 Monica F. Danilevicz1
Monica F. Danilevicz1 Mitchell Gill1
Mitchell Gill1 Robyn Anderson1
Robyn Anderson1 Jacqueline Batley1
Jacqueline Batley1 Mohammed Bennamoun2
Mohammed Bennamoun2 Philipp E. Bayer1
Philipp E. Bayer1 David Edwards1*
David Edwards1*- 1School of Biological Sciences and Institute of Agriculture, University of Western Australia, Perth, WA, Australia
- 2School of Physics, Mathematics and Computing, University of Western Australia, Perth, WA, Australia
Genomic prediction tools support crop breeding based on statistical methods, such as the genomic best linear unbiased prediction (GBLUP). However, these tools are not designed to capture non-linear relationships within multi-dimensional datasets, or deal with high dimension datasets such as imagery collected by unmanned aerial vehicles. Machine learning (ML) algorithms have the potential to surpass the prediction accuracy of current tools used for genotype to phenotype prediction, due to their capacity to autonomously extract data features and represent their relationships at multiple levels of abstraction. This review addresses the challenges of applying statistical and machine learning methods for predicting phenotypic traits based on genetic markers, environment data, and imagery for crop breeding. We present the advantages and disadvantages of explainable model structures, discuss the potential of machine learning models for genotype to phenotype prediction in crop breeding, and the challenges, including the scarcity of high-quality datasets, inconsistent metadata annotation and the requirements of ML models.
Introduction
The expansion of genome sequencing technology has led to a rapid growth in plant genomic resources, including advanced genome and pangenome assemblies, providing a better understanding of plant genetic variation. Genotyping methods including genome resequencing (Morrell et al., 2011; Hu et al., 2018; Juliana et al., 2018) and single nucleotide polymorphism (SNP) arrays (Wang et al., 2014; Mason et al., 2017) have supported the expansion of crop genomic resources, assisting the identification of genetic variations related to agronomic traits. The emergence of plant pangenomes as a reference better represents the genomic variability within a species (Bayer et al., 2020; Danilevicz et al., 2020; Golicz et al., 2020), allowing researchers to expand genotypes from SNPs and indels to include gene presence absence variation, a structural variation that has been associated with crop disease resistance and stress tolerance (Golicz et al., 2016; Montenegro et al., 2017). Plant breeders now have a wide variety of tools available to assess genetic variation, however the most efficient way to apply these for crop improvement is often unclear (Heffner et al., 2009; Scheben et al., 2017; Nuccio et al., 2018). Genomic selection (GS) using best linear unbiased prediction (BLUP) was developed for livestock breeding (Meuwissen et al., 2001), but has seen success in plant breeding (Cooper et al., 2014; Gaffney et al., 2015; Crossa et al., 2017; Lebedev et al., 2020; Ukrainetz and Mansfield, 2020). This approach takes a subpopulation of breeding material, and through linear modelling, estimates the contribution of each SNP to phenotypes of interest. Due to the simplicity of the modelling, BLUP is straightforward to implement, and the contribution of each SNP to the phenotype is relatively easy to calculate. However, compared to animal breeding, genomic selection in plants has to account for the greater genotype by environment interactions, and requires the addition of appropriate multi-environment trial data (Voss-Fels et al., 2019).
Machine Learning (ML) and Deep Learning (DL) algorithms are more complex than linear prediction models and can discover non-linear relationships within datasets. The use of complex ML and DL models to associate plant genotypes with phenotypes is gaining popularity, with an increasing number of publications predicting a wide variety of agronomic traits, including yield, days to heading and thousand kernel weight (Montesinos-López A. et al., 2018, Montesinos-López et al., 2018 O. A.; Ma et al., 2018; Crossa et al., 2019; Khaki et al., 2019; Grinberg et al., 2020). Random forests, support vector machines and artificial neural networks may more easily capture the complex relationships between genotype, phenotype and environment compared with previous methods due to their non-linearity, and ML and DL have significant potential to support plant breeding. Here we review the progress that has been made applying ML and DL methods, and how to interpret their results within a biological context.
Machine Learning Methods for Genotype to Phenotype Prediction
Genotype to phenotype prediction has expanded with the application of GS. Genomic Best Linear Unbiased Prediction (GBLUP), a linear based modelling system has been used extensively in GS (Meuwissen et al., 2001), as have Bayesian systems (Pérez et al., 2010). Despite the successes of linear methods in GS, they can run into challenges due to the high dimensionality of marker data versus the number of individuals, and the presence of complex relationships that are difficult to account for (Crossa et al., 2017).
To improve on linear models in GS, there has been an increased use of nonlinear methods such as ML and DLmodels to predict plant phenotypes (Montesinos-López et al., 2021a). Nonlinear methods have been theorised to be better able to capture small interactions between markers, account for environment interactions and generate predictions with higher accuracy for data with high dimensionality (Pérez-Rodríguez et al., 2012; Cuevas et al., 2016). The ML and DL architectures can also include multimodal data and data types that are not suited to simple tabular formats (Figure 1). For example, the ML method random forest can capture patterns in high dimensional data to deliver accurate predictions and can also take into account non-additive effects (Heslot et al., 2012). Its use as a model for genomic selection has also demonstrated superior performance in comparison to linear models such as Bayesian Least Absolute Shrinkage and Selection Operator (LASSO) (Ornella et al., 2014) and Ridge Regression BLUP (RR-BLUP), depending on the genetic architecture of the predicted trait (Spindel et al., 2015). Other ML models that have shown potential for genomic selection include convolutional neural networks and feed forward deep neural networks that can outperform linear methods with correct optimisation of hyperparameters (Ma et al., 2018; Sandhu et al., 2020). Multi-trait DL models can help understand the relationship between related traits for improved prediction (Montesinos-López O. A. et al., 2018), and ensemble models that are a powerful way of combining multiple ML methods that may produce weaker predictions by themselves (Jubair and Domaratzki, 2019; Banerjee et al., 2020).
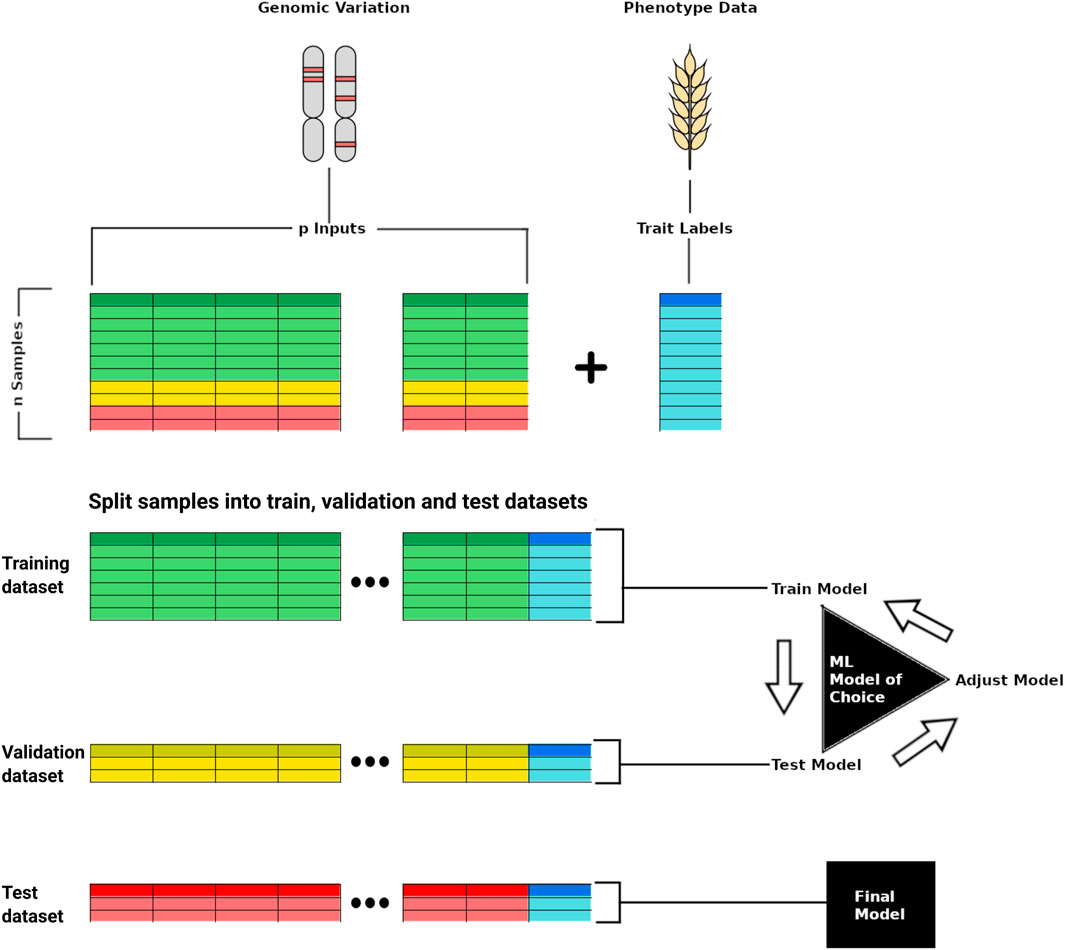
FIGURE 1. Scheme representing training a deep learning model for phenotype prediction based on genomic variation data. In the top, the genomic variation data of p samples is used as input whereas the phenotypic trait (e.g., yield) is used as a label for training the model. The genomic and phenotypic data is split into training, validation, and test datasets, with the first two employed for model development, and the last used last for testing the trained model capacity to predict a target trait from unseen input data.
