 Fuping Qian
Fuping Qian Xiaoge Jiang
Xiaoge Jiang Renjie Chai
Renjie Chai Dong Liu
Dong Liu- 1School of Life Sciences, Nantong University, Nantong, China
- 2Department of Rehabilitation Medicine, The Second People’s Hospital of Nantong, Affiliated Rehabilitation Hospital of Nantong University, Nantong, China
- 3State Key Laboratory of Bioelectronics, Jiangsu Province High-Tech Key Laboratory for Bio-Medical Research, Department of Otolaryngology Head and Neck Surgery, Zhongda Hospital, Southeast University, Nanjing, China
- 4Co-Innovation Center of Neuroregeneration, Nantong University, Nantong, China
- 5Institute for Stem Cell and Regeneration, Chinese Academy of Science, Beijing, China
- 6Beijing Key Laboratory of Neural Regeneration and Repair, Capital Medical University, Beijing, China
- 7Department of Otolaryngology Head and Neck Surgery, Sichuan Provincial People’s Hospital, University of Electronic Science and Technology of China, Chengdu, China
Solute carriers (SLCs) are important transmembrane transporters with members organized into 65 families. They play crucial roles in transporting many important molecules, such as ions and some metabolites, across the membrane, maintaining cellular homeostasis. SLCs also play important roles in hearing. It has been found that mutations in some SLC members are associated with hearing loss. In this review, we summarize SLC family genes related with hearing dysfunction to reveal the vital roles of these transporters in auditory function. This summary could help us understand the auditory physiology and the mechanisms of hearing loss and further guide future studies of deafness gene identification.
Introduction
SLCs are a large family of transporters and play vital roles in transporting many molecules, such as amino acids, glucose, ions, fatty acids, and neurotransmitters. There are 65 families of SLCs (SLC1–65), with more than 400 members (http://slc.bioparadigms.org/). Most of the SLC proteins have 12 transmembrane domains (Figure 1A) and have been found in many tissues. These SLCs play multiple roles in cellular ion homeostasis, cellular metabolism, and cell survival.
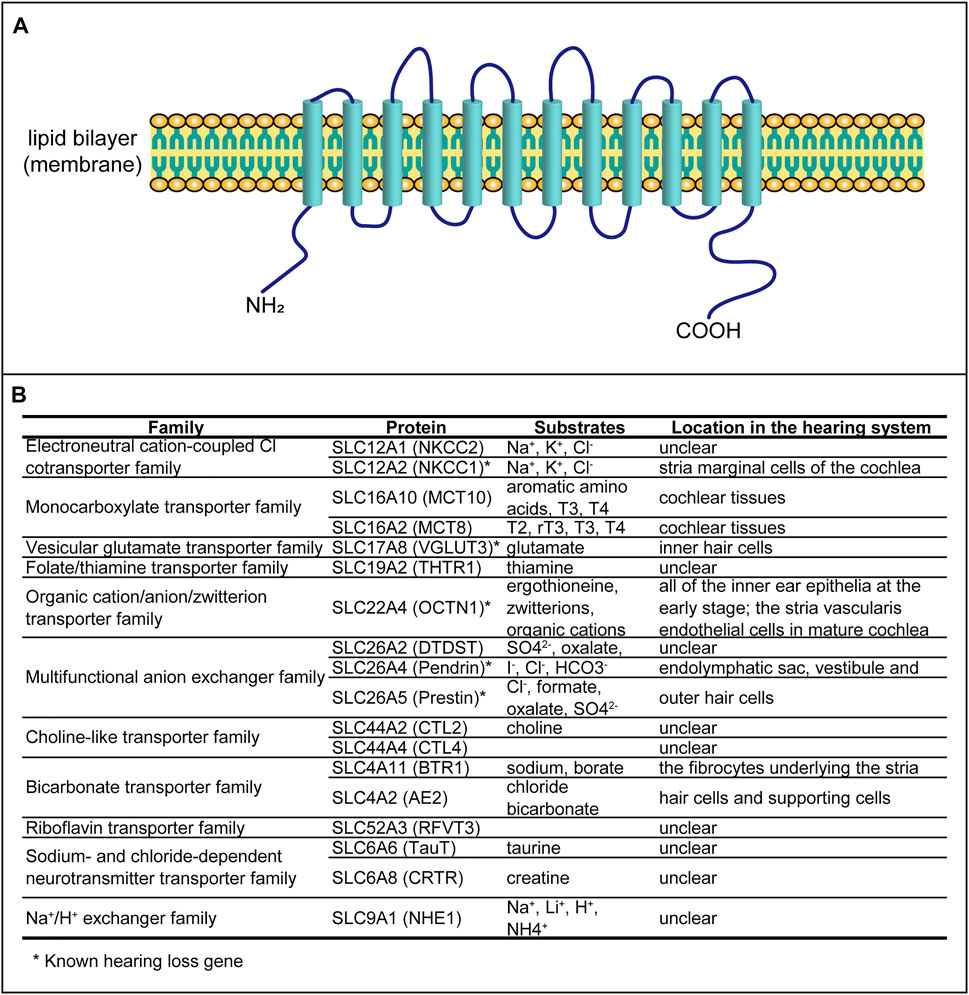
FIGURE 1. SLC proteins involved in auditory function. (A) Schematic structure of the transporter containing 12 transmembrane domains. (B) All SLC proteins associated with hearing dysfunction.
It has been reported that some of the SLCs are associated with hearing loss, including auditory organ development and hearing dysfunction. In this review, we summarize all SLCs related to hearing systematically (Figure 1B) in order to explore the expression patterns and possible functions.
Hearing is one of the most important sensory functions, and hearing loss would cause great inconvenience to the daily life of deaf people. The causes of hearing loss vary, from congenital to acquired impairments, but the defects of genes account for the majority. More than 200 hearing loss genes, including the syndromic and the nonsyndromic hearing loss genes, were identified in the last decades (https://hereditaryhearingloss.org/). Among these genes, five of them belong to the SLC family. Besides, another 13 SLC members were reported to be associated with hearing loss or involved in auditory organ development. These SLC genes, although most of them act as transporters, have different expression patterns in auditory organs and distinct function in hearing.
SLC12A2
The solute carrier family 12 (SLC12) gene, which encodes electroneutral cation-coupled chloride cotransporters, is very important in some physiological processes, such as cell volume regulation, modulation of intraneuronal chloride concentration, transepithelial ion movement, and blood pressure regulation (Arroyo et al., 2013). There are nine members in this family, namely, SLC12A1 to SLC12A9, and some members have been reported to be associated with human diseases. As is known, the mammalian cochlea is the auditory organ that is essential for hearing (Figure 2A). The solute carrier family 12 member 2 (SLC12A2) gene, encoding the Na–K–2Cl cotransporter-1 (NKCC1), is mainly expressed in the stria marginal cell of the cochlea (Crouch et al., 1997; Goto et al., 1997; Mizuta et al., 1997), which is critical for the maintenance of endocochlear potential because of its role in potassium recycling, keeping the endolymph at a high potassium concentration (Figure 2B). In mice, cochlear NKCC1 mRNA and protein decrease with increasing age (Liu et al., 2014), and knockout of the Slc12a2 gene results in complete collapse of Reissner’s membrane, and the Slc12a2−/− mice are deaf and exhibit classic shaker/waltzer behavior (Delpire et al., 1999). Besides, mutation in SLC12A2 also leads to sensorineural hearing loss in humans (Macnamara et al., 2019; McNeill et al., 2020; Mutai et al., 2020). Therefore, the loss of NKCC1 would be an important factor that causes age-related hearing loss (ARHL). It has been proved that aldosterone can enhance NKCC1 protein expression by increasing protein stability (Ding et al., 2014; Bazard et al., 2020), which provides a potential therapeutic for ARHL.
