 Fang Shen1
Fang Shen1 Yongjia Yang
Yongjia Yang Yu Zheng
Yu Zheng- 1The Laboratory of Genetics and Metabolism, Pediatrics Research Institute of Hunan Province, Hunan Children’s Hospital, Hengyang Medical School, University of South China, Changsha, China
- 2Emergency Research Institute of Hunan Province, Hunan People’s Hospital, Changsha, China
B3GALT6 is a well-documented disease-related gene. Several B3GALT6-recessive variants have been reported to cause Ehlers–Danlos syndrome (EDS). To the best of our knowledge, no dominant B3GALT6 variant that causes human disease has been reported. In 2012, we reported on a three-generation, autosomal-dominant family with multiple members who suffered from radioulnar joint rotation limitation, scoliosis, thick vermilion of both lips, and others, but the genetic cause was unknown. Here, exome sequencing of the family identified mutant B3GALT6 as the cause of the multiplex affected family. We observed that, in the compound heterozygous pattern (i.e., c.883C>T:p.R295C and c.510_517del:p.L170fs*268), mutant B3GALT6 led to severe consequences, and in the dominant pattern, an elongated B3GALT6 variant co-segregated with moderate phenotypes. The functional experiments were performed in vitro. The R295C variant led to subcellular mislocalization, whereas the L170fs*268 showed normal subcellular localization, but it led to an elongated protein. Given that most of the catalytic galactosyltransferase domain was disrupted for the L170fs*268 (it is unlikely that such a protein has activity), we propose that the L170fs*268 occupies the normal B3GALT6 protein position in the Golgi and exerts a dominant-negative effect.
Introduction
Glycosaminoglycan (GAG), a key component of the extracellular matrix, is essential for the development and maintenance of bone, cartilage, skin, and other tissues. GAG synthesis is initiated by the formation of a tetrasaccharide linker region attached to a serine residue in the proteoglycan core protein (Prydz and Dalen, 2000). Synthesis of the linker region involves four successive steps catalyzed by distinctive enzymes: xylosyltransferases I/II (encoded by XYLT1 and XYLT2) (Götting et al., 2000; Bui et al., 2014; Munns et al., 2015; Umair et al., 2018; LaCroix et al., 2019), galactosyltransferase I (β4GalT7, encoded by B4GALT7) (Guo et al., 2013; Cartault et al., 2015; Ritelli et al., 2017; Sandler-Wilson et al., 2019), galactosyltransferase II (β3GalT6, encoded by B3GALT6) (Malfait et al., 2013; Nakajima et al., 2013), and glucuronosyltransferase I (GlcAT-I, encoded by B3GAT3) (Yauy et al., 2018; Colman et al., 2019). Recessive pathogenic variants of any of these four genes lead to severe skeletal deformities and connective tissue disruptions.
B3GALT6 (NM_080605.3) is a single-exon gene on chromosome 1p36.33, which encodes beta-1,3-galactosyltransferase 6 (β3GalT6). The enzyme localizes predominantly in the Golgi apparatus and catalyzes the addition of a third galactose to the second galactose of the GAG linker region. In 2013, Malfait et al. (2013) and Nakajima et al. (2013) reported B3GALT6 pathogenic variants in human diseases. Since then, dozens of B3GALT6 variants in the recessive status have been identified in approximately 40 unrelated families with Ehlers–Danlos syndrome (EDS) (Sellars et al., 2014; Vorster et al., 2015; Alazami et al., 2016; Trejo et al., 2017; Ben-Mahmoud et al., 2018; Van Damme et al., 2018; Caraffi et al., 2019).
In 2012, we reported on an autosomal-dominant (AD), three-generation family with multiple members presenting with radioulnar limitation, scoliosis, thick vermilion of both lips, and a shortened and thickened femur neck (Zhu et al., 2012). Exome sequencing (ES) of the family was performed. Variant analysis and validation tests identified the following: 1) B3GALT6-recessive (compound heterozygous) variants led to severe phenotypes (EDS); 2) the frameshift-elongated variant (c.510_517del:p.L170fs*268) segregated with moderate deformities in three members of the family in a dominant manner. Functional experiments confirmed that the R295C variant was loss-of-function, but the elongated variant (p.L170fs*268) may exert a dominant-negative effect. This is the first report on B3GALT6-dominant variant leading to disease.
Materials and Methods
Study Subjects
The study was approved by the Academic Committee of Hunan Children’s Hospital (approval number: HCHLL58, Changsha City, Hunan Province, China). All family members provided written informed consent to participate in this study.
Exome Sequencing
ES was carried out on seven individuals (I:1, I:2, II:1, II:2, II:3, III:1, and III:2) (Figure 1A). The detailed ES pipelines have been reported previously (Yang et al., 2019). Briefly, genomic DNA was fragmented into 180–280 bp segments, libraries were prepared and captured using an Agilent SureSelect Human All Exon V6 kit for each individual, and the effective concentration of each sample was subsequently sequenced on an Illumina HiSeq X Ten Sequencing system (Illumina Inc., San Diego, CA, United States). The raw BCL file was then converted into a raw FASTQ file. Raw reads were filtered using FastQC to remove low-quality reads. Clean reads were then mapped to the reference genome GRCh37 using Bwa. After removing duplications, SNV and InDel were called and annotated using GATK. For each sample, 11.9 G bases were obtained. The average yield was 16.6 Gb with an error rate of <0.1%. Furthermore, >90% bases had a Phred quality score of ≥30 (Q30).