Whether ML and DL approaches improve prediction compared to traditional GBLUP for genomic selection is still unclear. Several publications compare DL models with linear ML models, but not with GBLUP directly, and many breeding methods use BLUP values, whereas ML and DL studies tend to predict single traits. In an example where GBLUP was compared to DL methods for multi-trait prediction, when the genotype by environment interaction term was included in the GBLUP modelling, it outperformed DL across 8 of the 9 datasets, whereas when it was not included, the DL model outperformed GBLUP in 6 of the 9 datasets (Montesinos-López A. et al., 2018). Another study observed that their DL model outperformed linear and Bayesian prediction models when predicting under strong epistatic interactions, using SNPs from octoploid strawberry and tetraploid blueberry (Zingaretti et al., 2020). As with many problems that involve algorithm choice, the no free lunch theorem suggests that no one algorithm will perform the best across all problems, and this seems to be the case for genomic prediction (Azodi et al., 2019). DL is an efficient method to extract representative features from large datasets, with the capacity to account for feature interaction effects. However, conventional ML methods and mixed linear models are still well suited to deal with limited datasets, in many cases generating more accurate predictions than DL models. We propose that phenotype prediction should be expanded beyond GBLUP methods to ensure sufficient models are evaluated for each genomic selection problem.
Genetic Variation Representation
The most common form of encoding whole genome SNP data for ML and DL, is to use one hot encoding, where each SNP position is represented by four columns, each one representing the four bases of DNA: A, T, C, and G. Presence of the base at each position is indicated by a 1 in this column, whereas absence is indicated by 0 (Zou et al., 2019). In this way a letter can be encoded as a binary representation, suitable for ML and DL processes, that only accept numeric input. SNP one hot encoding is one of the most common data representations for DNA sequence data in phenotype prediction (Montesinos-López A. et al., 2018, Montesinos-López et al., 2018 O. A.; Ma et al., 2018; Khaki et al., 2019; Grinberg et al., 2020).
In ML and DL, the number of features is usually lower than the number of samples, however it is common in trait association studies that the number of features significantly outnumbers the samples. Genotyping usually generates large numbers of genetic markers and phenotyping large cohorts of plants can be prohibitively expensive, restricting the number of samples. Feature selection, minor allele frequency and genome wide association study (GWAS) can be applied to reduce the dimensionality of datasets and remove excess information, presenting varying degrees of popularity in plant breeding. In human research, some studies have used minor allele frequency, position within promoter regions (Yin et al., 2019), selective SNP number reduction, and integrated transcription data (Zhou et al., 2019), where previous association results are used to strategically reduce SNP numbers. Other strategies to reduce SNP numbers include a focus on rare variants, where loss of function variants have been noted for their influence in phenotype (Monroe et al., 2020) and using GWAS to select regions of interest, though this has had mixed results (Spindel et al., 2016; Rice and Lipka, 2019; Jeong et al., 2020).
Genotype encoding in plants has been mostly limited to SNP encoding, however there are other forms of genomic variation, and different ways of encoding genetic variation data that could be employed for phenotype prediction. A recent article used methylation at cytosine positions to predict traits in Populus balsamifera, input cytosines were encoded using variable importance scores and individual methylation intensity, The results show that biomass and physiochemical wood traits can be modelled using methylation data and DL (Champigny et al., 2020). Outside of plant research, genomic k-mers were used to encode genomic variability and employed to predict antibiotic resistance in multiple species of pathogenic bacteria, using classification and regression trees and set covering machines algorithms for interpretability (Pérez et al., 2010; Drouin et al., 2019). In Chen et al. (2019), rare genetic variants from a Mycobacterium tuberculosis dataset were grouped by locus, using binary encoding to account for mutation presence/absence and feature labelling to encode the type of mutation (e.g., SNP, insertion/deletion, change in frameshift within a coding region). The dataset was used to predict antibiotic resistance in the pathogen. improving prediction for both logistic regression models and neural networks. The expansion of plant pangenome assemblies provides an opportunity to include gene presence/absence variation in trait prediction, encoded in a similar way to SNPs (Golicz et al., 2016; Jin et al., 2016; Hurgobin and Edwards, 2017; Gao et al., 2019). The way researchers choose to encode the genomic information and use the associated data (environmental or gene expression data) is an important aspect of trait association analysis, and a wider variety of encoding methods should be explored.
Integrating High Throughput Phenotyping Into Genotype to Phenotype Models
Genotype to phenotype models are commonly applied to sparsely collected phenotypic traits such as plant height and germination rate, and these traits are often hand-collected, potentially introducing bias and increasing experimental cost. In contrast, high throughput phenotyping allows monitoring hundreds or thousands of plants under field and greenhouse conditions (Tattaris et al., 2016; Shakoor et al., 2017), performing non-destructive measurement of traits such as biomass (Quirós Vargas et al., 2019; Marino and Alvino, 2020), plant height (Anderson et al., 2020), wheat spike count (Hasan et al., 2018; Xiong et al., 2019), and disease (De Castro et al., 2015; Nagasubramanian et al., 2018). The increased density of phenotypic data produced by high throughput phenotyping enables researchers to dynamically measure changes in plant growth, evaluating the impact of genomic variation at different developmental stages. For example, a study using images collected weekly by an unmanned aerial vehicle observed that two SNPs related to maize height had a larger effect during early growth (10–25 cm), with their contribution to this phenotype decreasing towards the end of the season (Adak et al., 2021). In rice, high throughput imagery was used to calculate six vegetation indexes that were used as input traits for the identification of eight quantitative trait loci (QTLs), one of which was corroborated by another analysis using ground-truth agronomic data (Naito et al., 2017). Similarly, vegetation indices and canopy temperature were employed in the identification of QTLs related to abscisic acid concentration and other physiological traits in cotton using inclusive composite interval mapping for multi-environment trials (Pauli et al., 2016). Vegetation indices and temperature were also used to support genomic selection for grain yield in wheat (Rutkoski et al., 2016). These studies show the potential of incorporating raw or extracted features from image data to detect genomic variation associated with desirable traits. Intermediate phenotypes such as transcriptome, proteome or metabolome data can also be associated in a multidimensional dataset, providing a more detailed description of the plant response to environmental conditions, and potentially increasing phenotype prediction accuracy (de Abreu E Lima et al., 2018; Voss-Fels et al., 2019; Scossa et al., 2021).
The addition of multidimensional datasets can increase the complexity of analysis exponentially, requiring algorithms that are able to uncover the relationships between the data types and the target trait. DL models have shown success in dealing with complex multimodal datasets, being effectively applied to generate image descriptions (Hossain et al., 2021), predict injuries in sports (Song et al., 2021) and disease detection from medical datasets (Khan et al., 2020; McKenzie et al., 2020; Fan et al., 2021). Recently, several studies were published using DL for trait prediction using high throughput plant phenotyping images as input. These take advantage of convolutional neural networks for extracting the spectral features of the leaf reflectance for disease classification (Picon et al., 2018; Nagasubramanian et al., 2019), wheat spike segmentation and counting (Misra et al., 2020), and QTLs related to root architecture traits (Pound et al., 2017). A wide diversity of DL architectures that have been developed to address problems in other areas of research can be adapted for use in crop breeding. For example, multimodal DL models are composed of multiple models, each trained using a single input type (e.g., rainfall, soil measurements, genetic data, hyperspectral imagery), or a single model trained on concatenated multimodal data (Figure 2). The different modalities contribute to enrich the available features for model learning, contributing to an improved final prediction (Baltrusaitis et al., 2019; Khaki et al., 2019; Gadiraju et al., 2020; Hoang Trong et al., 2020; Maimaitijiang et al., 2020). The use of multimodal models and other DL architectures, such as recurrent neural networks and graph neural networks, are still largely unexplored in genotype to phenotype predictions, but present a powerful alternative to traditional statistical methods.
