 Lihong Fan
Lihong Fan Longfei Ji2
Longfei Ji2
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 05 April 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.827560
This article is part of the Research Topic Advancing Our Understanding of the Genetic and Functional Basis of Skeletal Dysplasia View all 15 articles
Spondyloepiphyseal dysplasia congenital (SEDC) is a rare chondrodysplasia caused by dominant pathogenic variants in COL2A1. Here, we detected a novel variant c.3392G > T (NM_001844.4) of COL2A1 in a Chinese family with SEDC by targeted next-generation sequencing. To confirm the pathogenicity of the variant, we generated an appropriate minigene construct based on HeLa and HEK293T cell lines. Splicing assay indicated that the mutated minigene led to aberrant splicing of COL2A1 pre-mRNA and produced an alternatively spliced transcript with a skipping of partial exon 48, which generated a predicted in-frame deletion of 15 amino acids (p. Gly1131_Pro1145del) in the COL2A1 protein. Due to the pathogenicity of the variation, we performed prenatal diagnosis on the proband’s wife, which indicated that the fetus carried the same mutation.
Spondyloepiphyseal dysplasia congenital (SEDC) is a rare genetic disorder characterized by a short trunk, cervical spine subluxation, scoliosis, coxa vara, kyphosis, and metaphyseal changes (Xiong et al., 2018). Patients may also show extra-skeletal abnormalities such as myopia, hearing loss, and cleft palate (Deng et al., 2016; Zheng et al., 2020). The various mutations in the COL2A1 gene, which contains 57 exons are the genetic cause of SEDC (Anderson et al., 1990; Nishimura et al., 2005).
Until today, according to HGMD (professional 2020.4), at least 130 different COL2A1 mutations have been reported to be causally associated with SEDC. Most of these reported mutations were analyzed primarily at the genomic level (Dasa et al., 2019; Zheng et al., 2020). Only in a few studies have the effects of mutations been confirmed at both DNA and RNA levels (Bruni et al., 2021). A major concern is that we may detect many genomic DNA (gDNA) substitutions of unknown significance, and their pathogenicity is sometimes in urgent need of accurate assessment, especially when prenatal diagnosis and genetic counseling are required. A considerable number of studies have shown that exonic single-nucleotide variants can affect RNA splicing (Inoue et al., 2020; Wang et al., 2020). The most effective way to identify splicing alterations is to analyze the mRNA extracted from the patients’ relevant tissue. But in fact, this type of sample is not always available. In addition, it is often uneasy to analyze mRNA due to its instability and low expression level in peripheral leukocytes. Now minigene analysis has emerged as an alternative method to preliminarily assess whether a particular variant affects pre-mRNA splicing (Putscher et al., 2021).
In this study, we detected a novel variant in an SEDC family and further analyzed its pathogenicity by generating an appropriate minigene construct, followed by prenatal diagnosis and genetic counseling.
The proband (Ⅲ 1, Figure 1A) was a 36 years old man, with a height of 143 cm. He had an abnormal gait when he was 1 year old and then X-ray films showed hip dislocation (data not shown). Growth was markedly tardy from 13 years old and the pain in the hip appeared since the age of 25 years old, especially after long-distance walking. His eyesight, hearing, and intelligence were normal. X-ray films revealed flattened bilateral femoral heads, shortened femoral neck, flattened and narrowed articular surface of the lower femur and upper tibia (Figure 1B). His parents were healthy as neither of them displayed any signs of skeletal deformities or extra-skeletal deformities. As the proband’s wife was pregnant at the first visit, both the proband’s genetic diagnosis and prenatal diagnosis needed to be provided.
