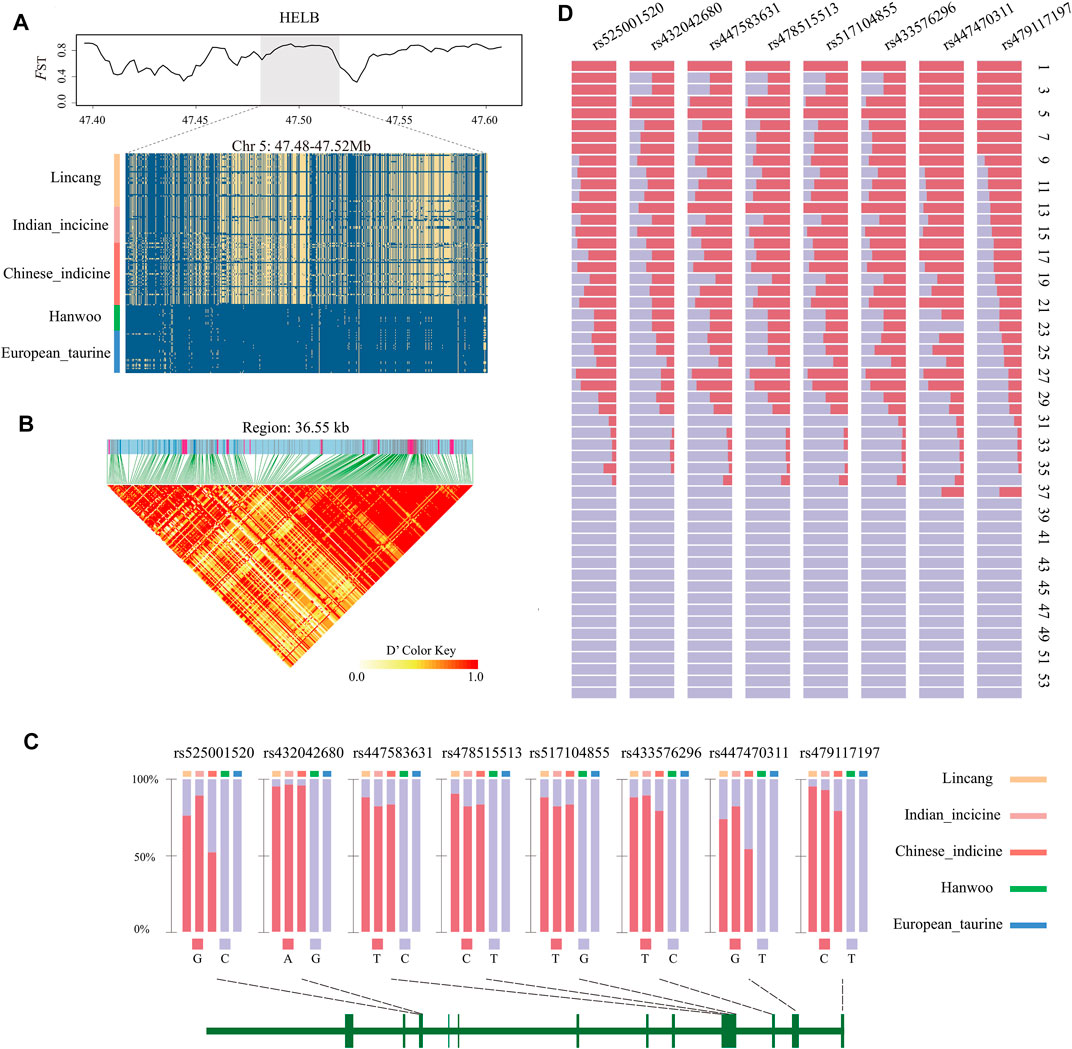
FIGURE 4. Characteristics of the HELB gene. (A) FST, TajimaD, and degree of haplotype sharing across populations. The major allele at each SNP position is colored in yellow. (B) Allele frequency of the eight missense SNPs in each group and the schematic structure of genes. The vertical bars in the structure diagram represent the exon region. (C) LD plot of SNPs. LD values (D′) between two loci are detailed in boxes (D′ = 0–1). D′ = 1 indicates perfect disequilibrium. (D) Allele frequency of the eight missense SNPs across the 54 populations in the BGVD. Population names associated with serial numbers are as follows. 1 Tharparkar; 2 Sahiwal; 3 Hariana; 4 Brahman; 5 Nelore; 6 Gir; 7 Dabieshan; 8 Wandong; 9 Jinjiang; 10 Lingnan; 11 Shorthorn Zebu; 12 Kenana; 13 Srilanka; 14 Bashan; 15 Dianzhong; 16 Wenshan; 17 Guangfeng; 18 Jian; 19 Weining; 20 Ogaden; 21 Leiqiong; 22 Luxi; 23 Nganda; 24 Boran; 25 JiaxianRed; 26 Kazakh; 27 Wannan; 28 Zaobei; 29 Nanyang; 30 Rashoki; 31 Tibetan; 32 Ankole; 33 NDama; 34 Chaidamu; 35 Mongolian; 36 BohaiBlack; 37 Yanbian; 38 Nsongora; 39 Mishima; 40 Kuchinoshima; 41 Hanwoo; 42 Jerse; 43 Simmental; 44 Gelbvieh; 45 Piedmontese; 46 Limousin; 47 Salers; 48 Charolais; 49 MaineAnjou; 50 Devon; 51 Holstein; 52 Hereford; 53 RedAngus; and 54 Angus.
Eight Missense Mutations in HELB to Indicine Cattle
The low diversity of the HELB gene haplotype in Lincang humped cattle (Figure 4A), corresponding to the LD heatmap, showed strong linkage disequilibrium (Figure 4B). Surprisingly, we detected eight missense mutations (rs433576296, rs517104855, rs478515513, rs447583631, rs432042680, rs479117197, rs447470311, and rs525001520) that were located within the HELB gene, indicating significant genomic differences between Lincang humped cattle and Hanwoo cattle. The allele frequencies of these missense mutations were estimated in the five major cattle populations. These missense mutations only occur in indicine cattle in our samples (Figure 4C), while one of the missense mutations (rs447470311) has been confirmed to be specific to indicine cattle (Naval-Sánchez et al., 2020).