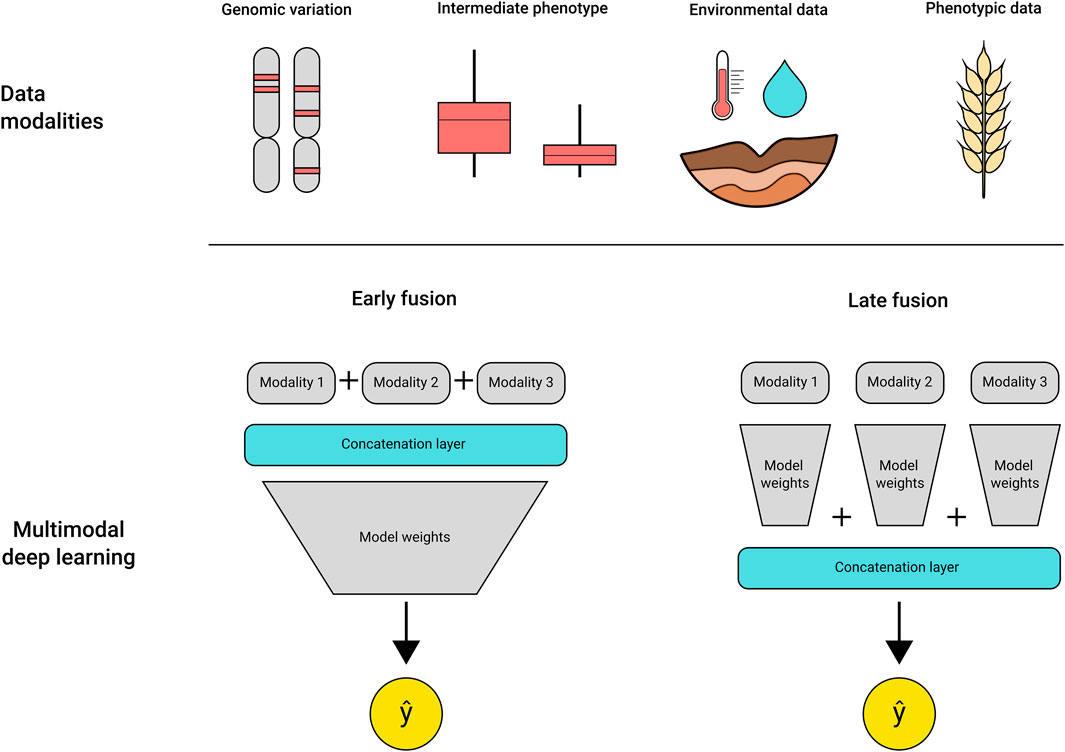
FIGURE 2. Multimodal DL models can use a variety of inputs as training data, as shown in “data modalities” at the top. The different data types can be fused early in the training process by using a concatenation layer that integrates the multiple data types in a single file per sample, called early fusion. Alternatively, the concatenation step may be deployed at the final layers of the model, merging the weights from each specialised module before outputting a prediction.
Challenges in deploying DL models emerge mainly from plant phenotypic plasticity, as plants present a wide range of phenotypes depending on the environmental conditions (Nicotra et al., 2010). The effectiveness of ML/DL models also depends on appropriately tuning the model hyperparameters to the target task, with many packages designed to assist tuning such as Optuna and HyperOpt (Bergstra et al., 2013; Akiba et al., 2019). Nonetheless, tuning hyperparameters for DL models tends to be more computationally intensive than conventional ML. Below we list some challenges that arise from phenotype variability that may need to be addressed to effectively use ML for genotype to phenotype prediction (Figure 3).
i) Consistent protocol for data collection and processing during training and model deployment (Hagiwara et al., 2020; Mårtensson et al., 2020). Because DL models learn directly from the dataset, varying the methods for data collection and processing can add noise causing the model to underperform. It is important to maintain a consistent protocol for data handling, as well as regular assessment to ensure that the model remains suitable for the task;
ii) Avoid the curse of dimensionality (Altman and Krzywinski, 2018). High throughput phenotyping platforms, hyperspectral cameras and pangenome assemblies can generate massive amounts of data, making it harder for the model to define which data points are representative of the trait. Feature selection algorithms can assist in selecting the most representative data subset for training the DLmodel (Cen et al., 2016; Khaki and Wang, 2019);
iii) Data imbalance. Scarcity of samples representing a specific genotype or environment can introduce bias to the model. This can be addressed by employing sampling methods such as over and under sampling, or generative DL to build an artificially augmented dataset (Radford et al., 2015);
iv) Changes in environmental conditions (interannual weather variation, differences in agroecological zone and crop management practices) may impact model performance due to plant phenotypic plasticity. Environmental effects on the phenotype should be considered when defining the model validation and future applicability, and can be addressed by collecting data that mimic the conditions that the model will see when predicting the phenotype (Montesinos-López O. A. et al., 2018; Khaki et al., 2019; Shook et al., 2021).
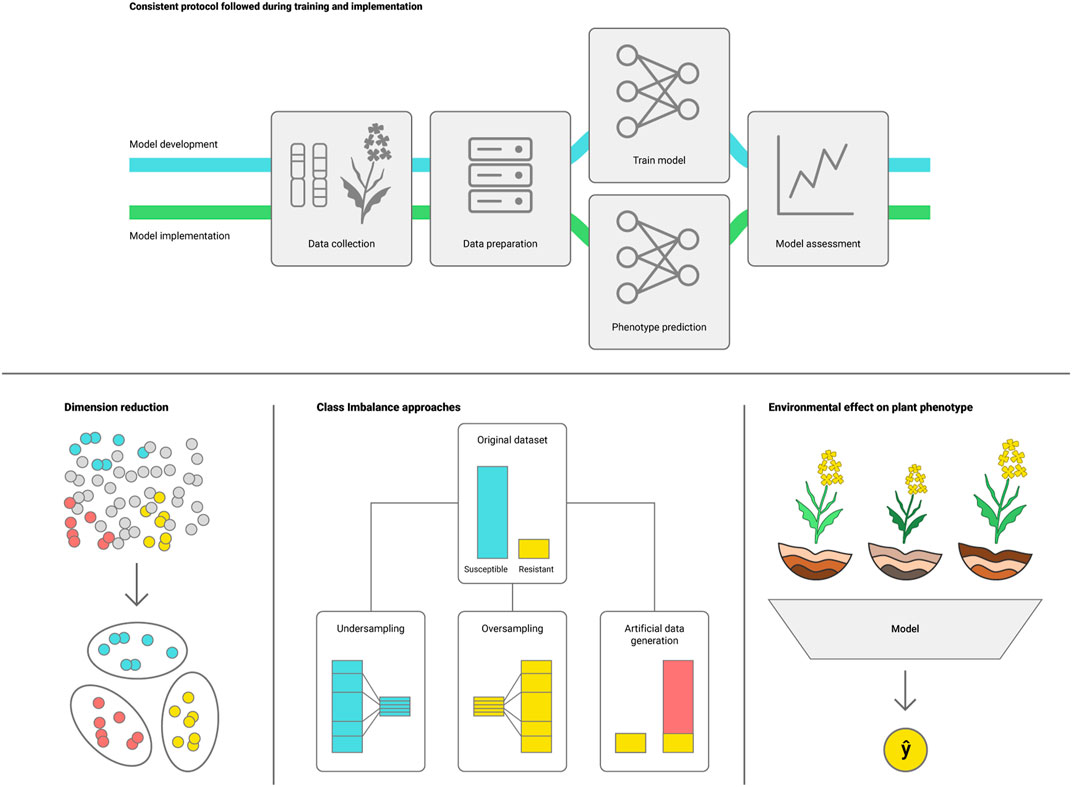
FIGURE 3. Potential challenges for deploying DL models for plant phenotype prediction: I) consistent protocols for development and implementation, ii) reduce dataset dimensionality, which may conceal valuable information, iii) reduce class representation imbalance and iv) account for environmental variations between conditions for plant growth in the training and deployment datasets.
As crop research advances, it is advisable that the models are updated with new data collected. ML and DL models are under intense research, with improvements and novel architectures being published regularly. Selecting the most appropriate architecture and addressing the challenges above will depend on the goal of the project and the data availability. Some data types benefit from complex DL models that draw complex non-linear relationships from the data, while others can achieve high performance using simpler tree-based models with greater potential for interpretability of the results. Nonetheless, the successful implementation of ML/DL workflows in crop breeding will require the familiarisation of breeders and other stakeholders to the models capabilities, so they can be used appropriately.
Data-Driven Breeding Requires Structured Datasets
The prediction accuracy of machine learning models is intrinsically related to the quality of the dataset employed during development (LeCun et al., 2015). ML models calibrate their internal weights based on the data provided, requiring a representative dataset with accurate annotation (Sasidharan Nair and Vihinen, 2013; Schaafsma and Vihinen, 2018). Consequently, a frequent challenge for training robust ML models is the lack of appropriate datasets with enough data points and sample variability. It has been suggested that the scarcity of plant phenotype datasets is because these are either inefficiently shared with the community due to missing information and difficulties to find the public repository in which it is stored (Zamir, 2013; Lobet, 2017), or because the data is maintained in data silos with restricted access (Bayer and Edwards, 2020). A few international consortia such as the AgBioData (Harper et al., 2018) and Breeding API (Selby et al., 2019) are making an effort to share and transform breeding datasets to become more findable, accessible, interoperable and reusable (FAIR) (Wilkinson et al., 2016). However, a centralised platform for hosting and managing phenotypic datasets is needed to make data more widely available, similar to approaches used to share genomic data. Another aspect that prevents researchers from employing previously published datasets is the lack of standardised metadata descriptions, encompassing experimental design, data collection protocol, field management, environmental variables, and other information. The observed plant phenotype is the result of the conditions that the plant experiences, thus reusing previously published data requires that all the factors influencing the target trait are described for the user. The Minimum information about a plant phenotype project (MIAPPE) (Papoutsoglou et al., 2020) offers a resource to guide researchers on how to annotate metadata to increase the usability and interoperability of the datasets, but the development and application of suitable standards need to be expanded to support the growth in ML/DL applications.