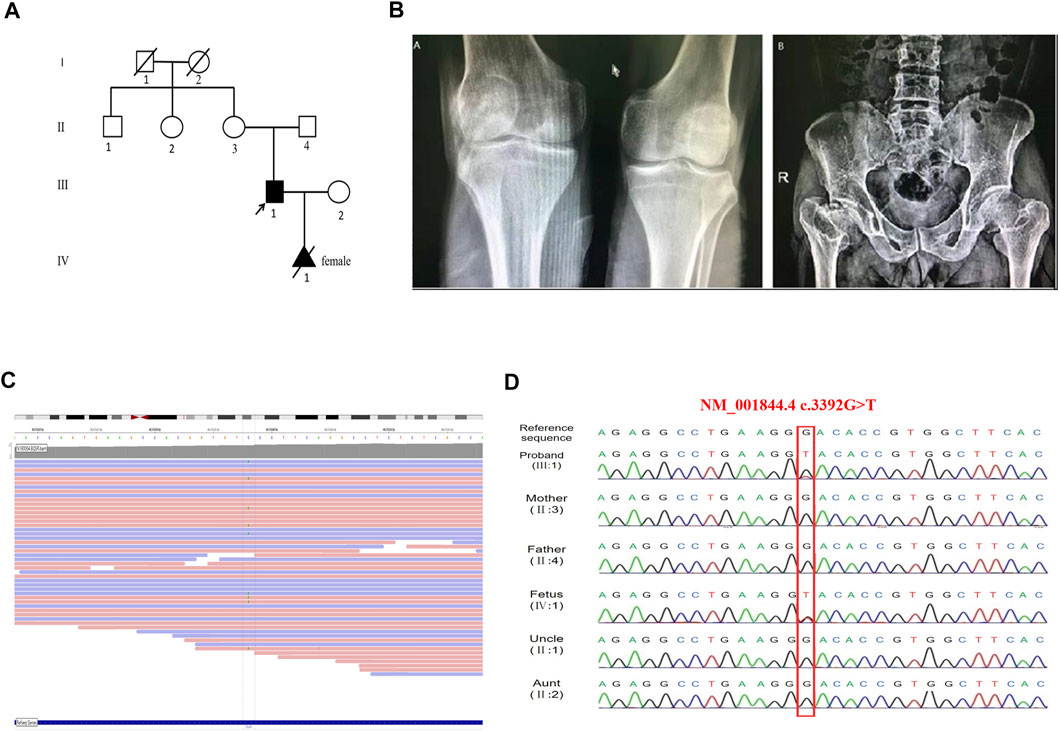
FIGURE 1. Feature of the proband and the COL2A1 mutation in this family. (A) The pedigree of the proband. (B) Radiographs of the proband. The lower femur and the upper tibia are flattened and narrowed (left panel), the bilateral femoral heads are flattened, and the femoral neck is shortened (right panel). (C) Screenshot of the Integrative Genomics Viewer. Alternative alleles at COL2A1 c.3392 position are marked. The total depth of sequencing coverage at this position was 44 reads. The allele frequency was 20% of tatal depth corresponding to the ref/alt was 35/9, suggesting mosaicism. (D) The Targeted next-generation sequencing and Sanger sequencing showed the proband and fetus carring the same mutation of c.3392G > T.
The current investigation was approved by the Ethics Committee of Huzhou Maternity & Child Health Care Hospital. All participants were provided their written informed consents.
A targeted next-generation sequencing panel was used to capture all exon sequences of 2,740 genes known to be associated with skeletal disorders. The peripheral blood sample of the proband was collected. Genomic DNA was then extracted using a Lab-Aid 820 DNA blood Mini Kit (Zeesan, China). Exome capture was performed using SureSelect Human All Exon V6 kit (Agilent Technologies, Santa Clara, CA, United States), followed by sequencing using an Illumina HiSeq2000 system (Illumina, San Diego, CA, United States). Whereafter, multiple database annotations (such as dbSNPs, ESP6500, GnomAD, 1,000 genomes, HGMD, LOVD 3.0, and ClinVar) were performed simultaneously to identify single nucleotide variants (SNVs). Finally, Sanger sequencing was performed to confirm the variant and evaluate the mode of inheritance.
To assess the presumptive effect of this variant on splicing, three different in silico prediction tools: SpliceAI (https://spliceailookup.broadinstitute.org/), Human Splicing Finder-Version 3.1 (HSF, http://www.umd.be/HSF3/HSF.shtml), and CBS were used.
To create hybrid minigene constructs we used two vectors including pcMINI and pcDNA3.1, which we developed previously. Two wild-type fragments, one containing partial intro47 (169bp)-Exon48 (108bp)-partial intro48 (218bp), another containing Exon47 (54bp)-intro47 (182bp)-Exon48 (108bp)-intro48 (244bp)-Exon49 (54bp) were amplified by NEST-PCR. Then we introduced mutations by site-directed mutagenesis using PrimeStar mutagenesis basal kit (Takara Bio Inc.), according to the manufacturer’s instructions. The wild-type and mutated amplified products were purified by a gel extraction kit and then respectively cloned into the pcMINI and the pcDNA3.1 expression vector at the KpnI and BamHI restriction sites to generate four minigene constructs: pcMINI-COL2A1-wt/mut, and pcDNA3.1-COL2A1-wt/mut. All the primers are shown in Supplementary Table S1.
After plasmid amplification in DH5α competent cells and plasmid extraction (SIMGEN), the sequences and correct orientations of all the recombinant vectors were checked by Sanger sequencing (Macrogen, Madrid, Spain). Then the hybrid minigenes were transfected into HEK293T and HeLa cells using Lipofectamine® 2000 (Thermo Fisher Scientific, Waltham, MA, CA). Total RNA was extracted from cells after 48h using the Trizol reagent (TaKaRa) according to the manufacturer’s instructions. Reverse transcription was performed with the Hifair® II 1st Strand cDNA Synthesis SuperMix for qPCR (gDNA digester plus) kit (Yeasen, Shanghai China) according to the manufacturer’s instructions. The cDNA was amplified by PCR, then PCR products were analyzed by electrophoresis on a 1.5% agarose gel and Sanger sequencing (Macrogen, Madrid, Spain).