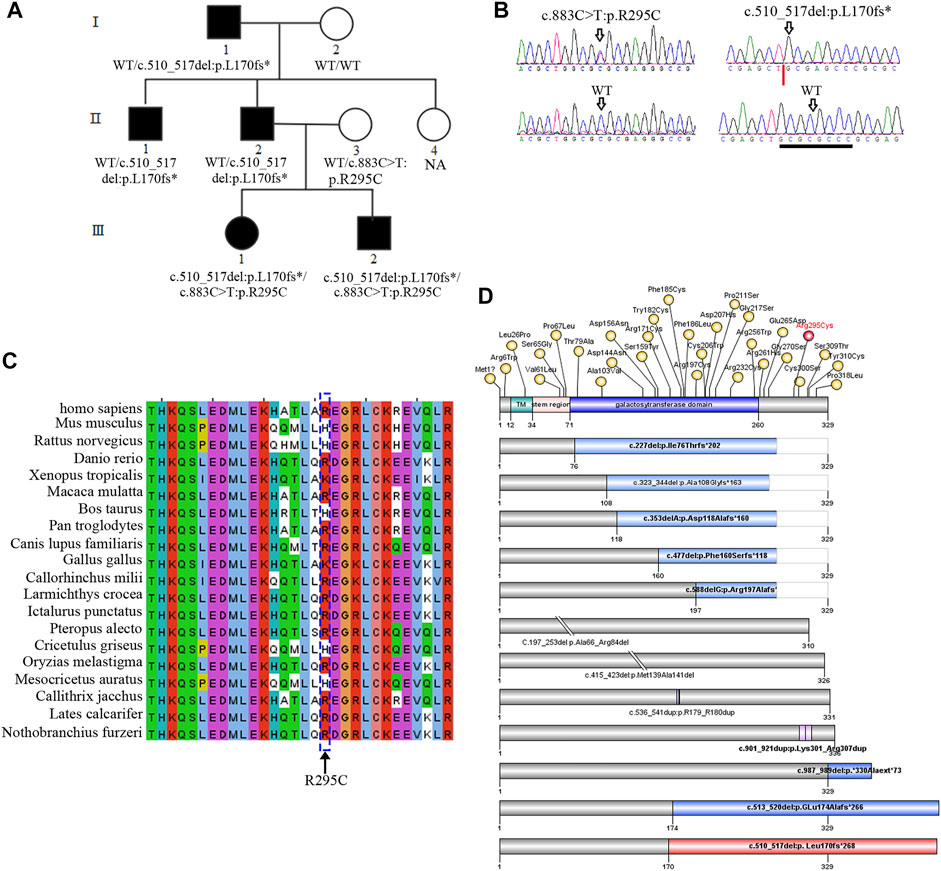
FIGURE 1. B3GALT6 mutated in an autosomal-dominant family. (A) Pedigree with five affected members. NA, genomic DNA was unavailable. (B) Two variants in the family, i.e., c.883C>T:p.R295C and c.510_517del:p. L170fs*268. Note: the trace figure of c.510_517del was identified by Sanger sequencing after the PCR fragment TA-clone experiment. (C) Protein sequence alignment of the residue 295 (arginine) across different species. (D) Schematic view of β3Galt6 protein and the position of the pathogenic variants reported so far. The upper bar represents the domain structure of wild-type (WT) β3Galt6. Missense variants identified so far were depicted above the WT-β3Galt6, and indel variants were depicted below the WT-β3Galt6. TM, transmembrane region. Note: variants depicted as red were found in this study.
Cell Transfection and Western Blot
The empty vector pCMV-14-3 ×flag or B3GALT6 expression constructs (WT, R295C, or L170fs*268) were separately transfected into HeLa cells using Lipofectamine 3000 (Invitrogen, L3000-015) for 48 h. Protein extracts were collected and separated by 10% SDS-PAGE, electrotransferred to a polyvinylidene fluoride (PVDF) membrane (0.45 µm, Merck Millipore Ltd.), blocked with buffer containing 5% non-fat milk, and incubated with mouse monoclonal anti-flag antibody (1:2500) (Sigma, F1804) overnight at 4°C and HRP-conjugated secondary antibodies for 1 h at room temperature and developed with an enhanced chemiluminescence HRP substrate kit (Millipore, WBKLS0500). The membrane was visualized using an iBright FL1500 imaging system (Invitrogen).