To check whether these mutations are also specific for indicine cattle in a wider cattle breed population, the frequencies of these eight missense mutations were searched among 432 individuals from 54 cattle breeds around the world (Figure 4D, Supplementary Table S5, S3), adopting BGVD (Bovine Genome Variation Database and Selective Signatures) (Chen et al., 2020) (Figure 4D, Supplementary Table S5, S3). It was observed that 37 breeds harbored rs479117197 and rs447470311 mutations, where 36 out of 37 were either indicine breeds or mixed with indicine. The remaining one was Yanbian cattle from Northeast Asia, which may have individual deviations. At the same time, in rs433576296, rs517104855, rs478515513, rs447583631, rs432042680, and rs525001520, only breeds with indicine ancestry showed the mutations, revealing a higher frequency of these mutations in indicine ancestry alone. It is worth mentioning that the species from Mongolian, Chaidamu, Kazakh from northwest China, and Tibetan depicted low-frequency mutations, probably due to minute indicine introgression (Chen et al., 2018).
Discussion
Lincang humped cattle is one of the groups of Yunnan high-humped cattle in the southern part of Lincang and Pu’er cities bordering Myanmar. Lincang humped cattle has become a valuable genetic resource in the region because of its good environmental adaptability and particularity to the Wa ethnicity (Gan, 2011). As cattle genetic resources are being exhausted (Y. Zhang, 2011), the genetic characterization of Lincang humped cattle is of great significance to this vital genetic resource.
The genetic diversity of genomes may reflect differences in cattle management or breed history. Compared to taurine cattle with long breeding histories, indicine cattle has a lower degree of selection history and higher genetic diversity, consistent with our results and matching those observed in earlier studies (Mei et al., 2018). The Myanmar border is a habitat of Indian indicine, whereas Lincang and Pu’er areas inhabit Lincang humped cattle admixed with Chinese and Indian indicines. Prior studies mark Yunnan as an admixture zone of Bos taurus and Bos indicus (R. Li et al., 2019), and the entry of Indian indicine in China is also proposed through the Yunnan border (Jia et al., 2007). Our research results indicated that Lincang humped cattle are composed of Indian–Chinese indicine cross genotypes in the genetic structure. This demonstrated that Indian indicine may have entered East Asia through the Lincang and Pu’er areas, which may be an important route for Indian indicine to migrate from the domestication sites (Utsunomiya et al., 2019).