Ultrasound-guided amniocentesis was performed at 19+2 weeks of gestation to obtain 30 ml of amniotic fluid, 15 ml for cell culture, a necessary step for conventional karyotype analysis, and another 15 ml for DNA extraction, which was performed for Sanger sequencing. The DNA extraction procedure is the same as described above.
A novel variant c.3392G > T (NM_001844.4) in exon 48 of COL2A1 was identified in the proband. A lower-than-expected number of reads supporting the variant suggested possible mosaicism (Figure 1C). This variant was not included in the normal population database (dbSNPs, ESP6500, GnomAD, and 1,000 genomes databases) or, as a pathogenic variant, in HGMD (professional 2020.2), LOVD 3.0, and ClinVar databases. Sanger sequencing of the mutation site observed in the proband confirmed the presence of the wild-type sequence in the unaffected parents (Figure 1D), thus identifying a de novo mutation in the proband. Functional predictions of the missense mutation by REVEL, Polyphen2, and ClinPred are shown in Table 1. In silico tools, HSF, SpliceAI, and CBS predicted that c.3392G > T may affect COL2A1 RNA splicing by creating a new donor site. The splicing score is also shown in Table 1.

TABLE 1. In silico analysis of the COL2A1 variant c.3392G > T.
Considering that the variant does not contain the splice acceptor site of exon 48 of COL2A1, we generated an appropriate minigene construct to explore the variant’s effect (Figure 2A, Figure 3A). By electrophoresis, the RT-PCR amplification products showed each mutant sample had two bands, one identical to the wild type and one slightly lower than the wild type (Figures 2B, 3B). Sanger sequencing of the long product showed that the wild type sequence came from the normal splicing, whereas the short product revealed a skipping of exon 48, which generated an in-frame deletion of 15 amino acids (p. Gly1131_Pro1145del) in the COL2A1 protein (Figures 2C,D, 3C,D).
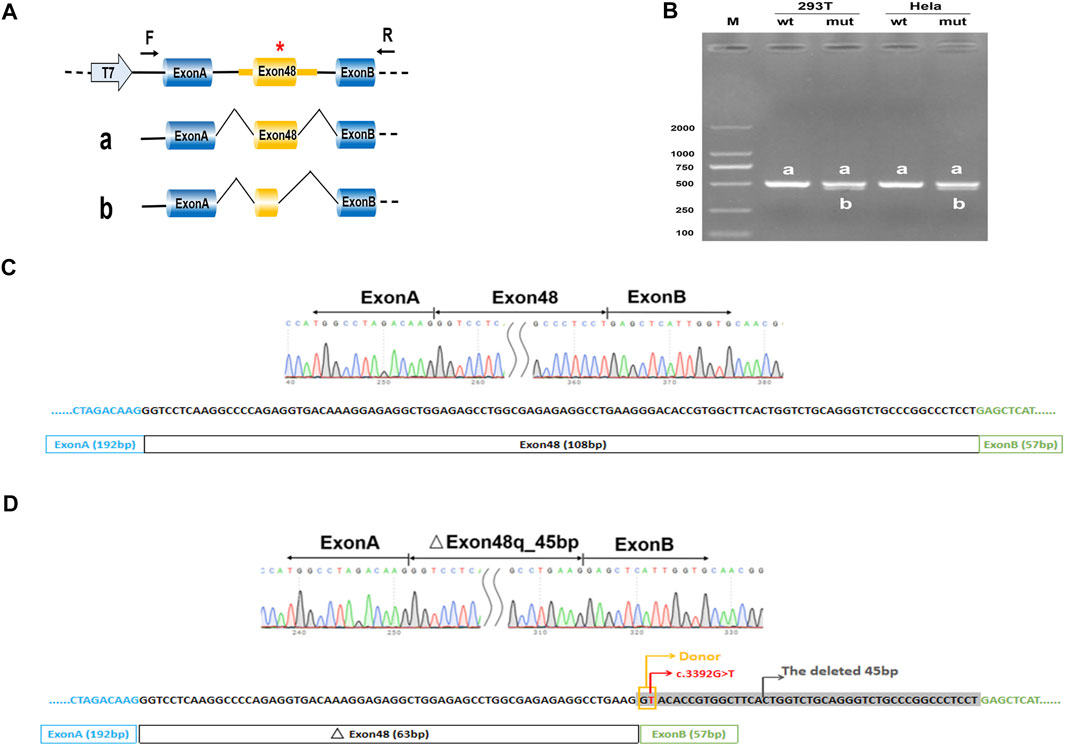
FIGURE 2. Minigene analysis based on pcDNA3.1-COL2A1-wt/mut recombinant vectors. (A), Schematic illustration of cloned vectors. Red * indicates the mutation position. (B) Gel electrophoresis of the RT-PCR products in both 293T and HeLa cell lines. M means the DNA marker. Wild type (wt) exhibited a single band, a (full); mutant type (mut) exhibited double bands, a (full) and b (45 bp deletion). (C) Sanger sequencing result of the normal transcript. (D) Sanger sequencing result of the transcript with cryptic splicing caused by the mutation. The two bases in the yellow rectangle form a new donor, and the sequences in grey shade represent the missing 45 bases.