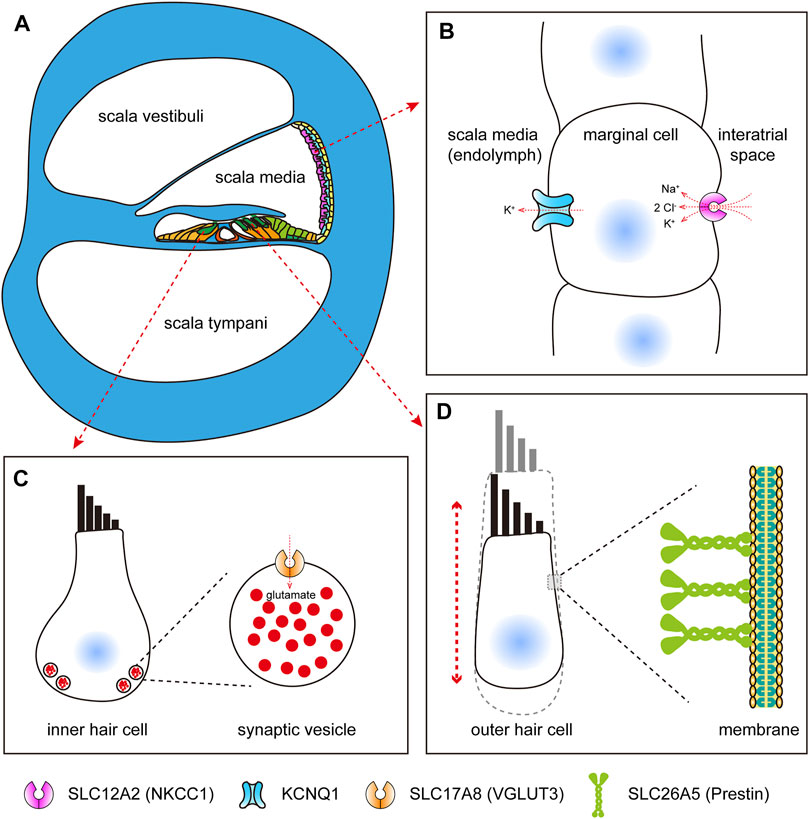
FIGURE 2. SLC proteins expressed in mammalian cochlear cells. (A) Schematic diagram of mammalian cochlea (cross section). (B) Schematic diagram of SLC12A2, which functions in potassium recycling in stria marginal cells, maintaining the endocochlear potential. (C) Schematic diagram of SLC17A8, which functions in transporting glutamate in inner hair cells. (D) Schematic diagram of SLC26A5, which acts as motor proteins, driving somatic electromotility in outer hair cells.
SLC17A8
Hair cells in the mammalian cochlea detect mechanical signals from the tectorial membrane and transmit them to the auditory neurons by releasing the transmitters into the synaptic cleft, which are then captured by the postsynaptic receptors. Glutamate, as the most abundant neurotransmitter in the central nervous system, plays a key role in the auditory function. Vesicular glutamate transporter 3 (VGLUT3), encoded by the slc17a8 gene, is exclusively expressed in hair cells and localized to the basal end of hair cells (Figure 2C), and the mutant slc17a8 hair cells showed reduced ribbon-associated synaptic vesicles and absent postsynaptic action currents in zebrafish (Obholzer et al., 2008). Similarly, in the cochlea of mice, the Slc17a8 gene is expressed in the inner hair cells, but not in the outer hair cells, and mice with Slc17a8 deletion lack auditory nerve responses to acoustic stimuli (Ruel et al., 2008; Seal et al., 2008), and also, the glutamate transmission deficit results in sensorineural deafness because of a mutation of SLC17A8 in humans (Ruel et al., 2008). In addition, more and more mutations within the SLC17A8 gene were identified in different families with hearing loss (Ryu et al., 2016; Ryu et al., 2017). A recent study has showed that tinnitus caused by sodium salicylate treatment was also due to the disruption of VGLUT3 in cochlear inner hair cells (Zhang et al., 2020).
SLC22A4
SLC22A4, also named as OCTN1, a 551-amino acid–long protein, is a pH-dependent organic cation transporter (Tamai et al., 1997) and has a broad expression in many organs or tissues, such as the colon (Peltekova et al., 2004; Meier et al., 2007), mammary glands (Lamhonwah et al., 2011), and airways (Horvath et al., 2007). It functions as an exchanger which carries organic cations or zwitterions across the plasma membrane through sodium-dependent or independent manners, and the substrates of this transporter include tetraethylammonium (TEA) (Yabuuchi et al., 1999), ergothioneine (ET) (Grundemann et al., 2005), and so on. The SLC22A4 gene was identified as a susceptibility gene for rheumatoid arthritis, and it was negatively regulated by RUNX1, a transcription factor which was also significantly associated with rheumatoid arthritis (Tokuhiro et al., 2003). In addition, the expression of SLC22A4 was regulated by nuclear factor-κB (NF-κB) and inflammatory cytokines, such as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) (Maeda et al., 2007). It was also reported that SLC22A4 deficiency increased the susceptibility to Crohn’s disease (Peltekova et al., 2004; Newman et al., 2005). Mice with Slc22a4 gene knockout exhibited greater susceptibility to intestinal inflammation under the ischemia and reperfusion model (Kato et al., 2010). All of the results demonstrated the important roles of SLC22A4 in chronic inflammation.
In recent years, the mutation of SLC22A4 was found to be associated with hereditary hearing loss in humans. In mammalian auditory organs, SLC22A4 is expressed in all of the inner ear epithelia at the early stage, including the hair cells and spiral ganglion neurons; however, the expression is restricted at the apical surface of stria vascularis (SV) endothelial cells in the mature cochlea (Ben Said et al., 2016), and the mutation of the SLC22A4 gene causes autosomal recessive nonsyndromic hearing loss, DFNB60, in humans (Ben Said et al., 2016; Chiereghin et al., 2021). However, the detailed physiological function of SLC22A4 in the hearing process and the underlying mechanisms of hearing loss caused by SLC22A4 variants are still unknown.
SLC26A4
The solute carrier family 26 member 4 (SLC26A4 or PDS) gene, encoding the protein pendrin, is the causal gene of Pendred syndrome, which is a recessively inherited disorder with hearing loss as the obvious feature (Everett et al., 1997; Li et al., 1998). In addition, mutation of the SLC26A4 gene is also associated with the enlargement of the vestibular aqueduct syndrome (EVAS) (Maciaszczyk and Lewiński, 2008). It was also reported that pendrin may regulate blood pressure because patients with SLC26A4 mutation are likely to be resistant to high blood pressure (Kim et al., 2017).