The smaller body size of Lincang humped cattle is associated with its ecological adaptation to hot and humid conditions (Gardner, Peters, Kearney, Joseph, & Heinsohn, 2011). Interestingly, our study detected genes related to skeletal muscle development (TCF12, SENP2, KIF1C, and PFN1). Skeletal muscle development is a complex biological process involving multiple key genes. The protein encoded by TCF12 acts as a complex to positively regulate itself during muscle development (Fu et al., 2020). Related pathways include extracellular signal-regulated kinase signal transduction and CDO in myogenesis (Parker et al., 2006). The SENP2 gene plays an essential role in the regulation of the muscle growth inhibitor expression and myogenesis, which encodes SUMO-specific protease 2 and is an important regulator of fatty acid metabolism in skeletal muscle (Koo et al., 2015). KIF1C plays a role in maintaining membrane circulation during myogenesis and adult muscle (Ginkel & Wordeman, 2000). PFN1 is a critical factor in skeletal development and regulates sternal bone development and endochondral bone formation (Miyajima et al., 2012). It should be noted that Lincang humped cattle weigh less than 300 kg and have an average height of 1 m (Y. Zhang, 2011). They were small body size cattle. The change in the body size can be explained as an adaptive response to the climate, which means positively selected genes associated with the body size may contribute Lincang humped cattle in humid and hot conditions.
The superior adaptability of Lincang humped cattle is partly attributed to their resistance to disease and parasites (Turner, 1980). Our study detected genes related to immune response and parasite resistance (LIPH, IRAK3, GZMM, and ELANE). A previous study identified the association of LIPH with cattle immunity (Zhuang et al., 2021). Similarly, the LIPH gene is also found in Dehong cattle (the same Yunnan high-humped cattle as Lincang humped cattle) (R. Li R et al., 2020). IRAK3 is thought to be a negative regulator of innate immune signaling (Lange, Nelen, Cohen, & Kulathu, 2021). In addition, this gene is also found in selective scans of other indicine cattle (Naval-Sánchez et al., 2020). GZMM can affect the killing efficacy against intracellular pathogens (S. Wang, Xia, Shi, & Fan, 2012). ELANE mutations may trigger neutrophil precursors’ death and lead to neutropenia (Garg et al., 2020). Furthermore, ELANE and GZMM have also been demonstrated in African N’Dama cattle, depicting multiple biological functions in parasitic infections (Ben-Jemaa et al., 2020).
For indicine cattle that have lived in tropical and subtropical climatic conditions, several reports have shown that the indicine breed exhibits stronger heat tolerance (Hansen, 2004; J.; Kim et al., 2017). This was also demonstrated by our screening of candidate genes for DNA damage repair and apoptosis associated with heat resistance (MED16, DNAJC8, HSPA4, FILIP1L, HELB, BCL2L1, and TPX2). MED16 is recruited as the HSP gene promoter in response to heat stress (S. Kim & Gross, 2013). Previous studies have established that knockdown of DNAJC8 decreases antioxidant defenses and increases oxidative damage in honeybees, while DNAJC8 has been shown to function significantly under heat stress in honeybees (G. Li G et al., 2020). Moreover, HSPA4 promotes cellular protection against thermal damage and prevents protein denaturation (Niu et al., 2006). Furthermore, several recent genome-wide analyses had detected selective scans for HSPA4 and highlighted it as a candidate gene for adaptation to hot climates in African indicine cattle (Edea et al., 2018; J.; Kim et al., 2017). FILIP1L interacts with HSF1 to modulate the heat shock response (Hu & Mivechi, 2011). BCL2L1 acts as an anti-apoptotic gene to control apoptosis inducers (Zinkel, Gross, & Yang, 2006), and it has been suggested that it may be a valuable candidate for heat stress studies in dairy cattle (Khan et al., 2020). TPX2 functions in the amplification of the DNA damage response (Neumayer, Belzil, Gruss, & Nguyen, 2014). Considering that Lincang humped cattle are well-adapted to hot climates, these genes may play a vital role in the thermal adaptability of Lincang humped cattle.