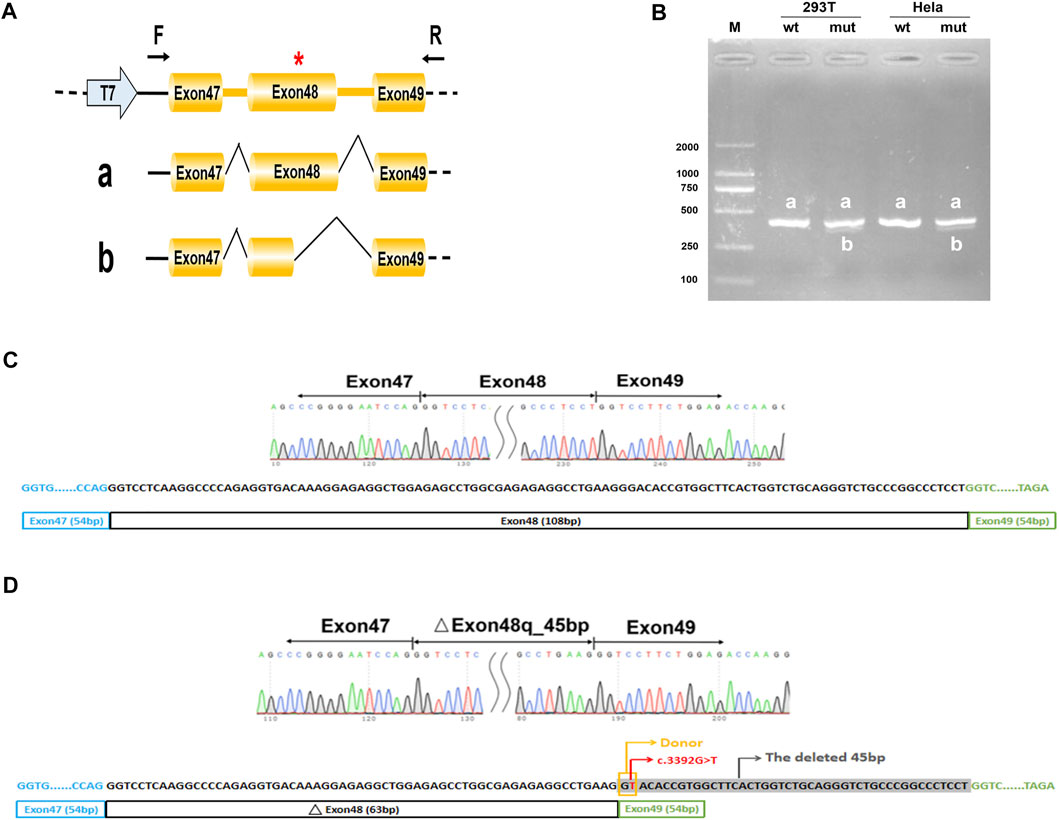
FIGURE 3. Minigene analysis based on pcMINI-COL2A1-wt/mut recombinant vectors. (A), Schematic illustration of cloned vectors. Red * indicates the mutation position. (B) Gel electrophoresis of the RT-PCR products in both 293T and HeLa cell lines. M means the DNA marker. Wild type (wt) exhibited a single band, a (full); mutant type (mut) exhibited double bands, a (full) and b (45 bp deletion). (C) Sanger sequencing result of the normal transcript. (D) Sanger sequencing result of the transcript with cryptic splicing caused by the mutation. The two bases in the yellow rectangle form a new donor, and the sequences in grey shade represent the missing 45 bases.
A comparison of the amino acid sequences indicates that these lost amino acids were highly evolutionary conserved among different species (Figures 4A,B). Moreover, the lost 15 amino acids locate in the triple helix region of COL2A1, predicating it may affect the protein spatial structure (Figure 4C). According to the American College of Medical Genetics and Genomics (ACMG) guidelines for interpretation of sequence variants (Richards et al., 2015), we consider this variant to be pathogenic (PS3+PM2_Supporting + PS4_Supporting + PP4+PS2).