A large number of high quality phenotypic datasets are generated each year during field trials led by industry, government or academic groups, however these often have controlled access to protect industry knowledge. An alternative approach to protect sensitive information while supporting collaboration towards data-driven breeding is the establishment of federated learning cohorts (Konečný et al., 2016). Within these, each participant institution trains the model with its own dataset and shares the updated model peer to peer or to a centralised server that will aggregate the models weights (Figure 4). The updated model parameters improve the baseline model, that is then shared among institutions (Yang et al., 2019). Federated learning has seen increasing application in digital health, where data sensitivity is a major issue (Brisimi et al., 2018; Lee et al., 2018; Rieke et al., 2020).
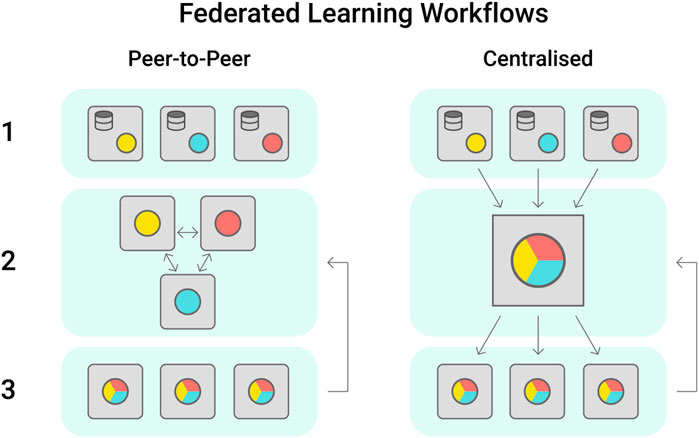
FIGURE 4. Federated learning workflows are most proposed in the peer-to-peer or centralised configurations. In Peer-to-peer, each institution trains a model locally using their own dataset, sharing the model trained weights with its partners. In this workflow, the models are aggregated by each institution as preferred. In the centralised scheme, the trained models are shared with one centralised cohort, which will aggregate the models received and share a singular version with the stakeholders.
Explainable (Interpretable) Machine Learning for Genotype to Phenotype Models
The increasing use of large scale data, such as genomic variation and high throughput imagery, have produced studies with highly accurate ML and non-ML prediction models (Montesinos-López et al., 2021b). However, building models capable of predicting a biological output can only be seen as one of the goals. Models should also attempt to address biological questions, which requires an understanding of how the models make predictions. The use of ML as a method to ask biological questions presents researchers with the problem of model explainability. Explainability in prediction models is a relatively new area for genomic prediction, as the main goal has often been to achieve optimum prediction performance, with model explainability less of a focus. This idea can be represented with GBLUP, one of the most widely implemented genomic selection methods. GBLUP offers researchers accurate prediction of breeding values but low explainability of its predictions, as estimating individual SNP effects can be difficult for genomic prediction datasets due to the “large-p little-n” problem (de Los Campos et al., 2013; Shen et al., 2013). As crop genomic prediction ability advances, it is becoming increasingly important for researchers to explain how a “black box” model has made predictions, to understand the biological implications, justify further research questions and support problem specific model improvement.
For genotype to phenotype prediction in crops, explainability provides the ability to identify important genomic markers and then apply these genomic markers to reduce the size of model inputs required to make further predictions. In Khaki and Wang (2019), analysis of feature importance determined that weather had a larger effect on crop yield than genotype, and models trained on a reduced selection of top ranking input features did not lead to a significant reduction in performance (Khaki and Wang, 2019). Soybean yield prediction models using genotype and other input data identified the importance of features from August-September, which coincides with crop reproduction (Shook et al., 2021). The identification of features from explainable models provides biological understanding and supports results from other methods that identify important features. For example, the application of saliency maps as a post-hoc method to a convolutional neural network for soybean trait prediction identified an overlap with significant SNPs found from a GWAS conducted with the same data (Liu et al., 2019). Model explainability should be a consideration for genomic prediction problems and included as a determining factor for evaluating optimum model performance.
When predicting plant phenotypes from genotype information, the use of an interpretable model offers an opportunity to select high ranking markers as a feature selection strategy, and there is evidence that selecting a subset of important markers can improve the prediction for a given phenotype (Oakey et al., 2016). This is due to the large number of SNPs acting as background noise for prediction, resulting in diminishing returns on performance unless a major proportion of included SNPs are associated with the trait (Pérez-Enciso et al., 2015). A tool such as CGBayesNets can be used to firstly select a sample of features that are informative for phenotype prediction (McGeachie et al., 2014). Harvestman is another tool that selects a representative and non-redundant subset of features with a specific focus on minimising overfitting issues, which is common in high dimensionality prediction tasks (Frisby et al., 2021). The best subset and encoding of features can then be used to train new models. Ensemble methods can also be implemented, where interpretable ML methods can be used for feature selection, and the high ranking features can then be input into another model, such as a DL architecture, to improve predictions (Azodi et al., 2019). A benefit of feature selection is that input feature reduction can reduce the computing resources and time required to train models.
Model interpretation is complex, as definitions of interpretability are variable (Lipton, 2018), and evaluation of these interpretations is non-standardised, making evidence based comparisons between interpretable methods is difficult (Doshi-Velez and Kim, 2017). Rudin (2019) argues that instead of extracting meaning from the ‘black box’ model after training, ML models should be built with interpretability in mind (Rudin, 2019). Less complex ML algorithms, such as decision tree-based models, are inherently interpretable, with inbuilt functions to determine feature importance, however it can be hard to quantify importance between different models as the importance of these features often uses a metric that measures relative feature importance against other input features for a given model (Lundberg et al., 2018). For example, extreme gradient boosting’s (XGBoost) inbuilt functions offer multiple methods of calculating feature importance (weight, cover, gain), each offering different importance scores and ranking of input features, leading to variable interpretation of results depending on the method used. Model-agnostic local explanation methods, such as Shapley additive explanations (SHAP) (Lundberg and Lee, 2017) and Local interpretable model-agnostic explanations (LIME) (Ribeiro et al., 2016) have the potential to overcome this issue due to how the methods consistently and transparently quantify the input’s effect on prediction across most model types (Lundberg et al., 2018). However, this leads back to the original criticism of describing the extraction of meaning post-hoc from the black box as a practice with potential bias [57], the ability to purposely engineer explanations (Slack et al., 2020) and the likelihood of false conclusions being made by inexperienced users (Chromik et al., 2021).
Explainability for genotype to phenotype prediction is a relatively new area in genomic prediction studies, and the interest in explainability and interpretability suggests that new DL algorithms may have enhanced interpretability (Montesinos-López et al., 2021b). This presents a challenge to evaluate new algorithms, as conclusion extracted from interpretable models can be erroneous if the model’s predictive performance is poor (Murdoch et al., 2019). Additionally, interpretable ML extracts associations through the correlation between features and outcomes (Azodi et al., 2020), but are often interpreted with the goal of guiding hypotheses to suggest possible causal relationships from features (Lipton, 2018; Murdoch et al., 2019). Placed in a genotype to phenotype context, this would mean the identification of important genomic regions for phenotype outcome would remain noncausal and associative until further work was conducted to determine causality. One such example of why further work is required is the possibility of an untested factor not included in the genotype data, such as epigenetic or environmental features, that cause both the genomic region and the predicted phenotype to be associated with each other, without the genomic region being the causal feature.
Discussion
ML models have the potential to predict complex phenotypic traits such as yield, biotic, and abiotic stress tolerance due to the way they capture the non-linear relationships between the genetic variation and environmental features extracted from the dataset. The growth of high throughput phenotyping and genotyping will continue to support crop breeding, however we need algorithms capable of handling data at this scale and complexity. Further research into the application of ML/DL models in crop breeding is required as the application of the model is highly dependent on representativeness of the training dataset, which may cause difficulties to apply the model in a real-world setting. Increased efforts to share datasets, or use data augmentation methods may help improve model generalisation. In addition, updating model weights as new datasets become available may also contribute to improve model prediction accuracy. Most genotype to phenotype studies currently lack interpretation of their predictions, which should be addressed to gain insight from the features. As researchers and breeders use advanced data analysis approaches, explainable ML models will help answer biological questions. Explainable MLwill be a key area for genotype to phenotype research, with an increasing focus on harnessing the potential of ML to generate accurate predictions combined with reliable interpretations.
Author Contributions
MD, MG, and RA wrote the manuscript with input and edits from JB, MB, PB, and DE.