HELB is involved in DNA damage response as a DNA end-excision inhibitor (Tkáč et al., 2016). Multiple mutations in HELB have been identified in mouse cell lines with temperature-sensitive DNA replication (Tada et al., 2001). Furthermore, the mutation rs447470311 in HELB revealed in tropical cattle may allow better adaptation to the environment (Naval-Sánchez et al., 2020). An important finding in our study was not only the identification of rs447470311 specific to cattle with indicine cattle pedigree but also the identification of seven missense mutations (rs479117197, rs433576296, rs517104855, rs478515513, rs447583631, rs432042680, and rs525001520) which were only found in cattle with indicine pedigree. Meanwhile, the strong linkage disequilibrium of HELB implied that the significant association of a few SNPs in the gene with the trait may be sufficient to indicate association with the majority of SNPs in the gene and implied a substantial enrichment of the biological function (Qanbari, 2019). Therefore, these results may support the hypothesis that missense mutations in HELB caused alterations in its DNA damage response function, making indicine cattle more adapted to the hot environment. Additional studies may be required in the future to fully understand the effects of HELB on adaptation in indicine cattle.
Conclusion
This study explored the genomic variation in the local cattle population at the China–Myanmar border for the first time via whole-genome resequencing data. The genomic diversity of Lincang humped cattle was explored and identified as indicine cattle. It is proposed that the Indian indicine might have migrated to southwestern China through the Lincang and Pu’er areas. In addition, we identified candidate genes associated with environmental adaptations such as the body size, immunity, and heat tolerance. Finally, we identified missense mutations in HELB that were specific to indicine cattle and were presumed to be associated with adaptation to hot environments. Overall, these results provided a basis for a proper genetic assessment of Lincang humped cattle and further studies on the relationship between HELB and heat tolerance in indicine cattle.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The study was approved by the Institutional Animal Care and Use Committee of Northwest A&F University, following the recommendation of the Regulations for the Administration of Affairs Concerning Experimental Animals of China. Specific consent procedures were not required for this study, following the recommendation of the Regulations for the Administration of Affairs Concerning Experimental Animals of China.
Author Contributions
NC and CL conceived and designed the experiments. LS and KQ performed the experiments. JZ, JL, and QS contributed analysis tools. LS wrote the manuscript. XM and QH revised the manuscript and provided suggestions. KQ, CL, and BH contributed in the funding for the research. All authors contributed to the manuscript and approved the submitted version.
Funding
This work was funded by the China Agriculture Research System of MOF and MARA (CARS-37), the Program of Yunling Scholar, and the Young and Middle-aged Academic Technology Leader Backup Talent Cultivation Program in Yunnan Province, China (No. 2018HB045), and Yunnan Provincial Major S and T Project (No. 2019ZG007), and the Doctoral Startup Project of Chuxiong Normal University (No. BSQD2101).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer declared an affiliation with several of the authors, LS, XM, NC, and CL to the handling editor at the time of review.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.833503/full#supplementary-material
Supplementary Figure S1 | Information for each group. (A) Locations for each cattle population. (B) The specific and shared SNPs for each group. The numbers in the circle components show specific SNPs for each group or overlapping SNPs among groups.
Supplementary Figure S2 | FST, TajimaD, nucleotide diversity, or degree of haplotype sharing across populations. The major allele at each SNP position is colored in yellow. (A) MED16 gene region (B) IRAK3 gene region (C) SENP2 gene region (D) FILIP1L gene region (E) HSPA4 gene region (F) BCL2L1 gene region (G) TPX2 gene region.
Supplementary Figure S3 | Allele frequency of the eight missense SNPs across the 432 individuals from 54 cattle breeds around the world by the BGVD.