Cellular Immunofluorescence
HeLa cells were seeded on coverslips in 24-well culture plates. When the cells reached 80% confluence, the B3GALT6 expression constructs were transfected for 48 h. Cells were blocked with 4% paraformaldehyde for 30 min, permeabilized with 0.2% Triton X-100 for 10 min, and blocked with 5% BSA for 1 h at room temperature. The solution was discarded, and the mouse monoclonal anti-flag antibody (1:500) (Sigma, F1804) and rabbit anti-GOLPH4 antibody (1:400) (Abcam, ab28049) were added onto the coverslips overnight at 4°C in a moist environmental box. The secondary antibody Cy3-conjugated goat anti-mouse IgG (Origene, TA130012) and FITC-conjugated goat anti-rabbit IgG (Origene, TA130021) were incubated for 1 h at room temperature in a dark place, and then DAPI was added onto coverslips for 5 min. Coverslips were mounted on the slide. Cell images were captured with laser scanning confocal microscopy LSM 800.
Results
B3GALT6 Variants in the Family
As previously reported, an autosomal-dominant family was investigated (Figure 1A; Table 1), and we suspected that the family was affected by Giuffre–Tsukahara syndrome at that time (Zhu et al., 2012). ES was successfully performed on seven family members (Figure 1A). After obtaining the variant list, we first focused on the loss-of-function variants (including canonical splicing variants, indels, or other truncating variants) as well as elongation frameshift variants. Because AD inheritance was observed in the index family (Figure 1A), and the disease is extremely rare, we focused on heterozygous, rare, coding variants with AF less than 0.0001 (Exac_eas; gnomAD_eas; 1000G_eas; gomAD_genome all). Then, we only considered the variants (absent in our 700 in-house ES data) that co-segregated with the affected status in the index family; one variant remained, c.510_517del:p. L170fs*268, at B3GALT6. Because B3GALT6 is a known gene for the connective tissue disorder (EDS), we reanalyzed all B3GALT6 rare variants with AF less than 0.001 (including missense variants) and identified another B3GALT6 missense variant, i.e., c.883C>T:p.R295C on three family members (Ⅱ:3, Ⅲ:1, and Ⅲ:2). Sanger sequencing validation confirmed that two family members with severe phenotypes (Ⅲ:1 and Ⅲ:2) had B3GALT6 compound heterozygous variants (c.883C>T:p.R295C/c.510_517del:p. L170fs*268) (Figures 1A,B; Table 1). Unaffected family member Ⅱ:3 was heterozygous for c.883C>T:p.R295C variant (Figures 1A,B; Table 1), and family members Ⅰ:1, Ⅱ:1, and Ⅱ:2 with moderate phenotypes suffered by c.510_517del:p.L170fs*268 heterozygous variant (Figures 1A,B; Table 1).
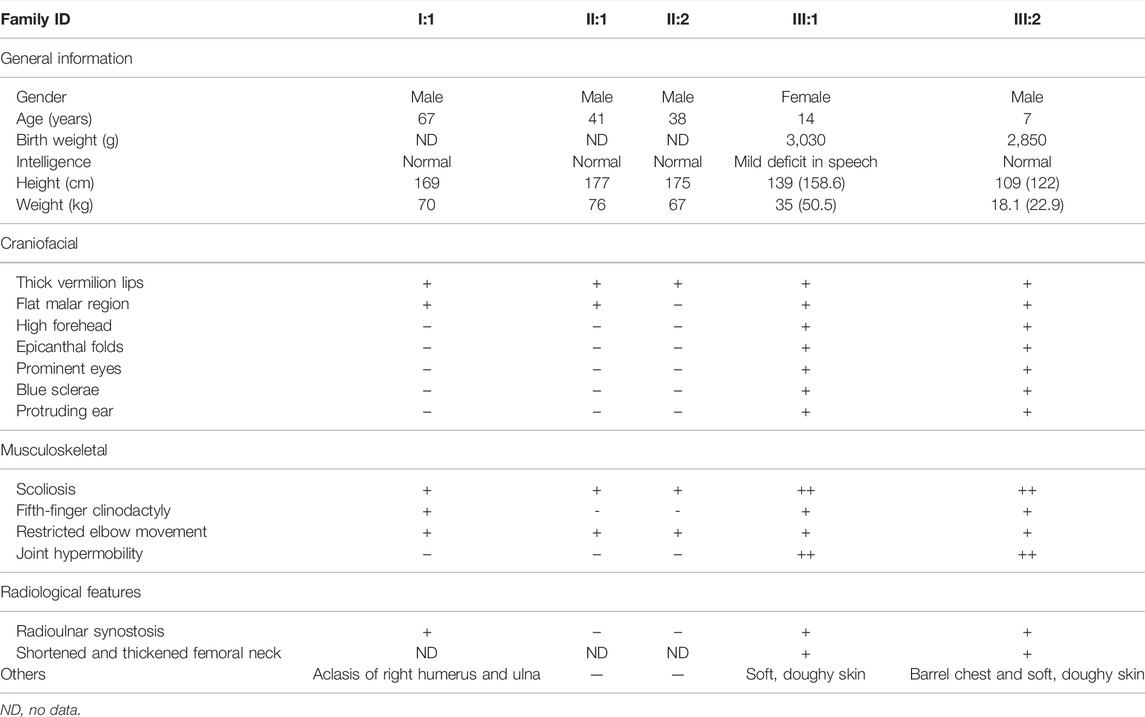
TABLE 1. Manifestations and radiographic findings of the individuals in the family.
