 Mauricio Flores
Mauricio Flores David W. Goodrich
David W. Goodrich- Roswell Park Comprehensive Cancer Center, Department of Pharmacology and Therapeutics, Buffalo, NY, United States
The retinoblastoma susceptibility gene (RB1) is the first tumor suppressor gene discovered and a prototype for understanding regulatory networks that function in opposition to oncogenic stimuli. More than 3 decades of research has firmly established a widespread and prominent role for RB1 in human cancer. Yet, this gene encodes but one of three structurally and functionally related proteins that comprise the pocket protein family. A central question in the field is whether the additional genes in this family, RBL1 and RBL2, are important tumor suppressor genes. If so, how does their tumor suppressor activity overlap or differ from RB1. Here we revisit these questions by reviewing relevant data from human cancer genome sequencing studies that have been rapidly accumulating in recent years as well as pertinent functional studies in genetically engineered mice. We conclude that RBL1 and RBL2 do have important tumor suppressor activity in some contexts, but RB1 remains the dominant tumor suppressor in the family. Given their similarities, we speculate on why RB1 tumor suppressor activity is unique.
Introduction
The RB1 tumor suppressor gene, discovered and isolated more than 30 years ago (Friend et al., 1986; Fung et al., 1987; Lee et al., 1987), has been the subject of extensive study due to its prominent role in cancer. Mutational loss of RB1 function is the primary cause of the pediatric cancer retinoblastoma. Retinoblastoma either clusters in families as a hereditary susceptibility or it can arise sporadically. As predicted by the “two-hit hypothesis (Knudson, 1971; Comings, 1973),” hereditary retinoblastoma patients inherit a mutationally inactivated RB1 allele from one parent while the remaining allele is inactivated somatically. Sporadic retinoblastoma, on the other hand, is associated with somatic genetic inactivation of both RB1 alleles with the lower probability of two genetic hits accounting for the delayed age at diagnosis for these cases. DNA sequencing of retinoblastoma tumors has demonstrated few, if any, additional genetic alterations beyond those in the RB1 gene (Zhang et al., 2012). This establishes RB1 as a rare example where mutation of a single human gene is sufficient, or at least rate limiting, to cause a human cancer. RB1 loss of function is involved in the development of other cancers as well. Hereditary retinoblastoma patients, for example, have increased risk of subsequent unrelated cancers (Schonfeld et al., 2021), indicating that mutational inactivation of RB1 contributes to tumorigenesis in tissues beyond the retina. Cancer genome sequencing studies have confirmed RB1 is genetically altered in a significant fraction of cases for many common adult cancers (see below). Experimental studies of mice genetically engineered to delete murine Rb1 have confirmed that its loss, often in conjunction with other gene deletions, drives tumorigenesis in multiple tissues (Jacks et al., 1992; Meuwissen et al., 2003; Chen et al., 2004; MacPherson et al., 2004; Zhang et al., 2004; Zhou et al., 2006; Berman et al., 2008).
Soon after the molecular cloning of RB1, it became apparent there are two additional mammalian genes that it shares significant DNA sequence homology with (Ewen et al., 1991; Cobrinik et al., 1993; Hannon et al., 1993). These genes are now named RBL1 (retinoblastoma-like 1) and RBL2 (retinoblastoma-like 2) and their encoded proteins p107 and p130, respectively. Given their structural similarity to RB1, a central question has been whether RBL1 and RBL2 also function as tumor suppressor genes (Wirt and Sage, 2010; Indovina et al., 2013). It has been some years since published evidence relevant to this question has been reviewed, prior to widespread accumulation of human cancer genome sequencing data. The goal of this review is to re-examine evidence relevant to the tumor suppressor activity of the retinoblastoma protein paralogues, with an emphasis on recent DNA sequencing data from human cancer clinical specimens and experimental studies in genetically engineered mice. This evidence supports the hypothesis that all the paralogues can exhibit tumor suppressor activity in some biological contexts. However, the data also highlights a unique and more prominent role for RB1. We will highlight the diversity of retinoblastoma protein paralogue tumor suppressor activity and speculate on why pRb has unique tumor suppressor activity despite its structural and functional similarities to p107 and p130.
Similarities in the Structure of Pocket Protein Paralogues
The pRb, p107, and p130 proteins are structurally related. The amino acid sequences most similar between them comprise a structural domain within the carboxy half of the proteins called the “pocket.” This pocket domain is the distinguishing feature of the family and is required for many of their known cellular and molecular functions. Indeed, human germline mutations conferring susceptibility to retinoblastoma disrupt this pocket structure. The pocket domain is also the most evolutionarily conserved feature among pRb-like orthologues across different species. Pocket proteins have been identified based on structural similarity and studied in diverse species across many eukaryotic phyla, including animals and plants. Functionally analogous proteins, although not structurally similar, have also been identified in yeasts (Hasan et al., 2013). Accompanying contributions to this research topic discuss pocket proteins in some of these other species.