Funding
This work was supported by the Australian Government through the Australian Research Council (Projects DP210100296, DP200100762 and DE210100398) and the Grains Research and Development Corporation (Projects 9177539 and 9177591). MG, RA, and MD are supported by Research Training Program scholarships. MD received further support from the Forrest Research Foundation. The funding bodies had no involvement in the study design or its development.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Felipe Casaprima for his assistance with designing the figures.
Abbreviations
DL, Deep Learning; GBLUP, Genomic Best Linear Unbiased Predictions; GS, Genomic Selection; GWAS, Genome-Wide Association Study; LASSO, Least Absolute Shrinkage and Selection Operator; LIME, Local Interpretable Model-agnostic Explanations; MIAPPE, Minimum Information About a Plant Phenotype Experiment; ML, Machine Learning; SHAP, SHapley Additive exPlanations; SNP, Single Nucleotide Polymorphism; RR, Ridge Regression; QTL, Quantitative Trait Loci.
References
Adak, A., Conrad, C., Chen, Y., Wilde, S. C., Murray, S. C., Anderson II, S. L., et al. (2021). Validation of Functional Polymorphisms Affecting maize Plant Height by Unoccupied Aerial Systems (UAS) Discovers Novel Temporal Phenotypes, 11. G3 (Bethesda). doi:10.1093/g3journal/jkab075
Akiba, T., Sano, S., Yanase, T., Ohta, T., and Koyama, M. (2019). “Optuna: A Next-Generation Hyperparameter Optimization Framework,” in Proceedings of the 25th ACM SIGKDD international conference on knowledge discovery & data mining, 2623–2631.
Altman, N., and Krzywinski, M. (2018). The Curse(s) of Dimensionality. Nat. Methods 15, 399–400. doi:10.1038/s41592-018-0019-x
Anderson, S. L., Murray, S. C., Chen, Y., Malambo, L., Chang, A., Popescu, S., et al. (2020). Unoccupied Aerial System Enabled Functional Modeling of maize Height Reveals Dynamic Expression of Loci. Plant Direct 4, e00223. doi:10.1002/pld3.223
Azodi, C. B., Bolger, E., McCarren, A., Roantree, M., de Los Campos, G., and Shiu, S.-H. (2019). Benchmarking Parametric and Machine Learning Models for Genomic Prediction of Complex Traits. G3 (Bethesda) 9, 3691–3702. doi:10.1534/g3.119.400498
Azodi, C. B., Tang, J., and Shiu, S.-H. (2020). Opening the Black Box: Interpretable Machine Learning for Geneticists. Trends Genet. 36, 442–455. doi:10.1016/j.tig.2020.03.005
Baltrusaitis, T., Ahuja, C., and Morency, L.-P. (2019). Multimodal Machine Learning: A Survey and Taxonomy. IEEE Trans. Pattern Anal. Mach. Intell. 41, 423–443. doi:10.1109/TPAMI.2018.2798607
Banerjee, R., Marathi, B., and Singh, M. (2020). Efficient Genomic Selection Using Ensemble Learning and Ensemble Feature Reduction. J. Crop Sci. Biotechnol. 23, 311–323. doi:10.1007/s12892-020-00039-4
Bayer, P. E., and Edwards, D. (2020). Machine Learning in Agriculture: from Silos to Marketplaces. Plant Biotechnol. J. 19, 648–650. doi:10.1111/pbi.13521
Bayer, P. E., Golicz, A. A., Scheben, A., Batley, J., and Edwards, D. (2020). Plant Pan-Genomes Are the New Reference. Nat. Plants 6, 914–920. doi:10.1038/s41477-020-0733-0
Bergstra, J., Yamins, D., and Cox, D. D. (2013). Hyperopt: A python Library for Optimizing the Hyperparameters of Machine Learning Algorithms. Proc. 12th Python Sci. Conf. 13, 20. doi:10.25080/majora-8b375195-003
Brisimi, T. S., Chen, R., Mela, T., Olshevsky, A., Paschalidis, I. C., and Shi, W. (2018). Federated Learning of Predictive Models from Federated Electronic Health Records. Int. J. Med. Inform. 112, 59–67. doi:10.1016/j.ijmedinf.2018.01.007
Cen, H., Lu, R., Zhu, Q., and Mendoza, F. (2016). Nondestructive Detection of Chilling Injury in Cucumber Fruit Using Hyperspectral Imaging with Feature Selection and Supervised Classification. Postharvest Biol. Tech. 111, 352–361. doi:10.1016/j.postharvbio.2015.09.027
Champigny, M. J., Unda, F., Skyba, O., Soolanayakanahally, R. Y., Mansfield, S. D., and Campbell, M. M. (2020). Learning from Methylomes: Epigenomic Correlates of Populus Balsamifera Traits Based on Deep Learning Models of Natural DNA Methylation. Plant Biotechnol. J. 18, 1361–1375. doi:10.1111/pbi.13299
Chen, M. L., Doddi, A., Royer, J., Freschi, L., Schito, M., Ezewudo, M., et al. (2019). Beyond Multidrug Resistance: Leveraging Rare Variants with Machine and Statistical Learning Models in Mycobacterium tuberculosis Resistance Prediction. EBioMedicine 43, 356–369. doi:10.1016/j.ebiom.2019.04.016
Chromik, M., Eiband, M., Buchner, F., Krüger, A., and Butz, A. (2021). “I Think I Get Your point, AI! the Illusion of Explanatory Depth in Explainable AI,” in 26th International Conference on Intelligent User Interfaces, New York, NY, USA (ACM), 307–317. doi:10.1145/3397481.3450644
Cooper, M., Messina, C. D., Podlich, D., Totir, L. R., Baumgarten, A., Hausmann, N. J., et al. (2014). Predicting the Future of Plant Breeding: Complementing Empirical Evaluation with Genetic Prediction. Crop Pasture Sci. 65, 311. doi:10.1071/CP14007
Crossa, J., Martini, J. W. R., Gianola, D., Pérez-Rodríguez, P., Jarquin, D., Juliana, P., et al. (2019). Deep Kernel and Deep Learning for Genome-Based Prediction of Single Traits in Multienvironment Breeding Trials. Front. Genet. 10, 1168. doi:10.3389/fgene.2019.01168
Crossa, J., Pérez-Rodríguez, P., Cuevas, J., Montesinos-López, O., Jarquín, D., de Los Campos, G., et al. (2017). Genomic Selection in Plant Breeding: Methods, Models, and Perspectives. Trends Plant Sci. 22, 961–975. doi:10.1016/j.tplants.2017.08.011
Cuevas, J., Crossa, J., Soberanis, V., Pérez‐Elizalde, S., Pérez‐Rodríguez, P., Campos, G. d. l., et al. (2016). Genomic Prediction of Genotype × Environment Interaction Kernel Regression Models. Plant Genome 9. doi:10.3835/plantgenome2016.03.0024
Danilevicz, M. F., Tay Fernandez, C. G., Marsh, J. I., Bayer, P. E., and Edwards, D. (2020). Plant Pangenomics: Approaches, Applications and Advancements. Curr. Opin. Plant Biol. 54, 18–25. doi:10.1016/j.pbi.2019.12.005
de Abreu E Lima, F., Willmitzer, L., and Nikoloski, Z. (2018). Classification-driven Framework to Predict maize Hybrid Field Performance from Metabolic Profiles of Young Parental Roots. PLoS ONE 13, e0196038. doi:10.1371/journal.pone.0196038
De Castro, A. I., Ehsani, R., Ploetz, R., Crane, J. H., and Abdulridha, J. (2015). Optimum Spectral and Geometric Parameters for Early Detection of laurel Wilt Disease in Avocado. Remote Sensing Environ. 171, 33–44. doi:10.1016/j.rse.2015.09.011
de Los Campos, G., Hickey, J. M., Pong-Wong, R., Daetwyler, H. D., and Calus, M. P. L. (2013). Whole-genome Regression and Prediction Methods Applied to Plant and Animal Breeding. Genetics 193, 327–345. doi:10.1534/genetics.112.143313
Doshi-Velez, F., and Kim, B. (2017). Towards A Rigorous Science of Interpretable Machine Learning, 08608. arXiv:1702.