Functional Characteristics of β3Galt6 Variants
The c.883C>T:p.R295C is not located in the catalytic galactosyltransferase domain (Figure 1D). Protein sequence alignment indicated that residue 295 (arginine) was not highly conserved across different species (Figure 1C). Meanwhile, in the gnomAD database, there was a very low frequency (AF = 0.0005) in the East Asian population, and 13/23 silico software predicted it to be benign or tolerant (Supplementary Table S1). The c.510_517del:p.L170fs*268 variant was not found in the ExAC, 1000G, and gnomAD databases, and it was not observed in 700 exome databases of our in-house datasheet that are unrelated to skeletal diseases (data not shown).
According to the first in-frame ATG in the reference sequence (NM_080605.4), the c.510_517del variant led to a frameshift mutation and introduced a termination codon delay. Consequently, it was predicted that the mutant-elongated protein contained 438 amino acids in contrast to the wild-type protein, which contained 329 amino acids (Figure 1; Supplementary Material S1).
β3Galt6 mainly functions in the Golgi (Bai et al., 2001). Previous studies have identified B3GALT6 variants exerting pathogenic effects by subcellular mislocalization or unstable structure of the mutant protein or by the unstable/incomplete transcript at the RNA level (Nakajima et al., 2013; Ben-Mahmoud et al., 2018). We then overexpressed β3Galt6 (WT, R295C, or L170fs*268) in the HeLa cells. Western blot analysis showed that: 1) the size of the R295C protein was the same as that of the wild-type protein, but the expression of R295C was increased significantly (Figures 2B,C); 2) in contrast to wild-type β3Galt6, a band with a larger molecular weight was observed for the frameshift mutant protein (L170fs*268), and a significantly reduced expression was observed (Figures 2B,C).
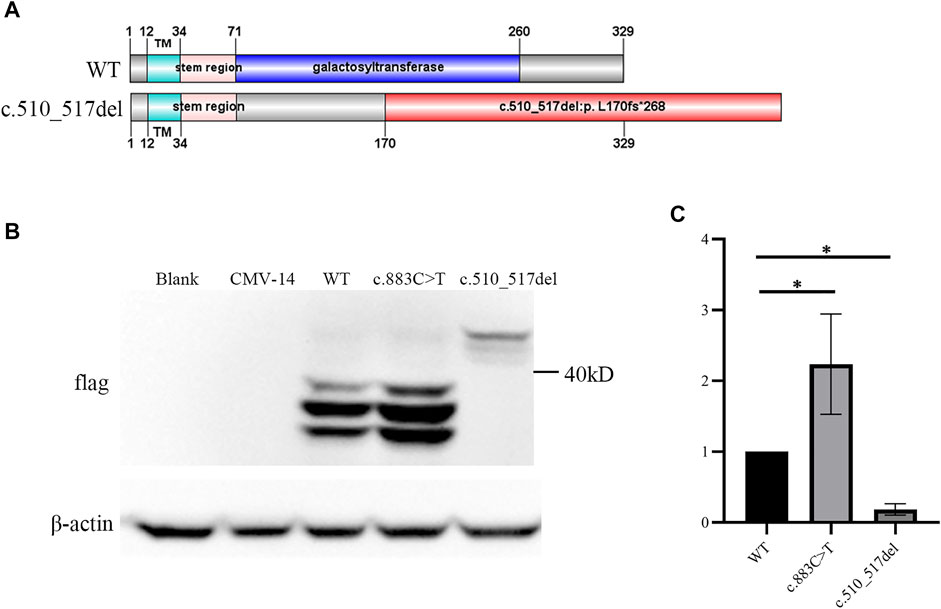
FIGURE 2. c.510_517del:p. L170fs*268 linked an elongated product. (A) Upper panel represents the wild-type (WT) β3Galt6. Lower panel represents the c.510_517del:p. L170fs*268 elongation variant. Note: in wild-type β3Galt6, the catalytic galactosyltransferase domain was depicted in blue; red: frameshift amino acid sequence caused by the deletion. (B) Western blot analysis of lysates from HeLa cells expressing WT and mutant B3GALT6. CMV-14, empty plasmid. (C) Relative density of β3Galt6. n = 4 per group, *p < 0.05.
Second, we checked the subcellular localization of β3Galt6 (WT, R295C, or L170fs*268). In contrast to WT-β3Galt6 expressed in the Golgi (as it co-localized with GOLPH4, which is a marker of the Golgi) (Figure 3), the mutant R295C protein was found in the cytoplasm but not in the Golgi (Figure 3), indicating that the mutant R295C protein was functionally null. In contrast, β3Galt6-L170fs*268 was located in the Golgi compartment, as it overlaps with GOLPH4. According to the ACMG/AMP criteria (Richards et al., 2015), both the variants were classified as pathogenic; the evaluation results of the L170fs*268 variant were PVS1, PM1, PM2, PP1, and PP4; and for the R295C variant, the evaluation results were PS3, PM2, PM3, PP1, and PP4.