The pocket domain is composed of two highly structured sub-domains separated by a less structured spacer. The structures of the subdomains have been solved by x-ray crystallography (Kim and Cho, 1997; Lee et al., 1998; Kim et al., 2001; Lee et al., 2002; Xiao et al., 2003; Liu and Marmorstein, 2007; Guiley et al., 2015) and both are reminiscent of cyclin box folds. There is also evidence that the amino half of pRb has a pocket-like structure composed of dual cyclin folds (Hassler et al., 2007). Consistent with cyclin folds in other proteins, the pocket protein cyclin folds provide surfaces that mediate key protein-protein interactions important for pocket protein function. Within the pocket domain itself, similarity between p107 and p130 (47% amino acid identity) is greater than between pRb and either p107 or p130 (<21% identity). The multiple dual cyclin folds with pocket proteins provides potential for multiple simultaneous protein interactions. While the pocket is the defining feature of these paralogues, it is important to note that both structured and unstructured regions outside the pocket also make important contributions to intra- and inter-molecular protein interactions and their regulation. In particular, the less structured carboxy terminal tail of pocket proteins also participates in important protein interactions, and the structure of this region is more distinct between pocket proteins (Rubin et al., 2005; Hirschi et al., 2010; Liban et al., 2017). Amino acid sequence divergence in protein regions outside of the pocket domains, including intrinsically disordered regions like the carboxy terminal tail, provide potential for functional diversity among the paralogues.
Similarities in the Function of Pocket Protein Paralogues
All pocket proteins exhibit a predominant nuclear localization, although they have been detected in other cellular compartments as well (Hilgendorf et al., 2013). The multiple protein interaction surfaces present coupled with the absence of demonstrated enzymatic or sequence specific DNA/RNA binding activity suggest pocket proteins function as molecular adaptors mediating protein complex assembly. This hypothesis is consistent with the ability of pocket proteins to interact with a large variety of cellular and viral proteins, a topic reviewed elsewhere (Morris and Dyson, 2001; Goodrich, 2006; Chinnam and Goodrich, 2011; Dyson, 2016). Hundreds of pRb protein interactions have been identified, although only a subset have been well validated. The proteins interacting with p107 and p130 have not been as thoroughly characterized, yet it is evident that the cellular proteome capable of interacting with each of the three paralogues are only partially overlapping. Thus the binding affinity of a given cellular protein for pocket protein paralogues can differ significantly. A key example is the E2F family of sequence specific DNA binding transcription factors. Interaction with E2F transcription factors is the canonical pocket protein function presumed to mediate most of their important cellular effects like cell cycle control (see below). E2F1-3 have a binding preference for pRb while E2F4-5 have a binding preference for p107 and p130. Pocket protein paralogues are thus localized to specific regions of the genome based on the DNA binding specificity of the transcription factors with which they physically interact. In turn, the pocket proteins recruit chromatin regulatory complexes to these genome locations thus influencing gene expression. With some exceptions, the pocket proteins recruit chromatin regulatory complexes that suppress RNA transcription from nearby genes. The partially overlapping protein interactions between paralogues is also reflected in the distinct transcriptional regulatory complexes they participate in and the resulting transcriptional outputs (Black et al., 2003; Litovchick et al., 2007; Pilkinton et al., 2007).
The canonical cellular function of pocket proteins is to negatively regulate the cell division cycle (Goodrich et al., 1991; Zhu et al., 1993; Claudio et al., 1994). It is of interest that the pocket proteins are differentially expressed throughout the cell cycle. RBL2 is most highly expressed in non-proliferating cells, quiescent or differentiated for example. RBL1 expression is highest in proliferating cells as cells enter S phase. RB1 expression is more uniform, expressed in both non-proliferating and proliferating cells throughout the cell cycle. E2F binding sites exist near many genes critical for the cell cycle, and pocket protein mediated silencing of RNA transcription from these genes enforces cell cycle regulation. The differential timing of pocket protein expression suggests they likely cooperate to enforce cell cycle control in different biological contexts. The broader expression of RB1 throughout the cell cycle foreshadows its broader impact on cancer. It has also been noted that RB1 has additional cancer relevant non-canonical functions that likely contribute to its tumor suppressor activity (Dick et al., 2018; Knudsen et al., 2019), a topic to which we return below.
Similarities in the Regulation of Pocket Protein Paralogues
Mitogenic signals normally activate kinases that phosphorylate the pocket proteins, cyclin dependent kinases (CDK) paramount among them. These phosphorylation events can relieve pocket protein mediated cell cycle suppression. Phosphorylation of pRb occurs on as many as 44 different amino acid residues (Hornbeck et al., 2015), although less than half of these are recurrently detected in the majority of phospho-proteome studies. The most commonly phosphorylated and evolutionarily conserved sites are proline directed, consistent with phosphorylation by CDKs or other proline directed kinases. All the pocket proteins exhibit an analogous overall phosphorylation pattern (Figure 1). Phosphorylation sites tend to cluster in the less structured regions of the proteins flanking the cyclin fold subdomains. Experimental evidence demonstrates that CDKs phosphorylate the pocket proteins directly and that this phosphorylation inhibits their cell cycle suppression activity (Connell-Crowley et al., 1997; Hansen et al., 2001; Leng et al., 2002; Tedesco et al., 2002). The structure of some phosphorylated pocket protein domains have been solved, identifying molecular interactions that regulate pocket protein function (Rubin et al., 2005; Burke et al., 2010; Hirschi et al., 2010; Burke et al., 2012). This regulation involves phosphorylation of less structured regions of the proteins that mediate conformational changes influencing both intra- and inter-molecular protein interactions. Intra-molecular interactions can regulate access to pocket contacts essential for E2F binding. Another structural model suggested by Liban et al. (2017) indicates phosphorylation interferes directly with inter-molecular contacts between the intrinsically unstructured carboxy terminus of the pocket proteins and E2F. This model also helps to explain how structural divergence in the carboxy terminal region among paralogues dictates binding preferences for specific E2F family members. Observations such as these confirm the notion that structural divergence between pocket protein paralogues can specify functional diversity.