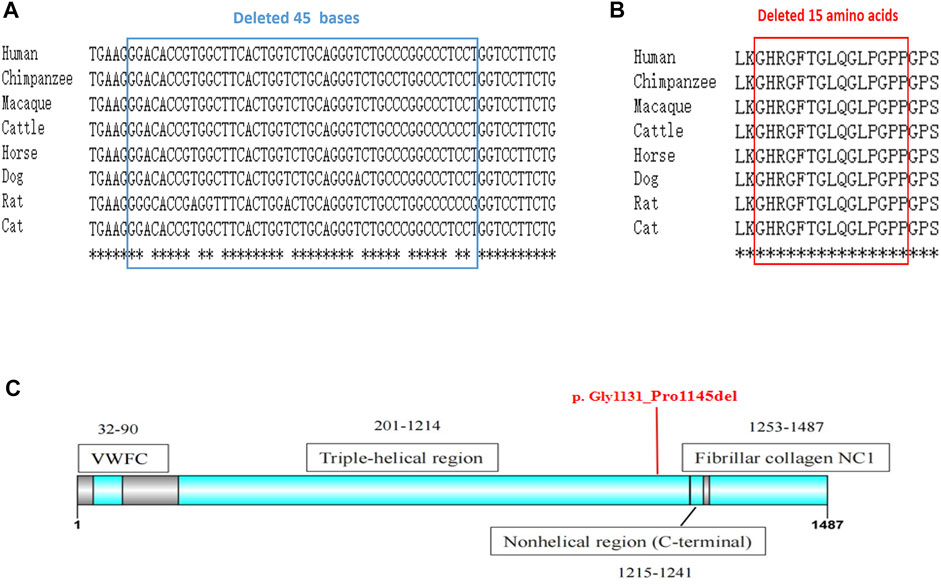
FIGURE 4. The conservation analysis and location of the mutation. (A) Alignment of COL2A1 DNA sequences from multiple species. The affected bases affected by our novel mutation are highly conserved during evolution. (B) Alignment of COL2A1 amino-acid sequences from multiple species., analyzed by Online clustal omega (https://www.ebi.ac.uk/Tools/msa/clustalo/). (C) The deleted 15 amino acids locate in the triple helix region of COL2A1. The secondary structure of the protein is obtained from the uniprot database.
The karyotype analysis revealed no abnormality. Sanger sequencing revealed that the fetus carried c.3392G > T in the heterozygous state (Figure 1D). The ultrasonography showed the fetus with corresponding structural malformations. At 22+3 weeks of gestation, head circumference, abdomen circumference, humerus length (HL), and femur length (FL) were at least lower than −2SD compared with the normal fetus at this gestational age (Figure 5). The couple chose to terminate the pregnancy due to molecular testing results and bone abnormalities.
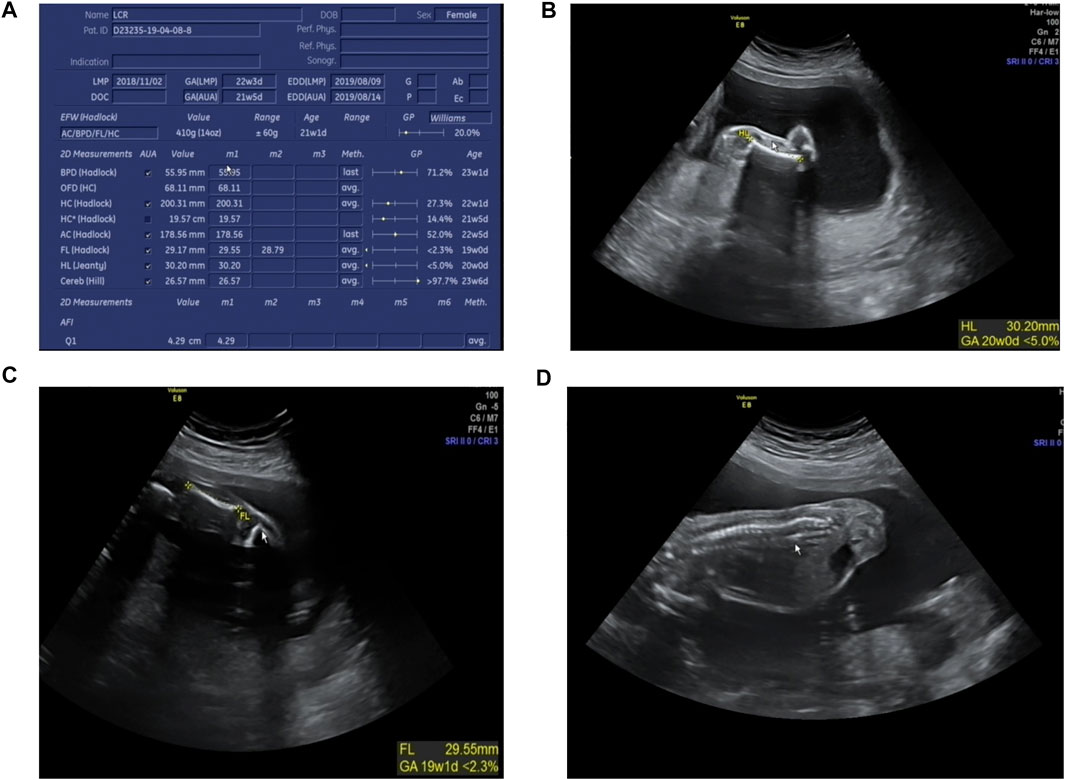
FIGURE 5. Radiographic features of the fetus (at 22+3 weeks’ gestation). (A) The fetus was generally smaller than the fetus at the same gestational age; (B) HL was equivalent to that of fetus at 20 weeks gestation; (C) FL was equivalent to that of fetus at 19+1 weeks’ gestation; (D) Fetus scoliosis could be seen.