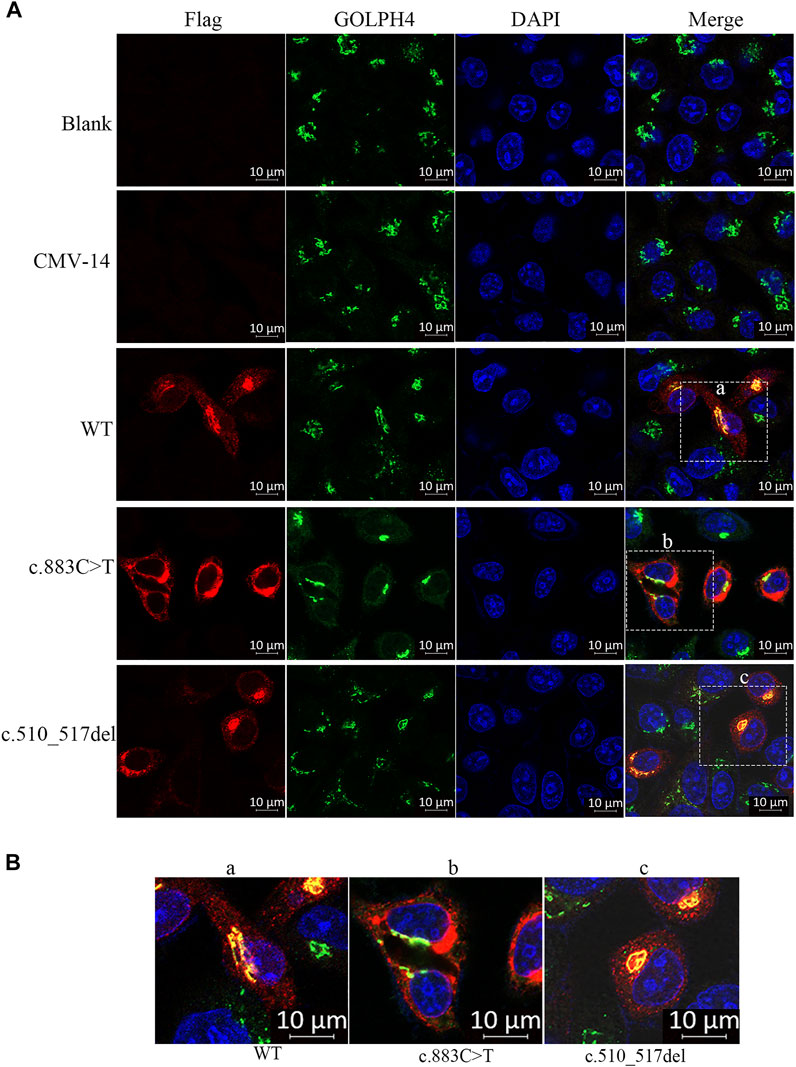
FIGURE 3. Subcellular localization of β3Galt6. (A) HeLa cells were transfected with plasmid carrying the wild-type (WT), the c.883C>T, or the c.510_517del variant. Cells were stained with anti-FLAG (red), anti-GOLPH4 (green), and DAPI (blue). (B) Enlarged images of (a–c) point of (A).
Heterozygous B3GALT6- L170fs*268 Leading to a Moderate Phenotype
The family members Ⅲ:1 and Ⅲ:2 exhibited severe phenotypes (which were consistent with EDS). Other individuals in the family (Ⅰ:1, Ⅱ:1, and Ⅱ:2) did not meet the criteria for EDS and clearly exhibited moderate phenotypes (Table 1 and ref 22). The in vitro study identified that a plasmid with c.510_517del:p. L170fs*26 variant can express an elongated protein (Figure 2), and the mutant elongated protein can be correctly localized in the Golgi. Given that the L170fs*26 frameshift variant resulted in about half of the catalytic galactosyltransferase domain being disrupted (Figure 2A), we propose that the elongated protein (having no catalytic function) occupies the Golgi position disrupting normal GAG production, resulting in a dominant-negative effect.
In brief, the present study identified two novel B3GALT6 pathogenic variants in a multiplex family: 1) in the recessive status, mutant B3GALT6 causing EDS; and 2) in the dominant status, the elongation-mutant β3Galt6 causing moderate phenotypes (although not reaching the level of a typical EDS).