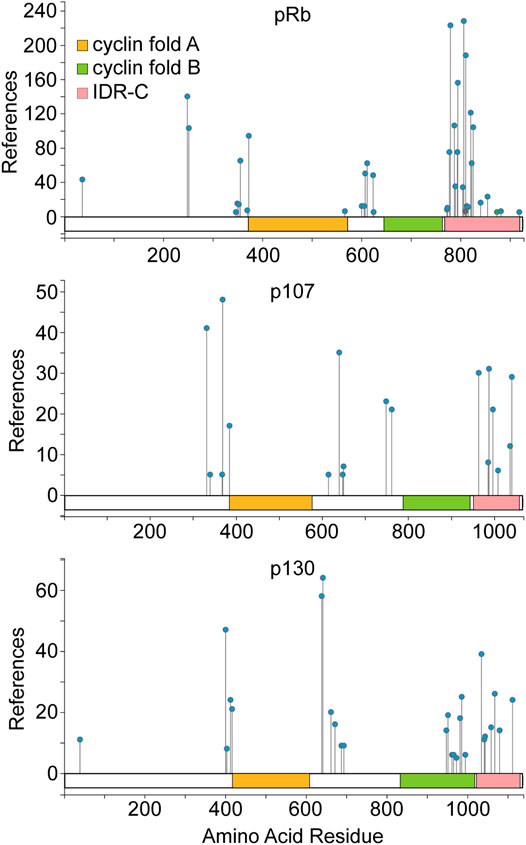
FIGURE 1. Pocket protein domain structure and regulation by phosphorylation. A schematic for each pocket protein paralogue is shown highlighting the amino acid residues comprising the dual cyclin fold pocket (indicated by orange and green shading) for each protein as well as the intrinsically disordered C terminal region. Pocket protein phosphorylation sites identified most commonly in the published literature are indicated, as compiled in the PhosphoSitePlus database from nearly 10,000 journal articles (Hornbeck et al., 2015). The number of independent studies within this compilation that identify a given site is plotted on the Y axis. Phosphorylation patterns of the pocket protein paralogues are broadly similar with phosphorylation sites clustering in less structured regions flanking the cyclin folds. The figure is adapted from PhosphoSitePlus.
Growth suppressive signals can maintain or induce pocket protein mediated cell cycle suppression by activating CDK inhibitory proteins (CDKi). There are at least eight proteins within this larger CDKi family, four encoded by the CDKN2A-D genes that target CDK4/6 enzymes preferentially and four encoded by CDKN1A-C plus the CDKN3 gene that target CDK2 enzymes. CDKi, CDKs and the pocket proteins together comprise the core pocket protein cell cycle regulatory pathway (Figure 2A). Early evidence suggested a quantitative regulatory model for this pathway wherein the balance of CDK and CDKi activity dictates how extensively the pocket proteins are phosphorylated. Accumulation of pocket protein phosphorylation events beyond a threshold would disrupt pocket protein mediated protein interactions causing de-repression of cell cycle genes and stimulating cell cycle progression. This model has been based on the observation that pRb “hyperphosphorylation,” recognized by a shift in protein mobility during SDS-PAGE, correlates with the G1 to S cell cycle phase transition and that ectopic cyclin/CDK expression can drive pRb hyperphosphorylation and the cell cycle (Buchkovich et al., 1989; Chen et al., 1989; DeCaprio et al., 1989). However, CDKs have their own pocket protein phosphorylation site preferences. Sites preferred by the growth factor responsive CDK4/6 enzymes appear to be particularly important for initiating regulation of pocket protein cell cycle suppression (Connell-Crowley et al., 1997; Hansen et al., 2001; Leng et al., 2002; Tedesco et al., 2002; Schade et al., 2019), but cooperation with CDK2 enzymes is likely necessary for hyperphosphorylation (Hatakeyama et al., 1994; Weinberg, 1995).