The SLC26A4 gene was found to be expressed in the thyroid at high levels, and it can help the thyroid follicular cells transport iodine (Everett et al., 1997; Maciaszczyk and Lewiński, 2008). In the inner ear, the SLC26A4 gene was detected in the endolymphatic sac, vestibule, and cochlea (Yoshino et al., 2006). However, pendrin may play different roles in the cochlear and vestibular systems because gene therapy of the Slc26a4 gene mutation restored the hearing phenotype but not the vestibular function in mice (Kim et al., 2019).
The protein pendrin contains 12 transmembrane domains, and it functions in sodium-dependent transportation of anions, such as iodides, chlorides, and bicarbonates (Dawson and Markovich, 2005; Maciaszczyk and Lewiński, 2008).
SLC26A5
The SLC16A5 (also named as prestin) protein, encoded by the solute carrier family 26 member 5 (SLC26A5) gene, is the most well-studied solute carrier in hearing-related research. In the mammalian inner ear, it is specifically expressed in the basolateral membrane of outer hair cells (Figure 2D), and deficiency of SLC26A5 results in nonsyndromic hearing loss (Liu et al., 2003). In nonmammalian vertebrates and insects, the homolog of SLC26A5 was also reported to be expressed in the auditory organs (Weber et al., 2003). The protein prestin is more than 700 amino acids in length, and nearly the full length of the protein is required for its proper expression and normal function (Zheng et al., 2005). Unlike most members of solute carrier family 26 (SLC26), which transport different anion substrates across the membrane, mammalian SLC26A5 functions as voltage-dependent motor proteins that drive somatic electromotility in outer hair cells (Zheng et al., 2000), which was thought to be crucial for frequency selectivity and sensitivity of mammalian hearing (Liberman et al., 2002; Liu et al., 2003; Cheatham et al., 2004; Dallos et al., 2008). Indeed, as for prestin, the motor function is an innovation of therians and is concurrent with diminished transporter capabilities (Tan et al., 2011). Nonmammalian prestin acts as an anion transporter; however, in mammals, prestin functions as both a motor protein (Zheng et al., 2000) and a weak transporter (Mistrík et al., 2012). Prestin can form higher order oligomers (Zheng et al., 2006) and interact with the cystic fibrosis transmembrane conductance regulator (CFTR) for activation (Homma et al., 2010). Moreover, it can be functionally regulated by calcium/calmodulin (Keller et al., 2014). Recently, the structure-based mechanism of prestin electromotive signal amplification was illustrated (Ge et al., 2021), providing a better understanding of the molecular basis of hearing and a crucial guidance for the treatment of hearing impairment.
Other SLCs
Except for the SLC members discussed above, which have been identified as hearing loss genes, some other SLCs, waiting to be verified as deafness genes, were reported to function in hearing-related processes.
SLC4A2 and SLC4A11
SLC4 family members are bicarbonate transporters and play vital roles in acid–base homeostasis (Romero et al., 2013). Among the 10 members (SLC4A1–5 and SLC4A7–11), two genes were reported to function in the hearing process.
The SLC4A2 gene, encoding HCO3−/Cl− anion exchangers, was reported to be expressed in mammalian inner ear cells, including hair cells and supporting cells (Stanković et al., 1997; Hosoya et al., 2016), and the gene mutant mice were virtually deaf (Gawenis et al., 2004). In our previous study, slc4a2b, the homolog of human SLC4A2, was proven to be required for hair cell development and function in zebrafish (Qian et al., 2020).
SLC4A11 gene mutation causes genetic corneal dystrophies. However, in addition to corneal disease, deficiency of this gene also leads to sensorineural deafness (Desir et al., 2007; Groger et al., 2010; Vilas et al., 2013). In the inner ear, the SLC4A2 gene is expressed in the fibrocytes underlying the stria vascularis, and SLC4A2-null fibrocytes manifest intracellular vacuolations and extracellular edemas, which cause reduced endocochlear potential and hearing threshold (Groger et al., 2010).
SLC6A6 and SLC6A8
SLC6 is a sodium- and chloride-dependent neurotransmitter transporter family, and it has more than 20 members, namely, SLC6A1–21 (Pramod et al., 2013). The substrates of these transporters include serotonin, dopamine, norepinephrine, GABA, taurine, creatine, and some amino acids. The SLC6 family genes are important for normal biological and physiological processes and related to a number of human diseases.
The SLC6A6 gene encodes the taurine transporter, and mice with Slc6a6 gene knockout develop multisystemic dysfunctions, including hearing impairment caused by loss of hair cells and spiral ganglion neurons (Warskulat et al., 2007). However, a homozygous SLC6A6 mutation in two boys with early-onset retinal degeneration did not cause hearing loss (Preising et al., 2019).
Another SLC6 family member, SLC6A8, encoding the creatine transporter, was also found to be associated with hearing loss. It was reported that a patient with double deletion of the SLC6A8 and BAP31 genes suffered from severe dystonia and sensorineural deafness (Osaka et al., 2012).
SLC9A1
The SLC9 family is mainly characterized by Na+/H+ exchangers (Donowitz et al., 2013). So far, the gene reported to be related to hearing loss in this family is SLC9A1, encoding Na+/H+ exchanger 1 (NHE1), which is important in maintaining intracellular pH homeostasis by exchanging one intracellular H+ for one extracellular Na+(Fliegel, 2009). Complete or near-complete loss of function of SLC9A1 causes the Lichtenstein–Knorr syndrome, which is characterized by cerebellar ataxia and sensorineural hearing loss (Guissart et al., 2015). However, deafness may not be an essential phenotypic feature of SLC9A1 mutation because other patients with variant SLC9A1 did not show hearing loss (Iwama et al., 2018).
SLC12A1
SLC12 is an electroneutral cation–coupled chloride cotransporter family (Arroyo et al., 2013). Except the known deafness gene SLC12A2 discussed earlier, disrupted SLC12A1 was also reported to be involved in hearing loss. In that case, translocation of the SLC12A1 and ATE1 (arginyltransferase 1) genes was found in a boy with nonsyndromic hearing loss; however, no hearing impairment occurred in his brother, father, and grandfather who have the same translocation (Vona et al., 2014). In another case, a homozygous missense mutation within the SLC12A1 gene caused type I antenatal Bartter syndrome (ABS), without hearing deficits (Halperin et al., 2019). All of these demonstrate that SLC12A1 has a role in hearing loss, but it is probably through polygenic or multifactorial ways.
SLC16A2 and SLC16A10
SLC16 family members are mainly responsible for the transport of monocarboxylates, such as lactate and pyruvate; therefore, they are called monocarboxylate transporters (MCTs) (Halestrap, 2013). Distinct with other members in this family, SLC16A2 (also named as MCT8) and SLC16A10 (also named as MCT10), which share similarities with each other, prefer to transport the iodothyronines (T4 and T3) (Friesema et al., 2008). As known to us, the thyroid hormone is required for hair cell survival and normal hearing (Rüsch et al., 2001; Mustapha et al., 2009; Ng et al., 2015). Unsurprisingly, SLC16A2 and SLC16A10 are expressed in the cochlear tissues (Sharlin et al., 2011), and they were also reported to have a role in the maintenance of cochlear hair cells and hearing through T3-dependent mechanisms (Sharlin et al., 2018).