Discussion
As a key enzyme in GAG synthesis, β3Galt6 is encoded by the B3GALT6 gene (Bai et al., 2001). In an autosomal-recessive manner, pathogenic variants of B3GALT6 lead to a multisystem disorder mainly comprising bone deformity and connective tissue disruption, that is, spondylodysplastic EDS (Malfait et al., 2013; Nakajima et al., 2013; Van Damme et al., 2018; Caraffi et al., 2019). Approximately, 60 patients from 40 families have been reported to have mutated B3GALT6 (Malfait et al., 2013; Nakajima et al., 2013; Sellars et al., 2014; Ritelli et al., 2015; Vorster et al., 2015; Alazami et al., 2016; Trejo et al., 2017; Ben-Mahmoud et al., 2018; Van Damme et al., 2018; Caraffi et al., 2019; Zhang et al., 2020; Descartes et al., 2021; Leoni et al., 2021). Among these patients, 41 B3GALT6 pathogenic variants have been reported (Figure 1C), including two start-codon loss (Nakajima et al., 2013; Van Damme et al., 2018), two in-frame deletion variants (Nakajima et al., 2013; Van Damme et al., 2018), two in-frame duplication variants (Alazami et al., 2016; Trejo et al., 2017), five frameshift variants that lead to truncation of the protein (Malfait et al., 2013; Nakajima et al., 2013; Ritelli et al., 2015; Van Damme et al., 2018; Caraffi et al., 2019), two frameshift variants that lead to elongation of the protein (Van Damme et al., 2018; Caraffi et al., 2019), and 28 missense variants (Malfait et al., 2013; Nakajima et al., 2013; Sellars et al., 2014; Ritelli et al., 2015; Vorster et al., 2015; Alazami et al., 2016; Trejo et al., 2017; Ben-Mahmoud et al., 2018; Van Damme et al., 2018; Caraffi et al., 2019; Zhang et al., 2020; Descartes et al., 2021; Leoni et al., 2021). Frameshift variants that lead to protein elongation (combined with other missense variants) have been reported in several severe sporadic patients (Van Damme et al., 2018; Caraffi et al., 2019).
Given that 41 B3GALT6 pathogenic variants (including elongation variants) have been reported on EDS as described previously, none of these variants caused a dominant phenotype. In the present study, we identified two novel B3GALT6 pathogenic variants, i.e., the c.883C>T:p.R295C and the c.510_517del:p.L170fs*268. The B3GALT6-prolonged variant (c.510_517del:p.L170fs*268) was detected in five family members. In two members (who combined with another point R295C variant), severe phenotypes (typical components of EDS) were observed, which is consistent with previous studies, that is, in the recessive status, B3GALT6 variants lead to EDS. The novelty of the present study is that in the other three family patients (Ⅰ:1, Ⅱ:1, and Ⅱ:2), the heterozygous L170fs*268 variant co-segregated with less severe but obvious phenotypes, such as radioulnar joint limitation, “S”-shaped scoliosis, and thick vermilion of the lips.
To test the functional consequences of the elongation variant, we expressed B3GALT6 L170fs*268 protein in HeLa cells. Western blot analysis identified an apparently elongated band (with a significantly reduced amount), which was consistent with the cell immunofluorescence assay in which the B3GALT6 L170fs*268 protein could be detected in the cytoplasm. However, in the immunofluorescence assay, we did not observe a reduction in the L170fs*268 protein. Such a difference in protein amount between the Western blot assay and the immunofluorescence assay may contribute to the L170fs*268 proteins having less stability when they are separated from the in vivo cells.
Similar to WT-β3Galt6, which is expressed in the Golgi, the β3Galt6-L170fs*268 is also located in the Golgi. Therefore, the previous pathomechanism (subcellular mislocalization or haploinsufficiency) cannot explain the heterozygous β3Galt6-L170fs*268 variant-causing phenotypes. Given that the heterozygous β3Galt6-L170fs*268 variant co-segregated with moderate phenotypes in the family produced an elongated protein, and the mutant protein could localize to the Golgi apparatus, we propose that the disease-causing mechanism for this elongation variant is the L170fs*268 that occupies the Golgi apparatus and disrupts the normal CAG production. Nevertheless, to confirm the novel β3Galt6-pathogenic mechanism, further studies, such as galactosyltransferase activity assay, GAG synthesis assay, and CS and HS chain quantifications, are warranted to elucidate the detailed mechanism.
In conclusion, we identified heterozygous B3GALT6-causing phenotypes that implicate a new dominant inheritance pattern of the mutated B3GALT6 or CAG syntheses.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The studies involving human participants were reviewed and approved by the Academic Committee of Hunan Children’s Hospital. Written informed consent to participate in this study was provided by the participants or the participants’ legal guardian/next of kin.