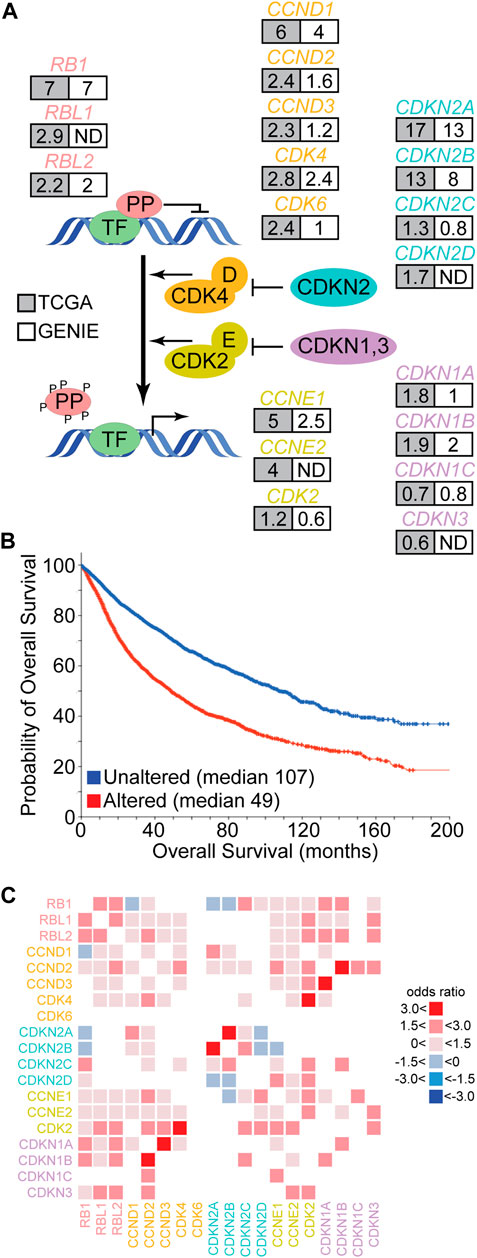
FIGURE 2. Genetic alteration of the pocket protein pathway in human cancer. (A) The schematic depicts the core pocket protein pathway and its regulation by CDK mediated phosphorylation. PP represent the pocket proteins and TF represent transcription factors like the E2F family. Each gene of the core PP pathway is listed with the frequency (%) of genetic alteration in cancer based on TCGA (grey) or GENIE (white) pan-cancer databases. ND indicates the gene was not measured. (B) Overall survival data collected by the TCGA was segregated into those cancers containing genetic alteration of at least one PP pathway member (red) or those cancers without alterations in the PP pathway (blue). The probability of overall survival is plotted over time with median survival times listed. Figure modified from cBioPortal (Cerami et al., 2012; Gao et al., 2013). (C) TCGA pan-cancer data was used to identify significant pairwise co-occurrence (red) or mutually exclusive (blue) interactions between PP pathway members within individual cancer samples (q < 0.05). The shade of red or blue is based on the odds ratio calculated for the given interaction.
More recent data has complicated this quantitative model by demonstrating that pRb is not progressively phosphorylated by CDK4 enzymes in G1 phase. Rather CDK4 appears to phosphorylate pRb once and only once at many different amino acid residues (Narasimha et al., 2014). This generates multiple monophosphorylated pRb isoforms that exhibit distinct protein interaction preferences and transcriptional outputs (Sanidas et al., 2019; Hattori et al., 2014). Once cyclin E/CDK2 is activated in late G1, pRb is more completely functionally inactivated by hyperphosphorylation. This is complicated further by the observation that other kinases can also phosphorylate pRb (Gubern et al., 2016; Inoue et al., 2007; Nair et al., 2009; Nath et al., 2003; Hou et al., 2002; Guo et al., 2005; Dasgupta et al., 2004; Ventura et al., 2018) and that additional pocket protein post-translational modification are apparent (Chan et al., 2001; Nguyen et al., 2004; Markham et al., 2006; Carr et al., 2011; Leduc et al., 2006; Cho et al., 2012; Meng et al., 2016; Ledl et al., 2005; Miwa et al., 2006; Kim et al., 2015; Saddic et al., 2010; Munro et al., 2010). It is thus increasingly clear that pRb is regulated both quantitatively and qualitatively. As p107 and p130 are not as extensively studied, it is not clear whether they are also monophosphorylated by CDK4/6 enzymes. However, it appears likely given the similarity in their overall phosphorylation patterns (Figure 1) (Hornbeck et al., 2015) and the observation that the mechanism of cyclin D CDK4/6 specific docking to pocket proteins is shared among the paralogues (Topacio et al., 2019).
In sum, these observations suggest the pocket protein paralogues share important structural, functional, and regulatory features. Yet each pocket protein paralogue diverges in these features as well, with p107 and p130 more similar to each other than either of them are to pRb. Given the interplay between post-translational modification, protein interactions, and pocket protein activity, pocket proteins are well situated to integrate output from cellular regulatory networks and translate them into changes in gene expression. Differences between pocket protein structure, regulation, and molecular interactions, therefore, likely account for their functional diversity.
Pocket Protein Pathway Genetic Alterations in Primary Human Cancers
Human cancer genome sequencing studies demonstrate that the core pocket protein pathway is altered recurrently across a wide range of cancer types (Figure 2A) (Knudsen et al., 2020). For example, the Cancer Genome Atlas (TCGA) has molecularly characterized more than 10,000 patient samples across 32 cancer types. In this data set, the frequency of alteration for different pocket protein pathway genes ranges from 0.6 to 17% of cases (Gao et al., 2013). The GENIE pan-cancer database contains DNA sequence data from more than 110,000 patients and 100 cancer types. The frequency of genetic alteration for pocket protein pathway genes in this dataset ranges from 0.6 to 13% (Gao et al., 2013). Genetic alterations in the core pocket protein pathway are clinically significant as TCGA pan-cancer data indicates patients whose cancers exhibit a genetic alteration in at least one member of the pathway have an overall survival less than half that of patients whose cancer does not contain an alteration in the pathway (Figure 2B) (Gao et al., 2013). The genetic link between pocket protein pathway genetic alteration and cancer is strengthened further by data from hereditary retinoblastoma patients. These patients inherit one mutationally inactivated RB1 allele and have increased risk for additional cancers unrelated to retinoblastoma later in life (Schonfeld et al., 2021). For example, hereditary retinoblastoma patients have significantly elevated risk of developing sarcomas. In the general population, pocket protein pathway alteration also correlates with reduced overall survival for patients suffering from sarcomas (log rank p < 0.05) (Table 1). However, the correlation between pathway alteration and shorter overall survival is not uniform across cancer types. Assuming these differences are not entirely due to bias in sample size, they suggest pocket protein pathway alterations have non-uniform, cancer context dependent effects.