References
Alexander, D. H., and Lange, K. (2011). Enhancements to the ADMIXTURE Algorithm for Individual Ancestry Estimation. BMC Bioinformatics 12, 246. doi:10.1186/1471-2105-12-246
Andersson, L., and Georges, M. (2004). Domestic-animal Genomics: Deciphering the Genetics of Complex Traits. Nat. Rev. Genet. 5 (3), 202–212. doi:10.1038/nrg1294
Barendse, W. (2017). Climate Adaptation of Tropical Cattle. Annu. Rev. Anim. Biosci. 5, 133–150. doi:10.1146/annurev-animal-022516-022921
Ben-Jemaa, S., Mastrangelo, S., Lee, S.-H., Lee, J. H., and Boussaha, M. (2020). Genome-wide Scan for Selection Signatures Reveals Novel Insights into the Adaptive Capacity in Local North African Cattle. Sci. Rep. 10 (1), 19466. doi:10.1038/s41598-020-76576-3
Chen, N., Cai, Y., Chen, Q., Li, R., Wang, K., Huang, Y., et al. (2018). Whole-genome Resequencing Reveals World-wide Ancestry and Adaptive Introgression Events of Domesticated Cattle in East Asia. Nat. Commun. 9 (1), 2337. doi:10.1038/s41467-018-04737-0
Chen, N., Fu, W., Zhao, J., Shen, J., Chen, Q., Zheng, Z., et al. (2020). BGVD: An Integrated Database for Bovine Sequencing Variations and Selective Signatures. Genomics, Proteomics & Bioinformatics 18 (2), 186–193. doi:10.1016/j.gpb.2019.03.007
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The Variant Call Format and VCFtools. Bioinformatics 27 (15), 2156–2158. doi:10.1093/bioinformatics/btr330
Decker, J. E., McKay, S. D., Rolf, M. M., Kim, J., Molina Alcalá, A., Sonstegard, T. S., et al. (2014). Worldwide Patterns of Ancestry, Divergence, and Admixture in Domesticated Cattle. Plos Genet. 10 (3), e1004254. doi:10.1371/journal.pgen.1004254
Dong, S.-S., He, W.-M., Ji, J.-J., Zhang, C., Guo, Y., and Yang, T.-L. (2021). LDBlockShow: a Fast and Convenient Tool for Visualizing Linkage Disequilibrium and Haplotype Blocks Based on Variant Call Format Files. Brief Bioinform 22 (4), 227. doi:10.1093/bib/bbaa227
Edea, Z., Dadi, H., Dessie, T., Uzzaman, M. R., Rothschild, M. F., Kim, E.-S., et al. (2018). Genome-wide Scan Reveals Divergent Selection Among Taurine and Zebu Cattle Populations from Different Regions. Anim. Genet. 49 (6), 550–563. doi:10.1111/age.12724
Fu, L., Wang, H., Liao, Y., Zhou, P., Xu, Y., Zhao, Y., et al. (2020). miR-208b Modulating Skeletal Muscle Development and Energy Homoeostasis through Targeting Distinct Targets. RNA Biol. 17 (5), 743–754. doi:10.1080/15476286.2020.1728102
Gan, S. L. (2011). Exploitation Status and Prospect Analysis of Cangyuan High Hump Yellow Cattle Cured-Beef Industry. China Cattle Sci. 37 (2), 66–68.