Author Contributions
YY, FS, and YiZ designed the research, analyzed the experimental data, and wrote the manuscript. YuZ, MT, LZ, ZL, and YF performed the sample collection and research. All authors contributed to the manuscript and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (31501017 to YY and 81271946 to YiZ) and Hunan Health Commission Research Fund (B2019019 to YY).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank the subjects for their participation in this research study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.824445/full#supplementary-material
References
Alazami, A. M., Al-Qattan, S. M., Faqeih, E., Alhashem, A., Alshammari, M., Alzahrani, F., et al. (2016). Expanding the Clinical and Genetic Heterogeneity of Hereditary Disorders of Connective Tissue. Hum. Genet. 135 (5), 525–540. doi:10.1007/s00439-016-1660-z
Bai, X., Zhou, D., Brown, J. R., Crawford, B. E., Hennet, T., and Esko, J. D. (2001). Biosynthesis of the Linkage Region of Glycosaminoglycans. J. Biol. Chem. 276 (51), 48189–48195. doi:10.1074/jbc.M107339200
Ben-Mahmoud, A., Ben-Salem, S., Al-Sorkhy, M., John, A., Ali, B. R., and Al-Gazali, L. (2018). A B3GALT6 Variant in Patient Originally Described as Al-Gazali Syndrome and Implicating the Endoplasmic Reticulum Quality Control in the Mechanism of Some β3GalT6-pathy Mutations. Clin. Genet. 93 (6), 1148–1158. doi:10.1111/cge.13236
Bui, C., Huber, C., Tuysuz, B., Alanay, Y., Bole-Feysot, C., Leroy, J. G., et al. (2014). Xylt1 Mutations in Desbuquois Dysplasia Type 2. Am. J. Hum. Genet. 94 (3), 405–414. doi:10.1016/j.ajhg.2014.01.020
Caraffi, S. G., Maini, I., Ivanovski, I., Pollazzon, M., Giangiobbe, S., Valli, M., et al. (2019). Severe Peripheral Joint Laxity Is a Distinctive Clinical Feature of Spondylodysplastic-Ehlers-Danlos Syndrome (EDS)-B4GALT7 and Spondylodysplastic-EDS-B3galt6. Genes 10, 799. doi:10.3390/genes10100799
Cartault, F., Munier, P., Jacquemont, M.-L., Vellayoudom, J., Doray, B., Payet, C., et al. (2015). Expanding the Clinical Spectrum of B4galt7 Deficiency: Homozygous P.R270c Mutation with Founder Effect Causes Larsen of Reunion Island Syndrome. Eur. J. Hum. Genet. 23 (1), 49–53. doi:10.1038/ejhg.2014.60
Colman, M., Van Damme, T., Steichen-Gersdorf, E., Laccone, F., Nampoothiri, S., Syx, D., et al. (2019). The Clinical and Mutational Spectrum of B3gat3 Linkeropathy: Two Case Reports and Literature Review. Orphanet J. Rare Dis. 14 (1), 138. doi:10.1186/s13023-019-1110-9
Descartes, M., Melenevsky, Y. V., Rudy, N., Smith, K., Callaway, K., and Parker, J. S. (2021). Keratoconus in a Patient with B3GALT6 ‐related Disorder. Clin. Genet. 99 (6), 849–850. doi:10.1111/cge.13940
Götting, C., Kuhn, J., Zahn, R., Brinkmann, T., and Kleesiek, K. (2000). Molecular Cloning and Expression of Human UDP-D-Xylose:Proteoglycan Core Protein β-d-Xylosyltransferase and its First Isoform XT-II. J. Mol. Biol. 304 (4), 517–528. doi:10.1006/jmbi.2000.4261
Guo, M. H., Stoler, J., Lui, J., Nilsson, O., Bianchi, D. W., Hirschhorn, J. N., et al. (2013). Redefining the Progeroid Form of Ehlers-Danlos Syndrome: Report of the Fourth Patient withB4GALT7deficiency and Review of the Literature. Am. J. Med. Genet. 161A (10), a–n. doi:10.1002/ajmg.a.36128
LaCroix, A. J., Stabley, D., Sahraoui, R., Adam, M. P., Mehaffey, M., Kernan, K., et al. (2019). Ggc Repeat Expansion and Exon 1 Methylation of Xylt1 Is a Common Pathogenic Variant in Baratela-Scott Syndrome. Am. J. Hum. Genet. 104 (1), 35–44. doi:10.1016/j.ajhg.2018.11.005
Leoni, C., Tedesco, M., Radio, F. C., Chillemi, G., Leone, A., Bruselles, A., et al. (2021). Broadening the Phenotypic Spectrum of Beta3GalT6 ‐associated Phenotypes. Am. J. Med. Genet. 185 (10), 3153–3160. doi:10.1002/ajmg.a.62399
Malfait, F., Kariminejad, A., Van Damme, T., Gauche, C., Syx, D., Merhi-Soussi, F., et al. (2013). Defective Initiation of Glycosaminoglycan Synthesis Due to B3galt6 Mutations Causes a Pleiotropic Ehlers-danlos-syndrome-like Connective Tissue Disorder. Am. J. Hum. Genet. 92 (6), 935–945. doi:10.1016/j.ajhg.2013.04.016
Munns, C. F., Fahiminiya, S., Poudel, N., Munteanu, M. C., Majewski, J., Sillence, D. O., et al. (2015). Homozygosity for Frameshift Mutations in Xylt2 Result in a Spondylo-Ocular Syndrome with Bone Fragility, Cataracts, and Hearing Defects. Am. J. Hum. Genet. 96 (6), 971–978. doi:10.1016/j.ajhg.2015.04.017