SLC19A2
SLC19 is a folate/thiamine transporter family, and there are three members (SLC19A1–3), of which SLC19A1 transports folates but not thiamine, and the other two transport thiamine but not folates (Zhao and Goldman, 2013). Mutations in the SLC19A2 gene, encoding thiamine transporter 1 (THTR1), were reported to be associated with thiamine-responsive megaloblastic anemia (TRMA) (Scharfe et al., 2000; Ozdemir et al., 2002; Ghaemi et al., 2013; Setoodeh et al., 2013; Sun et al., 2018; Amr et al., 2019), which is characterized by early-onset diabetes mellitus, anemia, and sensorineural deafness.
SLC26A2
The SLC26 family genes encode multifunctional anion exchangers and anion channels, and 11 members (SLC26A1–11) are included in this family (Alper and Sharma, 2013). In addition to the two members, SLC26A4 and SLC26A5, discussed above, another gene, SLC26A2, encoding for a sulfate/chloride transporter, may be associated with hearing loss. In zebrafish, the slc26a2 gene was proven to be critical for otic development and hair cell survival, and loss of function of slc26a2 led to defective auditory organ development and impaired hearing (Liu et al., 2015).
SLC44A2 and SLC44A4
The SLC44 family, with five members (SLC44A1–5), is a choline-like transporter family (Traiffort et al., 2013).
SLC44A2, also named choline transporter–like protein 2 (CTL2), is a transmembrane glycoprotein. It was identified as the target of the Kresge Hearing Research Institute-3 (KHRI-3) antibody, which can lead to autoimmune hearing loss by binding to SLC44A2 and blocking its transporter function (Nair, 2004). Moreover, knockout of the Slc44a2 gene also caused hair cell and spiral ganglion neuron loss, especially in the basal turn of the cochlea, and Slc44a2 null mice exhibited high-frequency hearing loss (Kommareddi et al., 2015).
SLC44A4 encodes the choline transport protein CTL4. Mutations in this gene were found in a Chinese family with postlingual nonsyndromic mid-frequency sensorineural hearing loss, and knockdown of the slc44a4 gene in zebrafish led to significant defects in the otic vesicle and lateral line neuromast development, accompanied by defective hearing (Ma et al., 2017). Further evidence showed that SLC44A4 mutation disrupted its choline uptake function and acetylcholine synthesis ability, leading to hearing loss.
SLC52A3
Brown–Vialetto–Van Laere (BVVL) syndrome is a rare neurodegenerative disease characterized by sensorineural hearing loss and a variety of cranial nerve palsies (Sathasivam, 2008; Yonezawa and Inui, 2013). Mutations in the SLC52A3 gene, encoding riboflavin transporter 3 (RFVT3), was found in the BVVL syndrome (Johnson et al., 2010; Bosch et al., 2011), and a high dose of riboflavin improved the syndrome (Anand et al., 2012), indicating that mutation of the SLC52A3 gene might be a cause of BVVL syndrome. However, the mechanisms by which SLC52A3 functions in the auditory system remain unclear.
Conclusion
According to the World Health Organization (WHO), one in four, about 2.5 billion, people worldwide will be living with some degree of hearing loss by 2050, and nearly 60% of hearing loss is caused by genetic factors. Therefore, hereditary hearing loss is a serious problem that needs attention. In the last decades, more than 200 human hearing loss genes have been identified by scientists all over the world.
The SLC family genes, with more than 400 members, encode different types of transporters that function in solute transporting. Deficiency of some of these genes would cause human diseases. Until now, 18 members, as listed in this review, in the SLC family were reported to be associated with the occurrence of hereditary deafness, including five known hearing loss genes, which indicated the crucial roles of solute carriers in hearing function. These SLCs have different expression patterns in the inner ear and have distinct function, and each of them is essential for normal hearing. Mutations in any of these genes would result in inner ear cell dysfunction or even cell death, leading to hearing impairment or hearing loss.
In this review, we systematically summarized all of the SLCs involved in hearing, from their expression to function to the underlying mechanisms. This study would be helpful in understanding the roles of solute transporting for normal hearing, and it can also provide ideas for deafness gene identification.
Author Contributions
FQ and XJ performed the literature review and wrote the manuscript, RC and DL guided the writing and review of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We acknowledge the support of the Scientific Research Project of Nantong Municipal Health Commission (No. QA2021034).
References
Alper, S. L., and Sharma, A. K. (2013). The SLC26 Gene Family of Anion Transporters and Channels. Mol. Aspects Med. 34 (2-3), 494–515. doi:10.1016/j.mam.2012.07.009
Amr, K., Pawlikowska, P., Aoufouchi, S., Rosselli, F., and El‐Kamah, G. (2019). Whole Exome Sequencing Identifies a New Mutation in the SLC19A2 Gene Leading to Thiamine‐responsive Megaloblastic Anemia in an Egyptian Family. Mol. Genet. Genomic Med. 7 (7), e00777. doi:10.1002/mgg3.777
Anand, G., Hasan, N., Jayapal, S., Huma, Z., Ali, T., Hull, J., et al. (2012). Early Use of High-Dose Riboflavin in a Case of Brown-Vialetto-Van Laere Syndrome. Dev. Med. Child. Neurol. 54 (2), 187–189. doi:10.1111/j.1469-8749.2011.04142.x