Gardner, J. L., Peters, A., Kearney, M. R., Joseph, L., and Heinsohn, R. (2011). Declining Body Size: a Third Universal Response to Warming? Trends Ecol. Evol. 26 (6), 285–291. doi:10.1016/j.tree.2011.03.005
Garg, B., Mehta, H. M., Wang, B., Kamel, R., Horwitz, M. S., and Corey, S. J. (2020). Inducible Expression of a Disease-Associated ELANE Mutation Impairs Granulocytic Differentiation, without Eliciting an Unfolded Protein Response. J. Biol. Chem. 295 (21), 7492–7500. doi:10.1074/jbc.RA120.012366
Ginkel, L. M., and Wordeman, L. (2000). Expression and Partial Characterization of Kinesin-Related Proteins in Differentiating and Adult Skeletal Muscle. MBoC 11 (12), 4143–4158. doi:10.1091/mbc.11.12.4143
Hansen, P. J. (2004). Physiological and Cellular Adaptations of Zebu Cattle to thermal Stress. Anim. Reprod. Sci. 82-83, 349–360. doi:10.1016/j.anireprosci.2004.04.011
Hoffmann, I. (2010). Climate Change and the Characterization, Breeding and Conservation of Animal Genetic Resources. Anim. Genet. 41 (Suppl. 1), 32–46. doi:10.1111/j.1365-2052.2010.02043.x
Hu, Y., and Mivechi, N. F. (2011). Promotion of Heat Shock Factor Hsf1 Degradation via Adaptor Protein Filamin A-Interacting Protein 1-like (FILIP-1L). J. Biol. Chem. 286 (36), 31397–31408. doi:10.1074/jbc.M111.255851
Huang, D. W., Sherman, B. T., and Lempicki, R. A. (2009). Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 4 (1), 44–57. doi:10.1038/nprot.2008.211
Jia, S., Chen, H., Zhang, G., Wang, Z., Lei, C., Yao, R., et al. (2007). Genetic Variation of Mitochondrial D-Loop Region and Evolution Analysis in Some Chinese Cattle Breeds. J. Genet. Genomics 34 (6), 510–518. doi:10.1016/s1673-8527(07)60056-3
Khan, A., Dou, J., Wang, Y., Jiang, X., Khan, M. Z., Luo, H., et al. (2020). Evaluation of Heat Stress Effects on Cellular and Transcriptional Adaptation of Bovine Granulosa Cells. J. Anim. Sci Biotechnol 11, 25. doi:10.1186/s40104-019-0408-8
Kim, J., Hanotte, O., Mwai, O. A., Dessie, T., Bashir, S., Diallo, B., et al. (2017). The Genome Landscape of Indigenous African Cattle. Genome Biol. 18 (1), 34. doi:10.1186/s13059-017-1153-y
Kim, S., and Gross, D. S. (2013). Mediator Recruitment to Heat Shock Genes Requires Dual Hsf1 Activation Domains and Mediator Tail Subunits Med15 and Med16. J. Biol. Chem. 288 (17), 12197–12213. doi:10.1074/jbc.M112.449553
Koo, Y. D., Choi, J. W., Kim, M., Chae, S., Ahn, B. Y., Kim, M., et al. (2015). SUMO-specific Protease 2 (SENP2) Is an Important Regulator of Fatty Acid Metabolism in Skeletal Muscle. Diabetes 64 (7), 2420–2431. doi:10.2337/db15-0115
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33 (7), 1870–1874. doi:10.1093/molbev/msw054
Lange, S. M., Nelen, M. I., Cohen, P., and Kulathu, Y. (2021). Dimeric Structure of the Pseudokinase IRAK3 Suggests an Allosteric Mechanism for Negative Regulation. Structure 29 (3), 238–251. doi:10.1016/j.str.2020.11.004
Li, G., Zhao, H., Guo, H., Wang, Y., Cui, X., Li, H., et al. (2020). Analyses of the Function of DnaJ Family Proteins Reveal an Underlying Regulatory Mechanism of Heat Tolerance in Honeybee. Sci. Total Environ. 716, 137036. doi:10.1016/j.scitotenv.2020.137036
Li, H., and Durbin, R. (2009). Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, R., Li, C., Chen, H., Li, R., Chong, Q., Xiao, H., et al. (2020). Genome‐wide Scan of Selection Signatures in Dehong Humped Cattle for Heat Tolerance and Disease Resistance. Anim. Genet. 51 (2), 292–299. doi:10.1111/age.12896