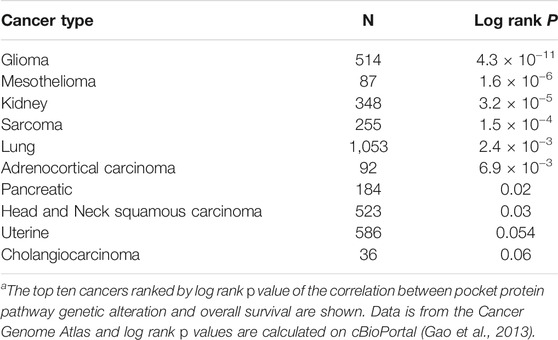
TABLE 1. Pocket protein pathway gene alterations and shorter overall survivala.
The pattern of alterations among pocket protein pathway genes is also informative. Analysis of pairwise interactions indicate that genetic alterations in different pathway members tend to co-occur within samples rather than to exhibit mutual exclusivity (Figure 2C). The only significant mutually exclusive interactions detected in TCGA data are between RB1 and CDKN2A/B or CCND1, between CDKN2D and CDKN2A/B, and between CCNE1 and CDKN2B. In most cases, therefore, genetic alteration of multiple pathway genes is predicted to cooperate in driving cancer development. In some cases, this prediction has been confirmed by experimental studies (see below). It is entirely unclear how this cooperation may occur, but two general and complementary explanations are possible. The simplest potential explanation is that multiple genetic alterations each contribute quantitatively to more complete pathway functional inactivation. Functional redundancies within the pathway, within the pocket proteins themselves for example, may require genetic alteration of multiple members for more complete functional inactivation. It is also possible that genetic alterations in different pathway members yield qualitatively distinct functional effects, and multiple alterations would be required to inactivate different aspects of tumor suppressor functions mediated by the pathway.
The most frequently mutated genes in the pathway are CDKN2A and CDKN2B. As CDKi represent the most proximal regulatory input into the core pathway, this could imply that broad deregulation of CDK4/6 activity and pocket protein phosphorylation is most critical for relieving tumor suppression. However, this hypothesis has important caveats. The high frequency of CDKN2A and CDKN2B alteration is likely biased by unique features of this genetic loci. CDKN2A and CDKN2B map adjacent to each other on chromosome 9, so copy number losses or structural alterations in this region often affect both genes (odds ratio >3.0, q-value < 0.001). Further, CDKN2A encodes two distinct proteins through use of alternative reading frames. One of these reading frames encodes the CDK4/6 CDKi p16 while the other encodes p14ARF. p14ARF is important for maintaining the stability and activity of p53, so genetic loss of CDKN2A function may be selected in cancer because it compromises both the pocket protein and p53 tumor suppressor pathways. There is evidence from clinical specimens indicating loss of these pathways synergize based on the observation that genetic alterations in RB1 and TP53 tend to co-occur in pan-cancer analysis (q < 0.001). There is also abundant experimental evidence indicating these pathways cooperate to control cancer relevant biology, with two examples including cellular senescence (Ben-Porath et al., 2005) and therapeutic resistance (Quintanal-Villalonga et al., 2020). Alteration of both pathways in mouse models often yield a cancer phenotype in situations where alteration of either pathway alone does not (Meuwissen et al., 2003; Zhou et al., 2006). Given the potential bias inherent in the CDKN2A/B locus, it remains unclear whether genetic inactivation of CDKN2A/B is the most efficient means to functionally inactivate pocket protein pathway activity (see below).
The frequency of RB1 alteration (7%) is next highest among the remaining pathway genes, higher than the frequency of RBL1 alteration (2.9%) or RBL2 alteration (2.2%). Interestingly, alterations in RBL1 and RBL2 include a significant number of gene amplifications. While the impact of these amplifications on pathway activity is unclear, if we assume they do not compromise pocket protein function and exclude them from the analysis, then the frequency of debilitating genetic alterations for RBL1 and RBL2 alterations decline to 1.7% and 1.9% respectively. The top 15 cancer types ranked by frequency of debilitating mutation for each pocket protein gene is different (Figure 3). For most cancers the frequency of debilitating RB1 alterations is greater than the frequency of RBL1 or RBL2 alterations. In some cancer types like sarcoma, this discrepancy is marked (RB1 = 25%, RBL1 = 0.4%, RBL2 = 2%). In other cancers like melanoma, the frequency of pocket protein paralogue alterations is comparable (melanoma RB1 = 5.6%, RBL1 = 5.9%, RBL2 = 3%). While the mutation frequency in RB1 is typically greater than for RBL1 or RBL2, there are some cancer subtypes where this is reversed. For example, mucinous adenocarcinomas of the colon exhibit few RB1 alterations (1.6%) but more frequent RBL1 (4.9%) or RBL2 (8.2%) alterations, although case numbers are relatively low. It is also notable that the nature of the genetic alterations affecting the pocket proteins can vary by cancer type. For example, prostate cancer is characterized by pocket protein gene deletions while genetic alterations in uterine cancer are primarily single nucleotide mutations (Figure 3). This likely reflects the nature of genome instability prevalent in a given cancer type but may also indicate selection for different modes of pocket protein pathway inactivation. For example, the fraction of cancers exhibiting gene deletions versus other genetic alterations is generally higher for RB1 than for RBL1 or RBL2, suggesting selective pressure may favor complete functional inactivation of RB1 alleles.