Nakajima, M., Mizumoto, S., Miyake, N., Kogawa, R., Iida, A., Ito, H., et al. (2013). Mutations in B3galt6, Which Encodes a Glycosaminoglycan Linker Region Enzyme, Cause a Spectrum of Skeletal and Connective Tissue Disorders. Am. J. Hum. Genet. 92 (6), 927–934. doi:10.1016/j.ajhg.2013.04.003
Prydz, K., and Dalen, K. T. (2000). Synthesis and Sorting of Proteoglycans. J. Cell. Sci. 113 (Pt 2), 193–205. doi:10.1242/jcs.113.2.193
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Ritelli, M., Chiarelli, N., Zoppi, N., Dordoni, C., Quinzani, S., Traversa, M., et al. (2015). Insights in the Etiopathology of Galactosyltransferase Ii (Galt-Ii) Deficiency from Transcriptome-wide Expression Profiling of Skin Fibroblasts of Two Sisters with Compound Heterozygosity for Two Novel B3galt6 Mutations. Mol. Genet. Metabolism Rep. 2, 1–15. doi:10.1016/j.ymgmr.2014.11.005
Ritelli, M., Dordoni, C., Cinquina, V., Venturini, M., Calzavara-Pinton, P., and Colombi, M. (2017). Expanding the Clinical and Mutational Spectrum of B4galt7-Spondylodysplastic Ehlers-Danlos Syndrome. Orphanet J. Rare Dis. 12 (1), 153. doi:10.1186/s13023-017-0704-3
Sandler-Wilson, C., Wambach, J. A., Marshall, B. A., Wegner, D. J., McAlister, W., Cole, F. S., et al. (2019). Phenotype and Response to Growth Hormone Therapy in Siblings with B4galt7 Deficiency. Bone 124, 14–21. doi:10.1016/j.bone.2019.03.029
Sellars, E. A., Bosanko, K. A., Lepard, T., Garnica, A., and Schaefer, G. B. (2014). A Newborn with Complex Skeletal Abnormalities, Joint Contractures, and Bilateral Corneal Clouding with Sclerocornea. Seminars Pediatr. Neurology 21 (2), 84–87. doi:10.1016/j.spen.2014.04.007
Trejo, P., Rauch, F., Glorieux, F. H., Ouellet, J., Benaroch, T., and Campeau, P. M. (2017). Spondyloepimetaphysial Dysplasia with Joint Laxity in Three Siblings with B3GALT6 Mutations. Mol. Syndromol. 8 (6), 303–307. doi:10.1159/000479672
Umair, M., Eckstein, G., Rudolph, G., Strom, T., Graf, E., Hendig, D., et al. (2018). Homozygous Xylt2 Variants as a Cause of Spondyloocular Syndrome. Clin. Genet. 93 (4), 913–918. doi:10.1111/cge.13179
Van Damme, T., Pang, X., Guillemyn, B., Gulberti, S., Syx, D., De Rycke, R., et al. (2018). Biallelic B3galt6 Mutations Cause Spondylodysplastic Ehlers-Danlos Syndrome. Hum. Mol. Genet. 27 (20), 3475–3487. doi:10.1093/hmg/ddy234
Vorster, A. A., Beighton, P., and Ramesar, R. S. (2015). Spondyloepimetaphyseal Dysplasia with Joint Laxity (Beighton Type); Mutation Analysis in Eight Affected South African Families. Clin. Genet. 87 (5), 492–495. doi:10.1111/cge.12413
Yang, Y., Zheng, Y., Li, W., Li, L., Tu, M., Zhao, L., et al. (2019). Smad6 Is Frequently Mutated in Nonsyndromic Radioulnar Synostosis. Genet. Med. 21 (11), 2577–2585. doi:10.1038/s41436-019-0552-8
Yauy, K., Tran Mau-Them, F., Willems, M., Coubes, C., Blanchet, P., Herlin, C., et al. (2018). B3gat3-Related Disorder with Craniosynostosis and Bone Fragility Due to a Unique Mutation. Genet. Med. 20 (2), 269–274. doi:10.1038/gim.2017.109
Zhang, J., Huang, K., and Dong, G. (2020). Clinical and Genetic Analysis of a Child with Spondyloepimetaphyseal Dysplasia Type 1 and Joint Laxity. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 37 (8), 887–890. doi:10.3760/cma.j.issn.1003-9406.2020.08.020
Keywords: B3GALT6, autosomal dominant, heterozygous variant, dominant-negative effect, EDS
Citation: Shen F, Yang Y, Zheng Y, Tu M, Zhao L, Luo Z, Fu Y and Zhu Y (2022) Mutant B3GALT6 in a Multiplex Family: A Dominant Variant Co-Segregated With Moderate Malformations. Front. Genet. 13:824445. doi: 10.3389/fgene.2022.824445
Received: 29 November 2021; Accepted: 25 April 2022;
Published: 06 June 2022.
Edited by:
Emiliano González Vioque, University Clinical Hospital of Santiago, SpainReviewed by:
Anupriya Kaur, Post Graduate Institute of Medical Education and Research (PGIMER), IndiaHongzhou Duan, Peking University, China
Bo Wang, Shanghai Children’s Medical Center, China
Copyright © 2022 Shen, Yang, Zheng, Tu, Zhao, Luo, Fu and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongjia Yang, eW9uZ2ppYTcyN0BhbGl5dW4uY29t; Yimin Zhu, eGlhbmdlcnlpeW1AYWxpeXVuLmNvbQ==