SEDC is a heterogeneous group of skeletal dysplasias, associated with COL2A1 mutations. Skeletal abnormalities in patients usually appear at birth and gradually progress (Dasa et al., 2019). Although therapeutic research has made progress in recent years, it is still incurable and may profoundly affect patients’ functional ability and quality of life. Prenatal diagnosis and genetic counseling are necessary for families with affected members.
Over the last decade, as gene sequencing has become widely used in clinical practice, thousands of rare variants have been identified. About 50% of these variants are missense variants, whose pathogenicity is generally difficult to assess accurately (Stenson et al., 2009). According to previous studies, approximately 15%–50% of exon single-nucleotide variations can affect protein function by affecting normal pre-mRNA splicing rather than substituting amino acids (Soukarieh et al., 2016; Baralle and Buratti, 2017). If only the DNA is examined, exonic variants far from the classical GT-AG splicing site might easily be misclassified as missense variants. Here, taking into account the patient’s typical clinical manifestations and that in silico splicing analysis showed the variant c.3392G > T in COL2A1 had a high probability of disrupting gene splicing, we further conducted a minigene splicing assay. The results showed that the c.3392G > T variant generated a new donor and resulted in cryptic splicing, leading to an in-frame deletion of 15 amino acids in the triple-helical domain, which is considered the most important domain of COL2A1 (Zhang et al., 2020). According to the previous analysis of the mutation types and phenotypes, non-substitutions (deletions, duplications, delins, and insertions) are more likely to cause mild phenotypes (Zhang et al., 2020). The explanation given is that the non-substitutions may result in significantly abnormal amino acid sequences that are easily recognized and degraded, while the normal allele without variants provide the essential functions of type II collagen to sustain the basic function of cartilage tissue. On the other hand, the size of the in-frame deletion is considered to have a very significant correlation with the severity of the phenotype (Bruni et al., 2021). Variations with missing amino acids equal to or less than 18, which correspond to the number of residues in a turn of the extended left-handed helix formed by each collagen chain in the collagen triple helix, are associated with less severe phenotypes (Gelse et al., 2003). Deletions of more than 18 amino acid residues may form more extended loops, introducing larger structural defects in heterotrimers, while shorter loops might be more easily accommodated, producing fewer irregular protein assemblies (Bruni et al., 2021).
Mosaicism has been previously reported in at least seven families with COL2A1-related disease (Winterpacht et al., 1993; Spranger et al., 1994; Forzano et al., 2007; Désir et al., 2012; Nagendran et al., 2012; Yamamoto et al., 2020; Muirhead et al., 2021). In these studies, parents in a somatic mosaic status with the COL2A1 mutation were clinically unaffected or showed a milder phenotype while offspring with heterozygous mutations tended to show a severe phenotype, the most severe cases even died neonatally (Forzano et al., 2007; Nagendran et al., 2012). Back to our study, the mutation c.3392G > T was detected in the proband at a low level although the sequencing result is not quantitative, and it has been proved as a loss of function variant, which is a common mechanism of COL2A1-related disorder. The features observed in the proband were slight, but fully consistent with SEDC, one type of COL2A1-related disorder. The fetus carried the heterozygous mutation and showed the key characteristics as early as 22+3 weeks. Consequently, we further clarified the genetic cause of SEDC in the family and speculated that the fetus would have more severe symptoms than the proband.
Before molecular defects in SEDC patients were reported, prenatal diagnosis of SEDC relied on ultrasound findings of shortened long bones, short femurs, a narrow chest, increased nuchal translucency, and so on (Patel and Filly, 1995; Chitty et al., 2006). According to previous studies, abnormal ultrasound findings of an SEDC fetus are not obvious until the second trimester of pregnancy. The earliest gestational age at which FL shortening could be detected is 16 weeks (Kirk and Comstock, 1990). In our study, the structural abnormalities of the fetus were first observed at 22+3 weeks gestation, which had exceeded the best period for prenatal diagnosis. Hence, for the families affected with SEDC, a prenatal molecular diagnosis should be completed as soon as possible. Obviously, confirmation of the proband’s pathogenic variation is a prerequisite for prenatal diagnosis and genetic counseling.