Arroyo, J. P., Kahle, K. T., and Gamba, G. (2013). The SLC12 Family of Electroneutral Cation-Coupled Chloride Cotransporters. Mol. Aspects Med. 34 (2-3), 288–298. doi:10.1016/j.mam.2012.05.002
Bazard, P., Ding, B., Chittam, H. K., Zhu, X., Parks, T. A., Taylor-Clark, T. E., et al. (2020). Aldosterone Up-Regulates Voltage-Gated Potassium Currents and NKCC1 Protein Membrane Fractions. Sci. Rep. 10 (1), 15604. doi:10.1038/s41598-020-72450-4
Ben Said, M., Grati, M. h., Ishimoto, T., Zou, B., Chakchouk, I., Ma, Q., et al. (2016). A Mutation in SLC22A4 Encoding an Organic Cation Transporter Expressed in the Cochlea Strial Endothelium Causes Human Recessive Non-syndromic Hearing Loss DFNB60. Hum. Genet. 135 (5), 513–524. doi:10.1007/s00439-016-1657-7
Bosch, A. M., Abeling, N. G. G. M., Ijlst, L., Knoester, H., van der Pol, W. L., Stroomer, A. E. M., et al. (2011). Brown-Vialetto-Van Laere and Fazio Londe Syndrome Is Associated with a Riboflavin Transporter Defect Mimicking Mild MADD: a New Inborn Error of Metabolism with Potential Treatment. J. Inherit. Metab. Dis. 34 (1), 159–164. doi:10.1007/s10545-010-9242-z
Cheatham, M. A., Huynh, K. H., Gao, J., Zuo, J., and Dallos, P. (2004). Cochlear Function inPrestinknockout Mice. J. Physiol. 560 (Pt 3), 821–830. doi:10.1113/jphysiol.2004.069559
Chiereghin, C., Robusto, M., Mauri, L., Primignani, P., Castorina, P., Ambrosetti, U., et al. (2021). SLC22A4 Gene in Hereditary Non-syndromic Hearing Loss: Recurrence and Incomplete Penetrance of the p.C113Y Mutation in Northwest Africa. Front. Genet. 12, 606630. doi:10.3389/fgene.2021.606630
Crouch, J. J., Sakaguchi, N., Lytle, C., and Schulte, B. A. (1997). Immunohistochemical Localization of the Na-K-Cl Co-transporter (NKCC1) in the Gerbil Inner Ear. J. Histochem. Cytochem. 45 (6), 773–778. doi:10.1177/002215549704500601
Dallos, P., Wu, X., Cheatham, M. A., Gao, J., Zheng, J., Anderson, C. T., et al. (2008). Prestin-based Outer Hair Cell Motility Is Necessary for Mammalian Cochlear Amplification. Neuron 58 (3), 333–339. doi:10.1016/j.neuron.2008.02.028
Dawson, P., and Markovich, D. (2005). Pathogenetics of the Human SLC26 Transporters. Cmc 12 (4), 385–396. doi:10.2174/0929867053363144
Delpire, E., Lu, J., England, R., Dull, C., and Thorne, T. (1999). Deafness and Imbalance Associated with Inactivation of the Secretory Na-K-2Cl Co-transporter. Nat. Genet. 22 (2), 192–195. doi:10.1038/9713
Desir, J., Moya, G., Reish, O., Van Regemorter, N., Deconinck, H., David, K. L., et al. (2007). Borate Transporter SLC4A11 Mutations Cause Both Harboyan Syndrome and Non-syndromic Corneal Endothelial Dystrophy. J. Med. Genet. 44 (5), 322–326. doi:10.1136/jmg.2006.046904
Ding, B., Frisina, R. D., Zhu, X., Sakai, Y., Sokolowski, B., and Walton, J. P. (2014). Direct Control of Na+-K+-2Cl−-Cotransport Protein (NKCC1) Expression with Aldosterone. Am. J. Physiology-Cell Physiol. 306 (1), C66–C75. doi:10.1152/ajpcell.00096.2013
Donowitz, M., Ming Tse, C., and Fuster, D. (2013). SLC9/NHE Gene Family, a Plasma Membrane and Organellar Family of Na+/H+ Exchangers. Mol. Aspects Med. 34 (2-3), 236–251. doi:10.1016/j.mam.2012.05.001
Everett, L. A., Glaser, B., Beck, J. C., Idol, J. R., Buchs, A., Heyman, M. a., et al. (1997). Pendred Syndrome Is Caused by Mutations in a Putative Sulphate Transporter Gene (PDS). Nat. Genet. 17 (4), 411–422. doi:10.1038/ng1297-411
Fliegel, L. (2009). Regulation of the Na+/H+exchanger in the Healthy and Diseased Myocardium. Expert Opin. Ther. Targets 13 (1), 55–68. doi:10.1517/14728220802600707
Friesema, E. C. H., Jansen, J., Jachtenberg, J.-w., Visser, W. E., Kester, M. H. A., and Visser, T. J. (2008). Effective Cellular Uptake and Efflux of Thyroid Hormone by Human Monocarboxylate Transporter 10. Mol. Endocrinol. 22 (6), 1357–1369. doi:10.1210/me.2007-0112
Gawenis, L. R., Ledoussal, C., Judd, L. M., Prasad, V., Alper, S. L., Stuart-Tilley, A., et al. (2004). Mice with a Targeted Disruption of the AE2 Cl−/HCO3− Exchanger Are Achlorhydric. J. Biol. Chem. 279 (29), 30531–30539. doi:10.1074/jbc.M403779200
Ge, J., Elferich, J., Dehghani-Ghahnaviyeh, S., Zhao, Z., Meadows, M., von Gersdorff, H., et al. (2021). Molecular Mechanism of Prestin Electromotive Signal Amplification. Cell 184, 4669–4679. doi:10.1016/j.cell.2021.07.034
Goto, S., Oshima, T., Ikeda, K., Ueda, N., and Takasaka, T. (1997). Expression and Localization of the Na-K-2Cl Cotransporter in the Rat Cochlea. Brain Res. 765 (2), 324–326. doi:10.1016/s0006-8993(97)00679-3
Gröger, N., Fröhlich, H., Maier, H., Olbrich, A., Kostin, S., Braun, T., et al. (2010). SLC4A11 Prevents Osmotic Imbalance Leading to Corneal Endothelial Dystrophy, Deafness, and Polyuria. J. Biol. Chem. 285 (19), 14467–14474. doi:10.1074/jbc.M109.094680
Grundemann, D., Harlfinger, S., Golz, S., Geerts, A., Lazar, A., Berkels, R., et al. (2005). Discovery of the Ergothioneine Transporter. Proc. Natl. Acad. Sci. 102 (14), 5256–5261. doi:10.1073/pnas.0408624102
Guissart, C., Li, X., Leheup, B., Drouot, N., Montaut-Verient, B., Raffo, E., et al. (2015). Mutation of SLC9A1, Encoding the Major Na+/H+ Exchanger, Causes Ataxia-Deafness Lichtenstein-Knorr Syndrome. Hum. Mol. Genet. 24 (2), 463–470. doi:10.1093/hmg/ddu461
Halestrap, A. P. (2013). The SLC16 Gene Family - Structure, Role and Regulation in Health and Disease. Mol. Aspects Med. 34 (2-3), 337–349. doi:10.1016/j.mam.2012.05.003
Halperin, D., Dolgin, V., Geylis, M., Drabkin, M., Yogev, Y., Wormser, O., et al. (2019). A Novel SLC12A1 Mutation in Bedouin kindred with Antenatal Bartter Syndrome Type I. Ann. Hum. Genet. 83 (5), 361–366. doi:10.1111/ahg.12317