Drouin, A., Letarte, G., Raymond, F., Marchand, M., Corbeil, J., and Laviolette, F. (2019). Interpretable Genotype-To-Phenotype Classifiers with Performance Guarantees. Sci. Rep. 9, 4071. doi:10.1038/s41598-019-40561-2
Fan, Z., Li, J., Zhang, L., Zhu, G., Li, P., Lu, X., et al. (2021). U-net Based Analysis of MRI for Alzheimer's Disease Diagnosis. Neural Comput. Applic 33, 13587–13599. doi:10.1007/s00521-021-05983-y
Frisby, T. S., Baker, S. J., Marçais, G., Hoang, Q. M., Kingsford, C., and Langmead, C. J. (2021). HARVESTMAN: a Framework for Hierarchical Feature Learning and Selection from Whole Genome Sequencing Data. BMC Bioinformatics 22, 174. doi:10.1186/s12859-021-04096-6
Gadiraju, K. K., Ramachandra, B., Chen, Z., and Vatsavai, R. R. (2020). “Multimodal Deep Learning Based Crop Classification Using Multispectral and Multitemporal Satellite Imagery,” in Proceedings of the 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, New York, NY, USA (ACM), 3234–3242. doi:10.1145/3394486.3403375
Gaffney, J., Schussler, J., Löffler, C., Cai, W., Paszkiewicz, S., Messina, C., et al. (2015). Industry-Scale Evaluation of Maize Hybrids Selected for Increased Yield in Drought-Stress Conditions of the US Corn Belt. Crop Sci. 55, 1608–1618. doi:10.2135/cropsci2014.09.0654
Gao, L., Gonda, I., Sun, H., Ma, Q., Bao, K., Tieman, D. M., et al. (2019). The Tomato Pan-Genome Uncovers New Genes and a Rare Allele Regulating Fruit Flavor. Nat. Genet. 51, 1044–1051. doi:10.1038/s41588-019-0410-2
Golicz, A. A., Bayer, P. E., Barker, G. C., Edger, P. P., Kim, H., Martinez, P. A., et al. (2016). The Pangenome of an Agronomically Important Crop Plant Brassica oleracea. Nat. Commun. 7, 13390. doi:10.1038/ncomms13390
Golicz, A. A., Bayer, P. E., Bhalla, P. L., Batley, J., and Edwards, D. (2020). Pangenomics Comes of Age: from Bacteria to Plant and Animal Applications. Trends Genet. 36, 132–145. doi:10.1016/j.tig.2019.11.006
Grinberg, N. F., Orhobor, O. I., and King, R. D. (2020). An Evaluation of Machine-Learning for Predicting Phenotype: Studies in Yeast, rice, and Wheat. Mach. Learn. 109, 251–277. doi:10.1007/s10994-019-05848-5
Hagiwara, A., Fujita, S., Ohno, Y., and Aoki, S. (2020). Variability and Standardization of Quantitative Imaging. Invest. Radiol. 55, 601–616. doi:10.1097/RLI.0000000000000666
Harper, L., Campbell, J., Cannon, E. K. S., Jung, S., Poelchau, M., Walls, R., et al. (2018). AgBioData Consortium Recommendations for Sustainable Genomics and Genetics Databases for Agriculture. Database (Oxford) 2018. doi:10.1093/database/bay088
Hasan, M. M., Chopin, J. P., Laga, H., and Miklavcic, S. J. (2018). Detection and Analysis of Wheat Spikes Using Convolutional Neural Networks. Plant Methods 14, 100. doi:10.1186/s13007-018-0366-8
Heffner, E. L., Sorrells, M. E., and Jannink, J.-L. (2009). Genomic Selection for Crop Improvement. Crop Sci. 49, 1–12. doi:10.2135/cropsci2008.08.0512
Heslot, N., Yang, H.-P., Sorrells, M. E., and Jannink, J.-L. (2012). Genomic Selection in Plant Breeding: A Comparison of Models. Crop Sci. 52, 146–160. doi:10.2135/cropsci2011.06.0297
Hoang Trong, V., Gwang-hyun, Y., Thanh Vu, D., and Jin-young, K. (2020). Late Fusion of Multimodal Deep Neural Networks for Weeds Classification. Comput. Elect. Agric. 175, 105506. doi:10.1016/j.compag.2020.105506
Hossain, M. Z., Sohel, F., Shiratuddin, M. F., Laga, H., and Bennamoun, M. (2021). Text to Image Synthesis for Improved Image Captioning. IEEE Access 9, 64918–64928. doi:10.1109/ACCESS.2021.3075579
Hu, H., Scheben, A., and Edwards, D. (2018). Advances in Integrating Genomics and Bioinformatics in the Plant Breeding Pipeline. Agriculture 8, 75. doi:10.3390/agriculture8060075
Hurgobin, B., and Edwards, D. (2017). SNP Discovery Using a Pangenome: Has the Single Reference Approach Become Obsolete? Biology 6, 21. doi:10.3390/biology6010021
Jeong, S., Kim, J.-Y., and Kim, N. (2020). GMStool: GWAS-Based Marker Selection Tool for Genomic Prediction from Genomic Data. Sci. Rep. 10, 19653. doi:10.1038/s41598-020-76759-y
Jin, M., Liu, H., He, C., Fu, J., Xiao, Y., Wang, Y., et al. (2016). Maize Pan-Transcriptome Provides Novel Insights into Genome Complexity and Quantitative Trait Variation. Sci. Rep. 6, 18936. doi:10.1038/srep18936
Jubair, S., and Domaratzki, M. (2019). “Ensemble Supervised Learning for Genomic Selection,” in 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) (IEEE), 1993–2000. doi:10.1109/BIBM47256.2019.8982998
Juliana, P., Singh, R. P., Poland, J., Mondal, S., Crossa, J., Montesinos‐López, O. A., et al. (2018). Prospects and Challenges of Applied Genomic Selection-A New Paradigm in Breeding for Grain Yield in Bread Wheat. Plant Genome 11, 180017. doi:10.3835/plantgenome2018.03.0017
Khaki, S., Wang, L., and Archontoulis, S. V. (2019). A CNN-RNN Framework for Crop Yield Prediction. Front. Plant Sci. 10, 1750. doi:10.3389/fpls.2019.01750
Khaki, S., and Wang, L. (2019). Crop Yield Prediction Using Deep Neural Networks. Front. Plant Sci. 10, 621. doi:10.3389/fpls.2019.00621
Khan, M. A., Ashraf, I., Alhaisoni, M., Damaševičius, R., Scherer, R., Rehman, A., et al. (2020). Multimodal Brain Tumor Classification Using Deep Learning and Robust Feature Selection: A Machine Learning Application for Radiologists. Diagnostics 10, 565. doi:10.3390/diagnostics10080565
Konečný, J., McMahan, H. B., Ramage, D., and Richtárik, P. (2016). Federated Optimization: Distributed Machine Learning for On-Device Intelligence. CoRR abs/1610.02527.
Lebedev, V. G., Lebedeva, T. N., Chernodubov, A. I., and Shestibratov, K. A. (2020). Genomic Selection for forest Tree Improvement: Methods, Achievements and Perspectives. Forests 11, 1190. doi:10.3390/f11111190
LeCun, Y., Bengio, Y., and Hinton, G. (2015). Deep Learning. Nature 521, 436–444. doi:10.1038/nature14539
Lee, J., Sun, J., Wang, F., Wang, S., Jun, C.-H., and Jiang, X. (2018). Privacy-Preserving Patient Similarity Learning in a Federated Environment: Development and Analysis. JMIR Med. Inform. 6, e20. doi:10.2196/medinform.7744
Lipton, Z. C. (2018). The Mythos of Model Interpretability. Queue 16, 31–57. doi:10.1145/3236386.3241340
Liu, Y., Wang, D., He, F., Wang, J., Joshi, T., and Xu, D. (2019). Phenotype Prediction and Genome-wide Association Study Using Deep Convolutional Neural Network of Soybean. Front. Genet. 10, 1091. doi:10.3389/fgene.2019.01091
Lobet, G. (2017). Image Analysis in Plant Sciences: Publish Then Perish. Trends Plant Sci. 22, 559–566. doi:10.1016/j.tplants.2017.05.002
Lundberg, S., and Lee, S.-I. (2017). A Unified Approach to Interpreting Model Predictions. Adv. Neural Inf. Process. Syst. 30, 4765–4774.
Lundberg, S. M., Erion, G. G., and Lee, S.-I. (2018). Consistent Individualized Feature Attribution for Tree Ensembles. arXiv:1802.03888.