In conclusion, we verified the pathogenicity of the novel COL2A1 variant c.3392G > T by next-generation sequencing and in vitro minigene assay, thus clarifying the cause of SEDC in the subject family and helping to guide for future pregnancy. In addition, our findings emphasize the importance of evaluating the impact of missense variants on pre-mRNA. The minigene assay may be a reliable and easy-to-use tool when a certain mutation is suspected to affect the normal splicing, and to collect the patient’s sample again for RNA transcription is impossible, or the mutant gene is not expressed or expressed too low in the blood.
The datasets presented in this study can be found in online repositories. The name of the repository and accession number can be found below: SRA, NCBI; PRJNA817328.
The studies involving human participants were reviewed and approved by the Ethics Committee of Huzhou Maternity and Child Health Care Hospital. The patients/participants provided their written informed consent to participate in this study.
XS designed the study and reviewed the manuscript. LF analyzed the data and wrote the manuscript. KT and GS collected the samples and performed the experiments. LJ and YX conducted the research and analyzed the results. ZL and SY analyzed the radiological results. All authors have read and approved the manuscript.
The work was supported by the Zhejiang medical and health technology program (2021KY1085) and Huzhou science and technology program (2020GY26).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the family for their participation in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.827560/full#supplementary-material
Anderson, I. J., Goldberg, R. B., Marion, R. W., Upholt, W. B., and Tsipouras, P. (1990). Spondyloepiphyseal Dysplasia Congenita: Genetic Linkage to Type II Collagen (COL2AI). Am. J. Hum. Genet. 46 (5), 896–901.
Baralle, D., and Buratti, E. (2017). RNA Splicing in Human Disease and in the Clinic. Clin. Sci. (Lond) 131 (5), 355–368. doi:10.1042/cs20160211
Bruni, V., Spoleti, C. B., La Barbera, A., Dattilo, V., Colao, E., Votino, C., et al. (2021). A Novel Splicing Variant of COL2A1 in a Fetus with Achondrogenesis Type II: Interpretation of Pathogenicity of In-Frame Deletions. Genes 12 (9), 1395. doi:10.3390/genes12091395
Chitty, L. S., Tan, A. W. C., Nesbit, D. L., Hall, C. M., and Rodeck, C. H. (2006). Sonographic Diagnosis of SEDC and Double Heterozygote of SEDC and Achondroplasia-A Report of Six Pregnancies. Prenat. Diagn. 26 (9), 861–865. doi:10.1002/pd.1525
Dasa, V., Eastwood, J. R. B., Podgorski, M., Park, H., Blackstock, C., Antoshchenko, T., et al. (2019). Exome Sequencing Reveals a Novel COL2A1 Mutation Implicated in Multiple Epiphyseal Dysplasia. Am. J. Med. Genet. 179 (4), 534–541. doi:10.1002/ajmg.a.61049
Deng, H., Huang, X., and Yuan, L. (2016). Molecular Genetics of the COL2A1-Related Disorders. Mutat. Research/Reviews Mutat. Res. 768, 1–13. doi:10.1016/j.mrrev.2016.02.003
Désir, J., Cassart, M., Donner, C., Coucke, P., Abramowicz, M., and Mortier, G. (2012). Spondyloperipheral Dysplasia as the Mosaic Form of Platyspondylic Lethal Skeletal Dyplasia torrance Type in Mother and Fetus with the sameCOL2A1mutation. Am. J. Med. Genet. 158a (8), 1948–1952. doi:10.1002/ajmg.a.35301
Forzano, F., Lituania, M., Viassolo, A., Superti-Furga, V., Wildhardt, G., Zabel, B., et al. (2007). A Familial Case of Achondrogenesis Type II Caused by a dominantCOL2A1 Mutation and "patchy" Expression in the Mosaic Father. Am. J. Med. Genet. 143a (23), 2815–2820. doi:10.1002/ajmg.a.32047
Gelse, K., Pöschl, E., and Aigner, T. (2003). Collagens-structure, Function, and Biosynthesis. Adv. Drug Deliv. Rev. 55 (12), 1531–1546. doi:10.1016/j.addr.2003.08.002
Inoue, T., Nagano, C., Matsuo, M., Yamamura, T., Sakakibara, N., Horinouchi, T., et al. (2020). Functional Analysis of Suspected Splicing Variants in CLCN5 Gene in Dent Disease 1. Clin. Exp. Nephrol. 24 (7), 606–612. doi:10.1007/s10157-020-01876-x