Li, R., Li, C., Chen, H., Liu, X., Xiao, H., and Chen, S. (2019). Genomic Diversity and Admixture Patterns Among Six Chinese Indigenous Cattle Breeds in Yunnan. Asian-australas J. Anim. Sci. 32 (8), 1069–1076. doi:10.5713/ajas.18.0605
Liu, Y., Xu, L., Yang, L., Zhao, G., Li, J., Liu, D., et al. (2020). Discovery of Genomic Characteristics and Selection Signatures in Southern Chinese Local Cattle. Front. Genet. 11, 533052. doi:10.3389/fgene.2020.533052
Loraine, A. E., Blakley, I. C., Jagadeesan, S., Harper, J., Miller, G., and Firon, N. (2015). Analysis and Visualization of RNA-Seq Expression Data Using RStudio, Bioconductor, and Integrated Genome Browser. Methods Mol. Biol. 1284, 481–501. doi:10.1007/978-1-4939-2444-8_24
Mei, C., Wang, H., Liao, Q., Wang, L., Cheng, G., Wang, H., et al. (2018). Genetic Architecture and Selection of Chinese Cattle Revealed by Whole Genome Resequencing. Mol. Biol. Evol. 35 (3), 688–699. doi:10.1093/molbev/msx322
Miyajima, D., Hayata, T., Suzuki, T., Hemmi, H., Nakamoto, T., Notomi, T., et al. (2012). Profilin1 Regulates Sternum Development and Endochondral Bone Formation. J. Biol. Chem. 287 (40), 33545–33553. doi:10.1074/jbc.M111.329938
Naval-Sánchez, M., Porto-Neto, L. R., Cardoso, D. F., Hayes, B. J., Daetwyler, H. D., Kijas, J., et al. (2020). Selection Signatures in Tropical Cattle Are Enriched for Promoter and Coding Regions and Reveal Missense Mutations in the Damage Response Gene HELB. Genet. Sel Evol. 52 (1), 27. doi:10.1186/s12711-020-00546-6
Neumayer, G., Belzil, C., Gruss, O. J., and Nguyen, M. D. (2014). TPX2: of Spindle Assembly, DNA Damage Response, and Cancer. Cell. Mol. Life Sci. 71 (16), 3027–3047. doi:10.1007/s00018-014-1582-7
Nielsen, R., Williamson, S., Kim, Y., Hubisz, M. J., Clark, A. G., and Bustamante, C. (2005). Genomic Scans for Selective Sweeps Using SNP Data. Genome Res. 15 (11), 1566–1575. doi:10.1101/gr.4252305
Niu, P., Liu, L., Gong, Z., Tan, H., Wang, F., Yuan, J., et al. (2006). Overexpressed Heat Shock Protein 70 Protects Cells against DNA Damage Caused by Ultraviolet C in a Dose-dependent Manner. Cell Stress Chaper 11 (2), 162–169. doi:10.1379/csc-175r.1
Parker, M. H., Perry, R. L. S., Fauteux, M. C., Berkes, C. A., and Rudnicki, M. A. (2006). MyoD Synergizes with the E-Protein HEBβ to Induce Myogenic Differentiation. Mol. Cel Biol 26 (15), 5771–5783. doi:10.1128/mcb.02404-05
Porto-Neto, L. R., Sonstegard, T. S., Liu, G. E., Bickhart, D. M., Da Silva, M. V., Machado, M. A., et al. (2013). Genomic Divergence of Zebu and Taurine Cattle Identified through High-Density SNP Genotyping. BMC Genomics 14 (1), 876. doi:10.1186/1471-2164-14-876
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., et al. (2007). PLINK: a Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 81 (3), 559–575. doi:10.1086/519795
Qanbari, S. (2019). On the Extent of Linkage Disequilibrium in the Genome of Farm Animals. Front. Genet. 10, 1304. doi:10.3389/fgene.2019.01304
Sabeti, P. C., Varilly, P., Varilly, P., Fry, B., Lohmueller, J., Hostetter, E., et al. (2007). Genome-wide Detection and Characterization of Positive Selection in Human Populations. Nature 449 (7164), 913–918. doi:10.1038/nature06250
Sun, L., Qu, K., Liu, Y., Ma, X., Chen, N., Zhang, J., et al. (2021). Assessing Genomic Diversity and Selective Pressures in Bashan Cattle by Whole-Genome Sequencing Data. Anim. Biotechnol., 1–12. doi:10.1080/10495398.2021.1998094