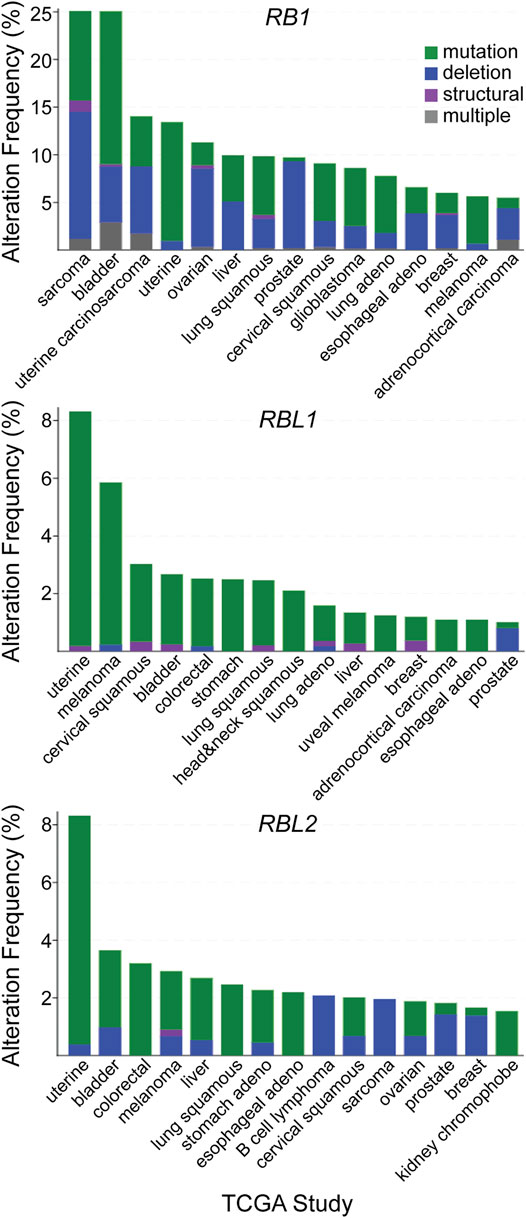
FIGURE 3. Patterns of pocket protein paralogue gene alteration in human cancer. The top fifteen TCGA cancer studies ranked by the frequency of debilitating genetic alterations observed for each individual pocket protein paralogue genes are listed. The nature of the debilitating genetic alteration is shown based on the indicated color code.
Pocket Protein Pathway Genetic Alterations in Advanced Human Cancers
Most cancer genome sequencing data from solid cancers has been obtained from surgically resected primary tumors because of the relative difficulty in accessing patient specimens from more advanced stages of disease. However, studies analyzing clinical specimens from advanced cancers, like metastatic disease, are emerging. In the limited number of pan-cancer studies that have been performed on metastatic solid tumors to date, pocket protein genetic alterations are also recurrent (Priestley et al., 2019; Robinson et al., 2017). Indeed the pocket protein and TP53 pathways appear to be two of the most frequently altered tumor suppressor pathways in metastatic solid cancers. The pattern of somatic pocket protein pathway mutations in metastatic cancers generally mimics that of primary tumors (Figure 4A). However, there are some interesting differences. As prostate cancers progress from primary tissue confined disease to metastatic disease, for example, there is a marked increase in the frequency of pocket protein pathway genetic mutations (Figure 4B). Metastatic prostate cancer can be treated successfully with androgen receptor signaling inhibitor therapy, although acquired therapeutic resistance is inevitable. One of the strongest genomic predictors of acquired therapeutic resistance and poor clinical outcome is RB1 loss of function alterations (Abida et al., 2019). RB1 status survives multivariate analysis as a significant predictor of time on therapy and overall survival. Thus pocket protein genetic alterations can occur late in prostate cancer progression, presumably to facilitate metastasis and acquired therapeutic resistance.