Homma, K., Miller, K. K., Anderson, C. T., Sengupta, S., Du, G.-G., Aguiñaga, S., et al. (2010). Interaction between CFTR and Prestin (SLC26A5). Biochim. Biophys. Acta (Bba) - Biomembranes 1798 (6), 1029–1040. doi:10.1016/j.bbamem.2010.02.001
Horvath, G., Schmid, N., Fragoso, M. A., Schmid, A., Conner, G. E., Salathe, M., et al. (2007). Epithelial Organic Cation Transporters Ensure pH-dependent Drug Absorption in the Airway. Am. J. Respir. Cell Mol Biol 36 (1), 53–60. doi:10.1165/rcmb.2006-0230OC
Hosoya, M., Fujioka, M., Kobayashi, R., Okano, H., and Ogawa, K. (2016). Overlapping Expression of Anion Exchangers in the Cochlea of a Non-human Primate Suggests Functional Compensation. Neurosci. Res. 110, 1–10. doi:10.1016/j.neures.2016.04.002
Iwama, K., Osaka, H., Ikeda, T., Mitsuhashi, S., Miyatake, S., Takata, A., et al. (2018). A Novel SLC9A1 Mutation Causes Cerebellar Ataxia. J. Hum. Genet. 63 (10), 1049–1054. doi:10.1038/s10038-018-0488-x
Johnson, J. O., Gibbs, J. R., Van Maldergem, L., Houlden, H., and Singleton, A. B. (2010). Exome Sequencing in Brown-Vialetto-Van Laere Syndrome. Am. J. Hum. Genet. 87 (4), 567–569. doi:10.1016/j.ajhg.2010.05.021
Kato, Y., Kubo, Y., Iwata, D., Kato, S., Sudo, T., Sugiura, T., et al. (2010). Gene Knockout and Metabolome Analysis of Carnitine/organic Cation Transporter OCTN1. Pharm. Res. 27 (5), 832–840. doi:10.1007/s11095-010-0076-z
Keller, J. P., Homma, K., Duan, C., Zheng, J., Cheatham, M. A., and Dallos, P. (2014). Functional Regulation of the SLC26-Family Protein Prestin by Calcium/calmodulin. J. Neurosci. 34 (4), 1325–1332. doi:10.1523/jneurosci.4020-13.2014
Kim, B. G., Yoo, T. H., Yoo, J. E., Seo, Y. J., Jung, J., and Choi, J. Y. (2017). Resistance to Hypertension and High Cl − Excretion in Humans with SLC26A4 Mutations. Clin. Genet. 91 (3), 448–452. doi:10.1111/cge.12789
Kim, M.-A., Kim, S. H., Ryu, N., Ma, J.-H., Kim, Y.-R., Jung, J., et al. (2019). Gene Therapy for Hereditary Hearing Loss by SLC26A4 Mutations in Mice Reveals Distinct Functional Roles of Pendrin in normal Hearing. Theranostics 9 (24), 7184–7199. doi:10.7150/thno.38032
Kommareddi, P., Nair, T., Kakaraparthi, B. N., Galano, M. M., Miller, D., Laczkovich, I., et al. (2015). Hair Cell Loss, Spiral Ganglion Degeneration, and Progressive Sensorineural Hearing Loss in Mice with Targeted Deletion of Slc44a2/Ctl2. Jaro 16 (6), 695–712. doi:10.1007/s10162-015-0547-3
Lamhonwah, A.-M., Mai, L., Chung, C., Lamhonwah, D., Ackerley, C., and Tein, I. (2011). Upregulation of Mammary Gland OCTNs Maintains Carnitine Homeostasis in Suckling Infants. Biochem. Biophysical Res. Commun. 404 (4), 1010–1015. doi:10.1016/j.bbrc.2010.12.100
Li, X. C., Everett, L. A., Lalwani, A. K., Desmukh, D., Friedman, T. B., Green, E. D., et al. (1998). A Mutation in PDS Causes Non-syndromic Recessive Deafness. Nat. Genet. 18 (3), 215–217. doi:10.1038/ng0398-215
Liberman, M. C., Gao, J., He, D. Z. Z., Wu, X., Jia, S., and Zuo, J. (2002). Prestin Is Required for Electromotility of the Outer Hair Cell and for the Cochlear Amplifier. Nature 419 (6904), 300–304. doi:10.1038/nature01059
Liu, F., Xia, W., Hu, J., Wang, Y., Yang, F., Sun, S., et al. (2015). Solute Carrier Family 26 Member A2 (Slc26a2) Regulates Otic Development and Hair Cell Survival in Zebrafish. PLoS One 10 (9), e0136832. doi:10.1371/journal.pone.0136832
Liu, X. Z., Ouyang, X. M., Xia, X. J., Zheng, J., Pandya, A., Li, F., et al. (2003). Prestin, a Cochlear Motor Protein, Is Defective in Non-syndromic Hearing Loss. Hum. Mol. Genet. 12 (10), 1155–1162. doi:10.1093/hmg/ddg127
Liu, Y., Chu, H., Chen, J., Zhou, L., Chen, Q., Yu, Y., et al. (2014). Age-related Change in the Expression of NKCC1 in the Cochlear Lateral wall of C57BL/6J Mice. Acta Oto-Laryngologica 134 (10), 1047–1051. doi:10.3109/00016489.2014.900704
Ma, Z., Xia, W., Liu, F., Ma, J., Sun, S., Zhang, J., et al. (2017). SLC44A4mutation Causes Autosomal Dominant Hereditary Postlingual Non-syndromic Mid-frequency Hearing Loss. Hum. Mol. Genet. 26 (2), ddw394–394. doi:10.1093/hmg/ddw394
Maciaszczyk, K., and Lewiński, A. (2008). Phenotypes of SLC26A4 Gene Mutations: Pendred Syndrome and Hypoacusis with Enlarged Vestibular Aqueduct. Neuro Endocrinol. Lett. 29 (1), 29–36.
Macnamara, E. F., Koehler, A. E., D'Souza, P., Estwick, T., Lee, P., Vezina, G., et al. (2019). Kilquist Syndrome: A Novel Syndromic Hearing Loss Disorder Caused by Homozygous Deletion of SLC12A2. Hum. Mutat. 40 (5), 532–538. doi:10.1002/humu.23722
Maeda, T., Hirayama, M., Kobayashi, D., Miyazawa, K., and Tamai, I. (2007). Mechanism of the Regulation of Organic Cation/carnitine Transporter 1 (SLC22A4) by Rheumatoid Arthritis-Associated Transcriptional Factor RUNX1 and Inflammatory Cytokines. Drug Metab. Dispos 35 (3), 394–401. doi:10.1124/dmd.106.012112
McNeill, A., Iovino, E., Mansard, L., Vache, C., Baux, D., Bedoukian, E., et al. (2020). SLC12A2 Variants Cause a Neurodevelopmental Disorder or Cochleovestibular Defect. Brain 143 (8), 2380–2387. doi:10.1093/brain/awaa176
Meier, Y., Eloranta, J. J., Darimont, J., Ismair, M. G., Hiller, C., Fried, M., et al. (2007). Regional Distribution of Solute Carrier mRNA Expression along the Human Intestinal Tract. Drug Metab. Dispos 35 (4), 590–594. doi:10.1124/dmd.106.013342
Mistrík, P., Daudet, N., Morandell, K., and Ashmore, J. F. (2012). Mammalian Prestin Is a Weak Cl−/HCO3−electrogenic Antiporter. J. Physiol. 590 (22), 5597–5610. doi:10.1113/jphysiol.2012.241448