Ma, W., Qiu, Z., Song, J., Li, J., Cheng, Q., Zhai, J., et al. (2018). A Deep Convolutional Neural Network Approach for Predicting Phenotypes from Genotypes. Planta 248, 1307–1318. doi:10.1007/s00425-018-2976-9
Maimaitijiang, M., Sagan, V., Sidike, P., Hartling, S., Esposito, F., and Fritschi, F. B. (2020). Soybean Yield Prediction from UAV Using Multimodal Data Fusion and Deep Learning. Remote Sensing Environ. 237, 111599. doi:10.1016/j.rse.2019.111599
Marino, S., and Alvino, A. (2020). Agronomic Traits Analysis of Ten Winter Wheat Cultivars Clustered by UAV-Derived Vegetation Indices. Remote Sensing 12, 249. doi:10.3390/rs12020249
Mårtensson, G., Ferreira, D., Granberg, T., Cavallin, L., Oppedal, K., Padovani, A., et al. (2020). The Reliability of a Deep Learning Model in Clinical Out-Of-Distribution MRI Data: A Multicohort Study. Med. Image Anal. 66, 101714. doi:10.1016/j.media.2020.101714
Mason, A. S., Higgins, E. E., Snowdon, R. J., Batley, J., Stein, A., Werner, C., et al. (2017). A User Guide to the Brassica 60K Illumina Infinium SNP Genotyping Array. Theor. Appl. Genet. 130, 621–633. doi:10.1007/s00122-016-2849-1
McGeachie, M. J., Chang, H.-H., and Weiss, S. T. (2014). CGBayesNets: Conditional Gaussian Bayesian Network Learning and Inference with Mixed Discrete and Continuous Data. Plos Comput. Biol. 10, e1003676. doi:10.1371/journal.pcbi.1003676
McKenzie, E. M., Santhanam, A., Ruan, D., O'Connor, D., Cao, M., and Sheng, K. (2020). Multimodality Image Registration in the Head‐and‐neck Using a Deep Learning‐derived Synthetic CT as a Bridge. Med. Phys. 47, 1094–1104. doi:10.1002/mp.13976
Meuwissen, T. H. E., Hayes, B. J., and Goddard, M. E. (2001). Prediction of Total Genetic Value Using Genome-wide Dense Marker Maps. Genetics 157, 1819–1829. doi:10.1093/genetics/157.4.1819
Misra, T., Arora, A., Marwaha, S., Chinnusamy, V., Rao, A. R., Jain, R., et al. (2020). SpikeSegNet-a Deep Learning Approach Utilizing Encoder-Decoder Network with Hourglass for Spike Segmentation and Counting in Wheat Plant from Visual Imaging. Plant Methods 16, 40. doi:10.1186/s13007-020-00582-9
Monroe, J. G., Arciniegas, J. P., Moreno, J. L., Sánchez, F., Sierra, S., Valdes, S., et al. (2020). The Lowest Hanging Fruit: Beneficial Gene Knockouts in Past, Present, and Future Crop Evolution. Curr. Plant Biol. 24, 100185. doi:10.1016/j.cpb.2020.100185
Montenegro, J. D., Golicz, A. A., Bayer, P. E., Hurgobin, B., Lee, H., Chan, C. K. K., et al. (2017). The Pangenome of Hexaploid Bread Wheat. Plant J. 90, 1007–1013. doi:10.1111/tpj.13515
Montesinos‐López, O. A., Montesinos‐López, A., Hernandez‐Suarez, C. M., Barrón‐López, J. A., and Crossa, J. (2021a). Deep‐learning Power and Perspectives for Genomic Selection. Plant Genome 14 (3), e20122. doi:10.1002/tpg2.20122
Montesinos-López, A., Montesinos-López, O. A., Gianola, D., Crossa, J., and Hernández-Suárez, C. M. (2018a). Multi-environment Genomic Prediction of Plant Traits Using Deep Learners with Dense Architecture. G3 (Bethesda) 8, 3813–3828. doi:10.1534/g3.118.200740
Montesinos-López, O. A., Montesinos-López, A., Crossa, J., Gianola, D., Hernández-Suárez, C. M., and Martín-Vallejo, J. (2018b). Multi-trait, Multi-Environment Deep Learning Modeling for Genomic-Enabled Prediction of Plant Traits. G3 (Bethesda) 8, 3829–3840. doi:10.1534/g3.118.200728
Montesinos-López, O. A., Montesinos-López, A., Pérez-Rodríguez, P., Barrón-López, J. A., Martini, J. W. R., Fajardo-Flores, S. B., et al. (2021b). A Review of Deep Learning Applications for Genomic Selection. BMC Genomics 22, 19. doi:10.1186/s12864-020-07319-x
Morrell, P. L., Buckler, E. S., and Ross-Ibarra, J. (2011). Crop Genomics: Advances and Applications. Nat. Rev. Genet. 13, 85–96. doi:10.1038/nrg3097
Murdoch, W. J., Singh, C., Kumbier, K., Abbasi-Asl, R., and Yu, B. (2019). Definitions, Methods, and Applications in Interpretable Machine Learning. Proc. Natl. Acad. Sci. U.S.A. 116, 22071–22080. doi:10.1073/pnas.1900654116
Nagasubramanian, K., Jones, S., Sarkar, S., Singh, A. K., Singh, A., and Ganapathysubramanian, B. (2018). Hyperspectral Band Selection Using Genetic Algorithm and Support Vector Machines for Early Identification of Charcoal Rot Disease in Soybean Stems. Plant Methods 14, 86. doi:10.1186/s13007-018-0349-9
Nagasubramanian, K., Jones, S., Singh, A. K., Sarkar, S., Singh, A., and Ganapathysubramanian, B. (2019). Plant Disease Identification Using Explainable 3D Deep Learning on Hyperspectral Images. Plant Methods 15, 98. doi:10.1186/s13007-019-0479-8
Nair, P. S., and Vihinen, M. (2013). VariBench: a Benchmark Database for Variations. Hum. Mutat. 34, 42–49. doi:10.1002/humu.22204
Naito, H., Ogawa, S., Valencia, M. O., Mohri, H., Urano, Y., Hosoi, F., et al. (2017). Estimating rice Yield Related Traits and Quantitative Trait Loci Analysis under Different Nitrogen Treatments Using a Simple tower-based Field Phenotyping System with Modified Single-Lens Reflex Cameras. ISPRS J. Photogrammetry Remote Sensing 125, 50–62. doi:10.1016/j.isprsjprs.2017.01.010
Nicotra, A. B., Atkin, O. K., Bonser, S. P., Davidson, A. M., Finnegan, E. J., Mathesius, U., et al. (2010). Plant Phenotypic Plasticity in a Changing Climate. Trends Plant Sci. 15, 684–692. doi:10.1016/j.tplants.2010.09.008
Nuccio, M. L., Paul, M., Bate, N. J., Cohn, J., and Cutler, S. R. (2018). Where Are the Drought Tolerant Crops? an Assessment of More Than Two Decades of Plant Biotechnology Effort in Crop Improvement. Plant Sci. 273, 110–119. doi:10.1016/j.plantsci.2018.01.020
Oakey, H., Cullis, B., Thompson, R., Comadran, J., Halpin, C., and Waugh, R. (2016). Genomic Selection in Multi-Environment Crop Trials. G3 (Bethesda) 6, 1313–1326. doi:10.1534/g3.116.027524
Ornella, L., Pérez, P., Tapia, E., González-Camacho, J. M., Burgueño, J., Zhang, X., et al. (2014). Genomic-enabled Prediction with Classification Algorithms. Heredity 112, 616–626. doi:10.1038/hdy.2013.144
Papoutsoglou, E. A., Faria, D., Arend, D., Arnaud, E., Athanasiadis, I. N., Chaves, I., et al. (2020). Enabling Reusability of Plant Phenomic Datasets with MIAPPE 1.1. New Phytol. 227, 260–273. doi:10.1111/nph.16544
Pauli, D., Andrade-Sanchez, P., Carmo-Silva, A. E., Gazave, E., French, A. N., Heun, J., et al. (2016). Field-Based High-Throughput Plant Phenotyping Reveals the Temporal Patterns of Quantitative Trait Loci Associated with Stress-Responsive Traits in Cotton. G3 (Bethesda) 6, 865–879. doi:10.1534/g3.115.023515
Pérez, P., Campos, G., Crossa, J., and Gianola, D. (2010). Genomic‐Enabled Prediction Based on Molecular Markers and Pedigree Using the Bayesian Linear Regression Package in R. Plant Genome 3, 106–116. doi:10.3835/plantgenome2010.04.0005
Pérez-Enciso, M., Rincón, J. C., and Legarra, A. (2015). Sequence- vs. Chip-Assisted Genomic Selection: Accurate Biological Information Is Advised. Genet. Sel. Evol. 47, 43. doi:10.1186/s12711-015-0117-5
Pérez-Rodríguez, P., Gianola, D., González-Camacho, J. M., Crossa, J., Manès, Y., and Dreisigacker, S. (2012). Comparison between Linear and Non-parametric Regression Models for Genome-Enabled Prediction in Wheat. G3 (Bethesda) 2, 1595–1605. doi:10.1534/g3.112.003665
Picon, A., Alvarez-Gila, A., Seitz, M., Ortiz-Barredo, A., Echazarra, J., and Johannes, A. (2019). Deep Convolutional Neural Networks for mobile Capture Device-Based Crop Disease Classification in the Wild. Comput. Elect. Agric. 161, 280–290. doi:10.1016/j.compag.2018.04.002
Pound, M. P., Atkinson, J. A., Townsend, A. J., Wilson, M. H., Griffiths, M., Jackson, A. S., et al. (2017). Deep Machine Learning Provides State-Of-The-Art Performance in Image-Based Plant Phenotyping. Gigascience 6, 1–10. doi:10.1093/gigascience/gix083
Quirós Vargas, J. J., Zhang, C., Smitchger, J. A., McGee, R. J., and Sankaran, S. (2019). Phenotyping of Plant Biomass and Performance Traits Using Remote Sensing Techniques in Pea (Pisum Sativum, L.). Sensors 19, 2031. doi:10.3390/s19092031
Radford, A., Metz, L., and Chintala, S. (2015). Unsupervised Representation Learning with Deep Convolutional Generative Adversarial Networks. arXiv:1511.06434.