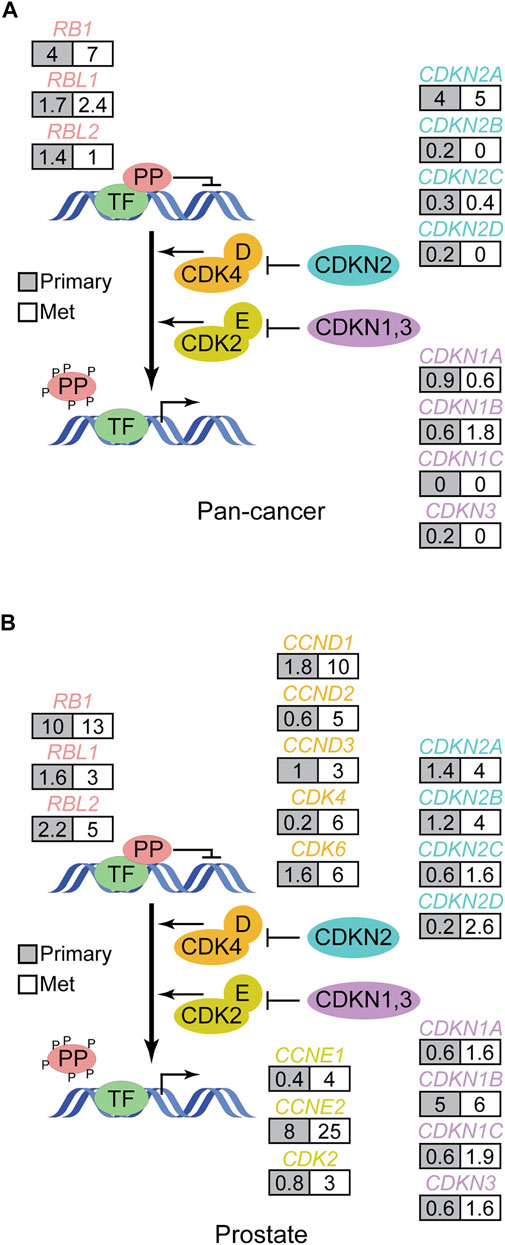
FIGURE 4. Pattern of pocket protein pathway gene alterations in metastatic human cancer. (A) The schematic in Figure 2 is modified to show how often (%) pocket protein pathway genes are genetically altered in pan-cancer genome wide DNA sequencing studies of metastatic solid cancers (Robinson et al., 2017; Priestley et al., 2019) compared to primary tumors (TCGA pan-cancer). Here grey boxes indicate primary tumors while white boxes reflect data from metastatic tumors. The data from metastatic cancers does not include copy number changes, so the analysis is restricted to debilitating mutations. (B) As in A but focused on primary and metastatic prostate cancer. Data is from Abida et al. (Abida et al., 2019) and TCGA and calculated in cBioPortal (Gao et al., 2013).
It is also becoming clear from such studies that RB1 and TP53 alterations are associated with prostate cancer lineage plasticity, transformation of prostate adenocarcinoma to an alternative lineage state no longer dependent on androgen receptor signaling. Neuroendocrine prostate cancer (NEPC) is one of the most obvious and common alternative lineage states that arise under pressure from androgen receptor signaling inhibitors, and this disease state arises by transdifferentiation from a pre-existing adenocarcinoma. The frequency of RB1 alteration increases markedly in NEPC compared to advanced prostate adenocarcinoma (Table 2) (Abida et al., 2019; Laudato et al., 2019). In NEPC, alteration of RB1 occurs more frequently than for alteration in any other core pocket protein pathway gene (Ku et al., 2017; Mu et al., 2017). It is noteworthy that alteration of the other pocket protein paralogues RBL1 and RBL2 is rare in both prostate adenocarcinoma and NEPC, suggesting loss of RB1 has a unique role in cancer lineage plasticity in the prostate. The functional role of RB1 alteration in NEPC transdifferentiation has been verified in experimental models of prostate cancer (Ku et al., 2017; Mu et al., 2017), but the potential role of the other paralogues has not been assessed.
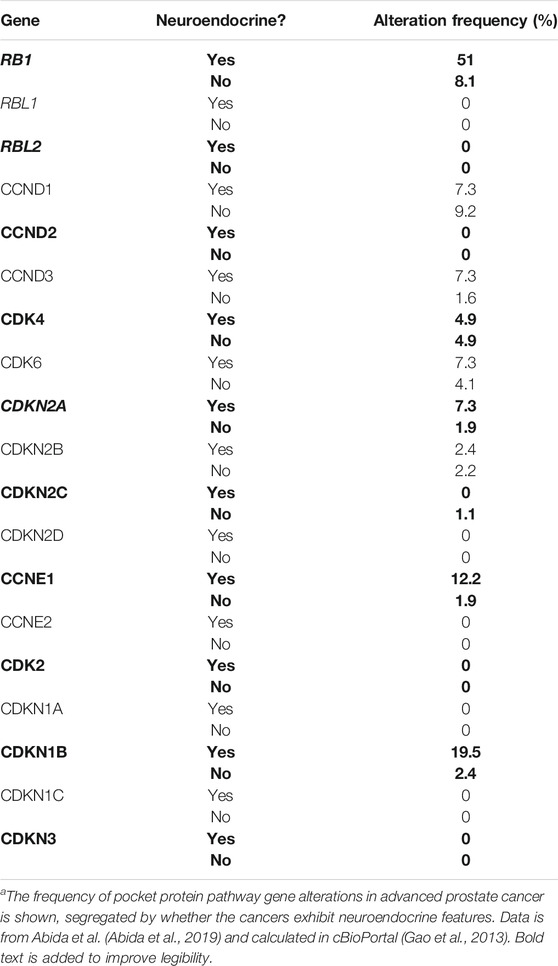
TABLE 2. Pocket protein pathway gene alterations in advanced prostate cancera
Analogous clinical observations have been made in other cancer types. Advanced EGFR mutant non-small cell lung cancers are now treated with EGFR tyrosine kinase inhibitors as standard of care. While very effective, nearly all patients will eventually relapse from this therapy. Alterations in the pocket protein pathway identify EGFR mutant lung cancer patients that have worse outcomes on this therapy (Niederst et al., 2015; Jiang et al., 2016; Blakely et al., 2017; Lee et al., 2017; Marcoux et al., 2019; Skoulidis and Heymach, 2019; Liu et al., 2021). Interestingly EGFR mutant lung cancer with concomitant RB1/TP53 alterations are more likely to undergo neuroendocrine transformation to a small cell lung cancer-like disease, analogous to the neuroendocrine transdifferentiation observed in prostate cancer (Quintanal-Villalonga et al., 2020). In fact, neuroendocrine lineage variants can arise in many solid tumors beyond the two examples detailed here, and these neuroendocrine variants converge on a similar histological and molecular phenotype, including ubiquitious alterations in RB1 and TP53 (Balanis et al., 2019). RBL1 and RBL2 genetic alterations are not common in these neuroendocrine cancers, again indicating RB1 alteration has a unique role in this cancer context.