Mizuta, K., Adachi, M., and Iwasa, K. H. (1997). Ultrastructural Localization of the Na-K-Cl Cotransporter in the Lateral wall of the Rabbit Cochlear Duct. Hear. Res. 106 (1-2), 154–162. doi:10.1016/s0378-5955(97)00010-5
Mustapha, M., Fang, Q., Gong, T.-W., Dolan, D. F., Raphael, Y., Camper, S. A., et al. (2009). Deafness and Permanently Reduced Potassium Channel Gene Expression and Function in Hypothyroid Pit1dw Mutants. J. Neurosci. 29 (4), 1212–1223. doi:10.1523/JNEUROSCI.4957-08.2009
Mutai, H., Wasano, K., Momozawa, Y., Kamatani, Y., Miya, F., Masuda, S., et al. (2020). Variants Encoding a Restricted Carboxy-Terminal Domain of SLC12A2 Cause Hereditary Hearing Loss in Humans. Plos Genet. 16 (4), e1008643. doi:10.1371/journal.pgen.1008643
Nair, T. S. (2004). Identification and Characterization of Choline Transporter-like Protein 2, an Inner Ear Glycoprotein of 68 and 72 kDa that Is the Target of Antibody-Induced Hearing Loss. J. Neurosci. 24 (7), 1772–1779. doi:10.1523/jneurosci.5063-03.2004
Newman, B., Gu, X., Wintle, R., Cescon, D., Yazdanpanah, M., Liu, X., et al. (2005). A Risk Haplotype in the Solute Carrier Family 22A4/22A5 Gene Cluster Influences Phenotypic Expression of Crohn's Disease. Gastroenterology 128 (2), 260–269. doi:10.1053/j.gastro.2004.11.056
Ng, L., Cordas, E., Wu, X., Vella, K. R., Hollenberg, A. N., and Forrest, D. (2015). Age-Related Hearing Loss and Degeneration of Cochlear Hair Cells in Mice Lacking Thyroid Hormone Receptor β1. Endocrinology 156 (10), 3853–3865. doi:10.1210/en.2015-1468
Nosrat, G., Martha, G., Alireza Baradaran, H., Rahim, V., and Vakili, R. (2013). Novel Mutation in the SLC19A2 Gene in an Iranian Family with Thiamine-Responsive Megaloblastic Anemia: a Series of Three Cases. Jcrpe 5 (3), 199–201. doi:10.4274/Jcrpe.969
Obholzer, N., Wolfson, S., Trapani, J. G., Mo, W., Nechiporuk, A., Busch-Nentwich, E., et al. (2008). Vesicular Glutamate Transporter 3 Is Required for Synaptic Transmission in Zebrafish Hair Cells. J. Neurosci. 28 (9), 2110–2118. doi:10.1523/JNEUROSCI.5230-07.2008
Osaka, H., Takagi, A., Tsuyusaki, Y., Wada, T., Iai, M., Yamashita, S., et al. (2012). Contiguous Deletion of SLC6A8 and BAP31 in a Patient with Severe Dystonia and Sensorineural Deafness. Mol. Genet. Metab. 106 (1), 43–47. doi:10.1016/j.ymgme.2012.02.018
Ozdemir, M. A., Akcakus, M., Kurtoglu, S., Gunes, T., and Torun, Y. A. (2002). TRMA Syndrome (Thiamine-responsive Megaloblastic Anemia): a Case Report and Review of the Literature. Pediatr. Diabetes 3 (4), 205–209. doi:10.1034/j.1399-5448.2002.30407.x
Peltekova, V. D., Wintle, R. F., Rubin, L. A., Amos, C. I., Huang, Q., Gu, X., et al. (2004). Functional Variants of OCTN Cation Transporter Genes Are Associated with Crohn Disease. Nat. Genet. 36 (5), 471–475. doi:10.1038/ng1339
Pramod, A. B., Foster, J., Carvelli, L., and Henry, L. K. (2013). SLC6 Transporters: Structure, Function, Regulation, Disease Association and Therapeutics. Mol. Aspects Med. 34 (2-3), 197–219. doi:10.1016/j.mam.2012.07.002
Preising, M. N., Görg, B., Friedburg, C., Qvartskhava, N., Budde, B. S., Bonus, M., et al. (2019). Biallelic Mutation of Human SLC6A6 Encoding the Taurine Transporter TAUT Is Linked to Early Retinal Degeneration. FASEB j. 33 (10), 11507–11527. doi:10.1096/fj.201900914RR
Qian, F., Wang, X., Yin, Z., Xie, G., Yuan, H., Liu, D., et al. (2020). The Slc4a2b Gene Is Required for Hair Cell Development in Zebrafish. aging 12 (19), 18804–18821. doi:10.18632/aging.103840
Romero, M. F., Chen, A.-P., Parker, M. D., and Boron, W. F. (2013). The SLC4 Family of Bicarbonate Transporters. Mol. Aspects Med. 34 (2-3), 159–182. doi:10.1016/j.mam.2012.10.008
Ruel, J., Emery, S., Nouvian, R., Bersot, T., Amilhon, B., Van Rybroek, J. M., et al. (2008). Impairment of SLC17A8 Encoding Vesicular Glutamate Transporter-3, VGLUT3, Underlies Nonsyndromic Deafness DFNA25 and Inner Hair Cell Dysfunction in Null Mice. Am. J. Hum. Genet. 83 (2), 278–292. doi:10.1016/j.ajhg.2008.07.008
Rüsch, A., Ng, L., Goodyear, R., Oliver, D., Lisoukov, I., Vennström, B., et al. (2001). Retardation of Cochlear Maturation and Impaired Hair Cell Function Caused by Deletion of All Known Thyroid Hormone Receptors. J. Neurosci. 21 (24), 9792–9800. doi:10.1523/jneurosci.21-24-09792.2001
Ryu, N., Lee, S., Park, H.-J., Lee, B., Kwon, T.-J., Bok, J., et al. (2017). Identification of a Novel Splicing Mutation within SLC17A8 in a Korean Family with Hearing Loss by Whole-Exome Sequencing. Gene 627, 233–238. doi:10.1016/j.gene.2017.06.040
Ryu, N., Sagong, B., Park, H.-J., Kim, M.-A., Lee, K.-Y., Choi, J. Y., et al. (2016). Screening of the SLC17A8 Gene as a Causative Factor for Autosomal Dominant Non-syndromic Hearing Loss in Koreans. BMC Med. Genet. 17, 6. doi:10.1186/s12881-016-0269-3
Sathasivam, S. (2008). Brown-Vialetto-Van Laere Syndrome. Orphanet J. Rare Dis. 3, 9. doi:10.1186/1750-1172-3-9
Scharfe, C., Hauschild, M., Klopstock, T., Janssen, A. J., Heidemann, P. H., Meitinger, T., et al. (2000). A Novel Mutation in the Thiamine Responsive Megaloblastic Anaemia Gene SLC19A2 in a Patient with Deficiency of Respiratory Chain Complex I. J. Med. Genet. 37 (9), 669–673. doi:10.1136/jmg.37.9.669
Seal, R. P., Akil, O., Yi, E., Weber, C. M., Grant, L., Yoo, J., et al. (2008). Sensorineural Deafness and Seizures in Mice Lacking Vesicular Glutamate Transporter 3. Neuron 57 (2), 263–275. doi:10.1016/j.neuron.2007.11.032