Kirk, J. S., and Comstock, C. H. (1990). Antenatal Sonographic Appearance of Spondyloepiphyseal Dysplasia Congenita. J. Ultrasound Med. 9 (3), 173–175. doi:10.7863/jum.1990.9.3.173
Muirhead, K. J., Clause, A. R., Schlachetzki, Z., Dubbs, H., Perry, D. L., Hagelstrom, R. T., et al. (2021). Genome Sequencing Identifies Three Molecular Diagnoses Including a Mosaic Variant in the COL2A1 Gene in an Individual with Pol III-Related Leukodystrophy and Feingold Syndrome. Cold Spring Harb Mol. Case Stud. 7 (6), a006143. doi:10.1101/mcs.a006143
Nagendran, S., Richards, A. J., McNinch, A., Sandford, R. N., and Snead, M. P. (2012). Somatic Mosaicism and the Phenotypic Expression of COL2A1 Mutations. Am. J. Med. Genet. 158a (5), 1204–1207. doi:10.1002/ajmg.a.35303
Nishimura, G., Haga, N., Kitoh, H., Tanaka, Y., Sonoda, T., Kitamura, M., et al. (2005). The Phenotypic Spectrum ofCOL2A1mutations. Hum. Mutat. 26 (1), 36–43. doi:10.1002/humu.20179
Patel, M. D., and Filly, R. A. (1995). Homozygous Achondroplasia: US Distinction between Homozygous, Heterozygous, and Unaffected Fetuses in the Second Trimester. Radiology 196 (2), 541–545. doi:10.1148/radiology.196.2.7617874
Putscher, E., Hecker, M., Fitzner, B., Lorenz, P., and Zettl, U. K. (2021). Principles and Practical Considerations for the Analysis of Disease-Associated Alternative Splicing Events Using the Gateway Cloning-Based Minigene Vectors pDESTsplice and pSpliceExpress. Ijms 22 (10), 5154. doi:10.3390/ijms22105154
Soukarieh, O., Gaildrat, P., Hamieh, M., Drouet, A., Baert-Desurmont, S., Frébourg, T., et al. (2016). Exonic Splicing Mutations Are More Prevalent Than Currently Estimated and Can Be Predicted by Using In Silico Tools. Plos Genet. 12 (1), e1005756. doi:10.1371/journal.pgen.1005756
Spranger, J., Menger, H., Mundlos, S., Winterpacht, A., and Zabel, B. (1994). Kniest Dysplasia Is Caused by Dominant Collagen II (COL2A1) Mutations: Parental Somatic Mosaicism Manifesting as Stickler Phenotype and Mild Spondyloepiphyseal Dysplasia. Pediatr. Radiol. 24 (6), 431–435. doi:10.1007/bf02011911
Stenson, P. D., Mort, M., Ball, E. V., Howells, K., Phillips, A. D., Thomas, N. S., et al. (2009). The Human Gene Mutation Database: 2008 Update. Genome Med. 1 (1), 13. doi:10.1186/gm13
Wang, S., Wang, Y., Wang, J., Liu, Z., Zhang, R., Shi, X., et al. (2020). Six Exonic Variants in the SLC5A2 Gene Cause Exon Skipping in a Minigene Assay. Front. Genet. 11, 585064. doi:10.3389/fgene.2020.585064
Winterpacht, A., Hilbert, M., Schwarze, U., Mundlos, S., Spranger, J., and Zabel, B. U. (1993). Kniest and Stickler Dysplasia Phenotypes Caused by Collagen Type II Gene (COL2A1) Defect. Nat. Genet. 3 (4), 323–326. doi:10.1038/ng0493-323
Xiong, Q., Liu, Y., Xue, Y., Liu, S., Wang, J., Li, P., et al. (2018). A Novel De Novo Mutation in COL2A1 Leading to Spondyloepiphyseal Dysplasia Congenita in a Chinese Family. Hum. Genome 5, 17059. doi:10.1038/hgv.2017.59
Yamamoto, K., Kubota, T., Takeyari, S., Kitaoka, T., Miyata, K., Nakano, Y., et al. (2020). Parental Somatogonadal COL2A1 Mosaicism Contributes to Intrafamilial Recurrence in a Family with Type 2 Collagenopathy. Am. J. Med. Genet. 182 (3), 454–460. doi:10.1002/ajmg.a.61422
Zhang, B., Zhang, Y., Wu, N., Li, J., Liu, H., and Wang, J. (2020). Integrated Analysis ofCOL2A1variant Data and Classification of Type II Collagenopathies. Clin. Genet. 97 (3), 383–395. doi:10.1111/cge.13680
Keywords: spondyloepiphyseal dysplasia congenital, COL2A1, next-generation sequencing, minigene, in-frame deletion, prenatal diagnosis
Citation: Fan L, Ji L, Xu Y, Shen G, Tang K, Li Z, Ye S and Shen X (2022) A Novel Mutation c.3392G>T of COL2A1 Causes Spondyloepiphyseal Dysplasia Congenital by Affecting Pre-mRNA Splicing. Front. Genet. 13:827560. doi: 10.3389/fgene.2022.827560
Received: 02 December 2021; Accepted: 22 March 2022;
Published: 05 April 2022.
Edited by:
Yi Zhang, Central South University, ChinaReviewed by:
May Thandar Aung-Htut, Murdoch University, AustraliaCopyright © 2022 Fan, Ji, Xu, Shen, Tang, Li, Ye and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xueping Shen, eHAwNTAyMDRAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.