Tada, S., Kobayashi, T., Omori, A., Kusa, Y., Okumura, N., Kodaira, H., et al. (2001). Molecular Cloning of a cDNA Encoding Mouse DNA Helicase B, Which Has Homology to Escherichia coli RecD Protein, and Identification of a Mutation in the DNA Helicase B from tsFT848 Temperature-Sensitive DNA Replication Mutant Cells. Nucleic Acids Res. 29 (18), 3835–3840. doi:10.1093/nar/29.18.3835
Tkáč, J., Xu, G., Adhikary, H., Young, J. T. F., Gallo, D., Escribano-Díaz, C., et al. (2016). HELB Is a Feedback Inhibitor of DNA End Resection. Mol. Cel 61 (3), 405–418. doi:10.1016/j.molcel.2015.12.013
Turner, J. W. (1980). Genetic and Biological Aspects of Zebu Adaptability. J. Anim. Sci. 50 (6), 1201–1205. doi:10.2527/jas1980.5061201x
Utsunomiya, Y. T., Milanesi, M., Fortes, M. R. S., Porto‐Neto, L. R., Utsunomiya, A. T. H., Silva, M. V. G. B., et al. (2019). Genomic Clues of the Evolutionary History of Bos indicus Cattle. Anim. Genet. 50 (6), 557–568. doi:10.1111/age.12836
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 38 (16), e164. doi:10.1093/nar/gkq603
Wang, S., Xia, P., Shi, L., and Fan, Z. (2012). FADD Cleavage by NK Cell Granzyme M Enhances its Self-Association to Facilitate Procaspase-8 Recruitment for Auto-Processing Leading to Caspase cascade. Cell Death Differ 19 (4), 605–615. doi:10.1038/cdd.2011.130
Weir, B. S., and Cockerham, C. C. (1984). Estimating F -Statistics for the Analysis of Population Structure. Evolution 38 (6), 1358–1370. doi:10.1111/j.1558-5646.1984.tb05657.x
Xia, X., Zhang, S., Zhang, H., Zhang, Z., Chen, N., Li, Z., et al. (2021). Assessing Genomic Diversity and Signatures of Selection in Jiaxian Red Cattle Using Whole-Genome Sequencing Data. BMC Genomics 22 (1), 43. doi:10.1186/s12864-020-07340-0
Zhang, C., Dong, S.-S., Xu, J.-Y., He, W.-M., and Yang, T.-L. (2019). PopLDdecay: a Fast and Effective Tool for Linkage Disequilibrium Decay Analysis Based on Variant Call Format Files. Bioinformatics 35 (10), 1786–1788. doi:10.1093/bioinformatics/bty875
Zhang, Y. (2011). Animal Genetic Resources in China Bovines. Beijing, China: China Agriculture Press.
Zhuang, H., Chen, X., Wang, Y., Huang, S., Chen, B., Zhang, C., et al. (2021). Identification of LIPH as an Unfavorable Biomarkers Correlated with Immune Suppression or Evasion in Pancreatic Cancer Based on RNA-Seq. Cancer Immunol. Immunother. 71, 601–612. doi:10.1007/s00262-021-03019-x
Keywords: whole-genome resequencing, Lincang humped cattle, genetic characteristics, selection signatures, HELB
Citation: Sun L, Qu K, Ma X, Hanif Q, Zhang J, Liu J, Chen N, Suolang Q, Lei C and Huang B (2022) Whole-Genome Analyses Reveal Genomic Characteristics and Selection Signatures of Lincang Humped Cattle at the China–Myanmar Border. Front. Genet. 13:833503. doi: 10.3389/fgene.2022.833503
Received: 11 December 2021; Accepted: 01 March 2022;
Published: 22 March 2022.
Edited by:
El Hamidi Hay, Agricultural Research Service (USDA), United StatesReviewed by:
Ran Li, Northwest A&F University, ChinaYue Yaojing, International Livestock Research Institute, Ethiopia
Xia Zhou, Soochow University, China
Copyright © 2022 Sun, Qu, Ma, Hanif, Zhang, Liu, Chen, Suolang, Lei and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chuzhao Lei, bGVpY2h1emhhbzExMThAMTI2LmNvbQ==; Bizhi Huang, aGJ6QHluYnAuY24=