Setoodeh, A., Haghighi, A., Saleh-Gohari, N., Ellard, S., and Haghighi, A. (2013). Identification of a SLC19A2 Nonsense Mutation in Persian Families with Thiamine-Responsive Megaloblastic Anemia. Gene 519 (2), 295–297. doi:10.1016/j.gene.2013.02.008
Sharlin, D. S., Ng, L., Verrey, F., Visser, T. J., Liu, Y., Olszewski, R. T., et al. (2018). Deafness and Loss of Cochlear Hair Cells in the Absence of Thyroid Hormone Transporters Slc16a2 (Mct8) and Slc16a10 (Mct10). Sci. Rep. 8 (1), 4403. doi:10.1038/s41598-018-22553-w
Sharlin, D. S., Visser, T. J., and Forrest, D. (2011). Developmental and Cell-specific Expression of Thyroid Hormone Transporters in the Mouse Cochlea. Endocrinology 152 (12), 5053–5064. doi:10.1210/en.2011-1372
Stanković, K. M., Brown, D., Alper, S. L., and Adams, J. C. (1997). Localization of pH Regulating Proteins H+ATPase and Cl-/HCO3- Exchanger in the guinea Pig Inner Ear. Hear. Res. 114 (1-2), 21–34. doi:10.1016/s0378-5955(97)00072-5
Sun, C., Pei, Z., Zhang, M., Sun, B., Yang, L., Zhao, Z., et al. (2018). Recovered Insulin Production after Thiamine Administration in Permanent Neonatal Diabetes Mellitus with a Novel Solute Carrier Family 19 Member 2 ( SLC19A2 ) Mutation. J. Diabetes 10 (1), 50–58. doi:10.1111/1753-0407.12556
Tamai, I., Yabuuchi, H., Nezu, J.-i., Sai, Y., Oku, A., Shimane, M., et al. (1997). Cloning and Characterization of a Novel Human pH-dependent Organic Cation Transporter, OCTN1. FEBS Lett. 419 (1), 107–111. doi:10.1016/s0014-5793(97)01441-5
Tan, X., Pecka, J. L., Tang, J., Okoruwa, O. E., Zhang, Q., Beisel, K. W., et al. (2011). From Zebrafish to Mammal: Functional Evolution of Prestin, the Motor Protein of Cochlear Outer Hair Cells. J. Neurophysiol. 105 (1), 36–44. doi:10.1152/jn.00234.2010
Tokuhiro, S., Yamada, R., Chang, X., Suzuki, A., Kochi, Y., Sawada, T., et al. (2003). An Intronic SNP in a RUNX1 Binding Site of SLC22A4, Encoding an Organic Cation Transporter, Is Associated with Rheumatoid Arthritis. Nat. Genet. 35 (4), 341–348. doi:10.1038/ng1267
Traiffort, E., O’Regan, S., and Ruat, M. (2013). The Choline Transporter-like Family SLC44: Properties and Roles in Human Diseases. Mol. Aspects Med. 34 (2-3), 646–654. doi:10.1016/j.mam.2012.10.011
Vilas, G. L., Loganathan, S. K., Liu, J., Riau, A. K., Young, J. D., Mehta, J. S., et al. (2013). Transmembrane Water-Flux through SLC4A11: a Route Defective in Genetic Corneal Diseases. Hum. Mol. Genet. 22 (22), 4579–4590. doi:10.1093/hmg/ddt307
Vona, B., Neuner, C., El Hajj, N., Schneider, E., Farcas, R., Beyer, V., et al. (2014). Disruption of the ATE1 and SLC12A1 Genes by Balanced Translocation in a Boy with Non-syndromic Hearing Loss. Mol. Syndromol 5 (1), 3–10. doi:10.1159/000355443
Warskulat, U., Heller‐Stilb, B., Oermann, E., Zilles, K., Haas, H., Lang, F., et al. (2007). Phenotype of the Taurine Transporter Knockout Mouse. Methods Enzymol. 428, 439–458. doi:10.1016/s0076-6879(07)28025-5
Weber, T., Gopfert, M. C., Winter, H., Zimmermann, U., Kohler, H., Meier, A., et al. (2003). Expression of Prestin-Homologous Solute Carrier (SLC26) in Auditory Organs of Nonmammalian Vertebrates and Insects. Proc. Natl. Acad. Sci. 100 (13), 7690–7695. doi:10.1073/pnas.1330557100
Yabuuchi, H., Tamai, I., Nezu, J., Sakamoto, K., Oku, A., Shimane, M., et al. (1999). Novel Membrane Transporter OCTN1 Mediates Multispecific, Bidirectional, and pH-dependent Transport of Organic Cations. J. Pharmacol. Exp. Ther. 289 (2), 768–773.
Yonezawa, A., and Inui, K.-i. (2013). Novel Riboflavin Transporter Family RFVT/SLC52: Identification, Nomenclature, Functional Characterization and Genetic Diseases of RFVT/SLC52. Mol. Aspects Med. 34 (2-3), 693–701. doi:10.1016/j.mam.2012.07.014
Yoshino, T., Sato, E., Nakashima, T., Teranishi, M., Yamamoto, H., Otake, H., et al. (2006). Distribution of Pendrin in the Organ of Corti of Mice Observed by Electron Immunomicroscopy. Eur. Arch. Otorhinolaryngol. 263 (8), 699–704. doi:10.1007/s00405-006-0045-7
Zhang, W., Peng, Z., Yu, S., Song, Q.-L., Qu, T.-F., Liu, K., et al. (2020). Exposure to Sodium Salicylate Disrupts VGLUT3 Expression in Cochlear Inner Hair Cells and Contributes to Tinnitus. Physiol. Res. 69 (1), 181–190. doi:10.33549/physiolres.934180
Zhao, R., and Goldman, I. D. (2013). Folate and Thiamine Transporters Mediated by Facilitative Carriers (SLC19A1-3 and SLC46A1) and Folate Receptors. Mol. Aspects Med. 34 (2-3), 373–385. doi:10.1016/j.mam.2012.07.006
Zheng, J., Du, G.-G., Anderson, C. T., Keller, J. P., Orem, A., Dallos, P., et al. (2006). Analysis of the Oligomeric Structure of the Motor Protein Prestin. J. Biol. Chem. 281 (29), 19916–19924. doi:10.1074/jbc.M513854200
Zheng, J., Du, G.-G., Matsuda, K., Orem, A., Aguiñaga, S., Deák, L., et al. (2005). The C-Terminus of Prestin Influences Nonlinear Capacitance and Plasma Membrane Targeting. J. Cell Sci 118 (Pt 13), 2987–2996. doi:10.1242/jcs.02431
Keywords: solute carrier, SLC, transporter, hereditary hearing loss, deafness gene
Citation: Qian F, Jiang X, Chai R and Liu D (2022) The Roles of Solute Carriers in Auditory Function. Front. Genet. 13:823049. doi: 10.3389/fgene.2022.823049
Received: 29 November 2021; Accepted: 03 January 2022;
Published: 26 January 2022.
Edited by:
Daniel Grinberg, University of Barcelona, SpainReviewed by:
An-Ping Chen, Cytovia Therapeutics, United StatesLiman Liu, University of Kentucky Medical Center, United States
Copyright © 2022 Qian, Jiang, Chai and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renjie Chai, cmVuamllY0BzZXUuZWR1LmNu; Dong Liu, bGl1ZG9uZ3RvbUBnbWFpbC5jb20=
†These authors have contributed equally to this work