Ribeiro, M. T., Singh, S., and Guestrin, C. (2016). “"Why Should I Trust You?",” in Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, New York, New York, USA (ACM Press), 1135–1144. doi:10.1145/2939672.2939778
Rice, B., and Lipka, A. E. (2019). Evaluation of RR‐BLUP Genomic Selection Models that Incorporate Peak Genome‐Wide Association Study Signals in Maize and Sorghum. Plant Genome 12, 180052. doi:10.3835/plantgenome2018.07.0052
Rieke, N., Hancox, J., Li, W., Milletarì, F., Roth, H. R., Albarqouni, S., et al. (2020). The Future of Digital Health with Federated Learning. Npj Digit. Med. 3, 119. doi:10.1038/s41746-020-00323-1
Rudin, C. (2019). Stop Explaining Black Box Machine Learning Models for High Stakes Decisions and Use Interpretable Models Instead. Nat. Mach. Intell. 1, 206–215. doi:10.1038/s42256-019-0048-x
Rutkoski, J., Poland, J., Mondal, S., Autrique, E., Pérez, L. G., Crossa, J., et al. (2016). Canopy Temperature and Vegetation Indices from High-Throughput Phenotyping Improve Accuracy of Pedigree and Genomic Selection for Grain Yield in Wheat. G3 (Bethesda) 6, 2799–2808. doi:10.1534/g3.116.032888
Sandhu, K. S., Lozada, D. N., Zhang, Z., Pumphrey, M. O., and Carter, A. H. (2020). Deep Learning for Predicting Complex Traits in spring Wheat Breeding Program. Front. Plant Sci. 11, 613325. doi:10.3389/fpls.2020.613325
Schaafsma, G. C. P., and Vihinen, M. (2018). Representativeness of Variation Benchmark Datasets. BMC Bioinformatics 19, 461. doi:10.1186/s12859-018-2478-6
Scheben, A., Wolter, F., Batley, J., Puchta, H., and Edwards, D. (2017). Towards CRISPR/Cas Crops - Bringing Together Genomics and Genome Editing. New Phytol. 216, 682–698. doi:10.1111/nph.14702
Scossa, F., Alseekh, S., and Fernie, A. R. (2021). Integrating Multi-Omics Data for Crop Improvement. J. Plant Physiol. 257, 153352. doi:10.1016/j.jplph.2020.153352
Selby, P., Abbeloos, R., Backlund, J. E., Basterrechea Salido, M., Bauchet, G., Benites-Alfaro, O. E., et al. (2019). BrAPI-an Application Programming Interface for Plant Breeding Applications. Bioinformatics 35, 4147–4155. doi:10.1093/bioinformatics/btz190
Shakoor, N., Lee, S., and Mockler, T. C. (2017). High Throughput Phenotyping to Accelerate Crop Breeding and Monitoring of Diseases in the Field. Curr. Opin. Plant Biol. 38, 184–192. doi:10.1016/j.pbi.2017.05.006
Shen, X., Alam, M., Fikse, F., and Rönnegård, L. (2013). A Novel Generalized ridge Regression Method for Quantitative Genetics. Genetics 193, 1255–1268. doi:10.1534/genetics.112.146720
Shook, J., Gangopadhyay, T., Wu, L., Ganapathysubramanian, B., Sarkar, S., and Singh, A. K. (2021). Crop Yield Prediction Integrating Genotype and Weather Variables Using Deep Learning. Plos one 16 (6), e0252402. doi:10.1371/journal.pone.0252402
Slack, D., Hilgard, S., Jia, E., Singh, S., and Lakkaraju, H. (2020). “Fooling LIME and SHAP,” in Proceedings of the AAAI/ACM Conference on AI. New York, NY, USA. Ethics, and SocietyACM, 180–186. doi:10.1145/3375627.3375830
Song, H., xiu-ying Han, x. y., Montenegro-Marin, C. E., and krishnamoorthy, S. (2021). Secure Prediction and Assessment of Sports Injuries Using Deep Learning Based Convolutional Neural Network. J. Ambient Intell. Hum. Comput 12, 3399–3410. doi:10.1007/s12652-020-02560-4
Spindel, J., Begum, H., Akdemir, D., Virk, P., Collard, B., Redoña, E., et al. (2015). Genomic Selection and Association Mapping in rice (Oryza Sativa): Effect of Trait Genetic Architecture, Training Population Composition, Marker Number and Statistical Model on Accuracy of rice Genomic Selection in Elite, Tropical rice Breeding Lines. Plos Genet. 11, e1004982. doi:10.1371/journal.pgen.1004982
Spindel, J. E., Begum, H., Akdemir, D., Collard, B., Redoña, E., Jannink, J.-L., et al. (2016). Genome-wide Prediction Models that Incorporate De Novo GWAS Are a Powerful New Tool for Tropical rice Improvement. Heredity 116, 395–408. doi:10.1038/hdy.2015.113
Tattaris, M., Reynolds, M. P., and Chapman, S. C. (2016). A Direct Comparison of Remote Sensing Approaches for High-Throughput Phenotyping in Plant Breeding. Front. Plant Sci. 7, 1131. doi:10.3389/fpls.2016.01131
Ukrainetz, N. K., and Mansfield, S. D. (2020). Prediction Accuracy of Single-step BLUP for Growth and wood Quality Traits in the lodgepole pine Breeding Program in British Columbia. Tree Genet. Genomes 16, 64. doi:10.1007/s11295-020-01456-w
Voss-Fels, K. P., Cooper, M., and Hayes, B. J. (2019). Accelerating Crop Genetic Gains with Genomic Selection. Theor. Appl. Genet. 132, 669–686. doi:10.1007/s00122-018-3270-8
Wang, S., Wong, D., Forrest, K., Allen, A., Chao, S., Huang, B. E., et al. (2014). Characterization of Polyploid Wheat Genomic Diversity Using a High‐density 90 000 Single Nucleotide Polymorphism Array. Plant Biotechnol. J. 12, 787–796. doi:10.1111/pbi.12183
Wilkinson, M. D., Dumontier, M., Aalbersberg, I. J., Appleton, G., Axton, M., Baak, A., et al. (2016). The FAIR Guiding Principles for Scientific Data Management and Stewardship. Sci. Data 3, 160018. doi:10.1038/sdata.2016.18
Xiong, H., Cao, Z., Lu, H., Madec, S., Liu, L., and Shen, C. (2019). TasselNetv2: In-Field Counting of Wheat Spikes with Context-Augmented Local Regression Networks. Plant Methods 15, 150. doi:10.1186/s13007-019-0537-2
Yang, Q., Liu, Y., Chen, T., and Tong, Y. (2019). Federated Machine Learning. ACM Trans. Intell. Syst. Technol. 10, 1–19. doi:10.1145/3298981
Yin, B., Balvert, M., van der Spek, R. A. A., Dutilh, B. E., Bohté, S., Veldink, J., et al. (2019). Using the Structure of Genome Data in the Design of Deep Neural Networks for Predicting Amyotrophic Lateral Sclerosis from Genotype. Bioinformatics 35, i538–i547. doi:10.1093/bioinformatics/btz369
Zamir, D. (2013). Where Have All the Crop Phenotypes Gone? Plos Biol. 11, e1001595. doi:10.1371/journal.pbio.1001595
Zhou, J., Park, C. Y., Theesfeld, C. L., Wong, A. K., Yuan, Y., Scheckel, C., et al. (2019). Whole-genome Deep-Learning Analysis Identifies Contribution of Noncoding Mutations to Autism Risk. Nat. Genet. 51, 973–980. doi:10.1038/s41588-019-0420-0
Zingaretti, L. M., Gezan, S. A., Ferrão, L. F. V., Osorio, L. F., Monfort, A., Muñoz, P. R., et al. (2020). Exploring Deep Learning for Complex Trait Genomic Prediction in Polyploid Outcrossing Species. Front. Plant Sci. 11, 25. doi:10.3389/fpls.2020.00025
Keywords: machine learning, plant phenotyping, phenotype prediction, plant breeding, big data
Citation: Danilevicz MF, Gill M, Anderson R, Batley J, Bennamoun M, Bayer PE and Edwards D (2022) Plant Genotype to Phenotype Prediction Using Machine Learning. Front. Genet. 13:822173. doi: 10.3389/fgene.2022.822173
Received: 25 November 2021; Accepted: 07 March 2022;
Published: 18 May 2022.
Edited by:
Pasquale Tripodi, Council for Agricultural and Economics Research (CREA), ItalyReviewed by:
Chuang Ma, Northwest A&F University, ChinaOsval Antonio Montesinos-López, Universidad de Colima, Mexico
Copyright © 2022 Danilevicz, Gill, Anderson, Batley, Bennamoun, Bayer and Edwards. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Edwards, ZGF2ZS5lZHdhcmRzQHV3YS5lZHUuYXU=