Pocket Protein Functional Activity in Cancers
An increasingly common approach for measuring pocket protein pathway functional activity is the use of gene expression signatures. These signatures are identified by comparing RNA transcription genome wide in experimental or clinical specimens with known pathway status, most commonly based on RB1 status. The signatures are defined by comparisons in training sets and are then used to assess pathway activity by analysis of RNA-seq data from additional clinical or experimental specimens (Chen et al., 2019; Knudsen et al., 2020). As expected, these signatures correlate with genetic RB1 inactivation, but they also identify samples with low pathway activity that retain wild type RB1. These expression-based measures of pathway activity predict cancer outcomes just like genetic alterations in the pocket protein pathway. Low pathway activity tends to correlate with worse clinical outcome. Comparison of gene expression signatures in samples with different alterations in the pathway suggest that pathway alterations other than RB1 can yield transcriptional changes similar to those observed upon genetic RB1 loss, primarily with respect to cell cycle regulatory genes. Yet it is also evident that functional suppression of pRb through alteration of upstream genes controlling CDK activity is not equivalent to genetic RB1 inactivation as assessed by transcriptional output (McNair et al., 2018), and these differences generally affect the expression of genes not obviously relevant to the cell cycle.
DNA and RNA sequencing data from human cancer clinical specimens are consistent with the notion that all the pocket paralogues have tumor suppressor activity. However, the pattern of pocket protein genetic alterations among the paralogues is dependent on cancer type. Among paralogues, RB1 loss of function alterations are more broadly observed across human cancer. In some contexts like neuroendocrine transdifferentiation, RB1 loss is uniquely observed. The pattern of RBL1 and RBL2 genetic alteration across cancer types is generally more similar to each other than to RB1 (Figure 3), consistent with structural similarities between the paralogues. This observational data is consistent with the notion that pocket protein paralogues have partially overlapping tumor suppressor functions, likely based on their overlapping role in cell cycle control, but that RB1 has a more dominant and unique role in tumor suppression.
Perhaps the most rigorous means to experimentally assess tumor suppressor functional activity is to evaluate effects of specific genetic mutations on spontaneous cancer initiation and progression in mice. Below we will review examples of such studies that yield more definitive insight on whether debilitating pocket protein gene mutations can drive tumor development, with a focus on experiments in which the effects of pocket protein paralogues have been compared (Figure 5).
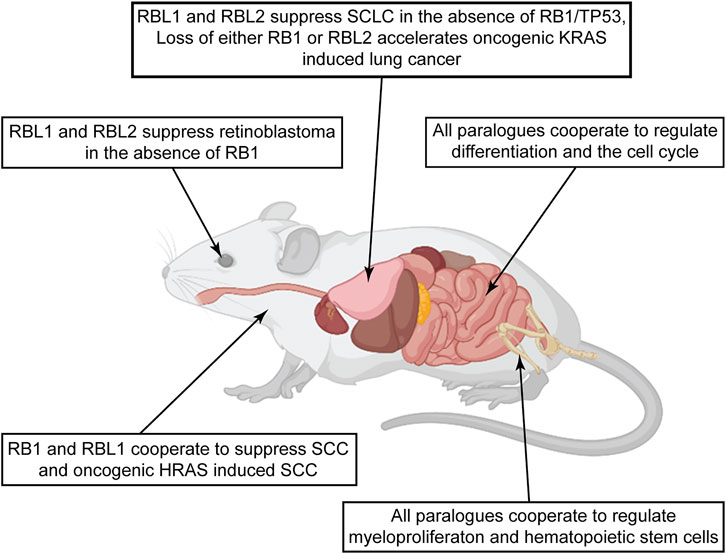
FIGURE 5. Pocket protein paralogues display diverse tumor suppressor activities across mouse tissues. The figure summarizes studies comparing potential tumor suppressor activities of the pocket protein paralogues in genetically engineered mice. Observations in different tissues highlight evidence of diverse tumor suppressor functions relevant to early tumor initiation or later tumor progression. Of notable interest are examples in the lung and epidermis studies, where the pocket protein paralogues curb specific oncogenic signaling. Created with BioRender.com.
Pocket Protein Tumor Suppressor Activity in the Retina
Experimental evidence implicating RBL1 and RBL2 in tumor suppression has roots in attempts to model retinoblastoma in mice. In contrast to humans, loss of Rb1 is insufficient to cause murine retinoblastoma. However, ablation of Rb1 plus one additional pocket protein paralogue is sufficient (Chen et al., 2004; Dannenberg et al., 2004; Zhang et al., 2004). The phenotype of resulting retinoblastoma tumors are different depending on the combination of pocket protein paralogues deleted. Ablation of Rb1/Rbl2 leads to rapid development of multifocal retinoblastoma while ablation of Rb1/Rbl1 causes more delayed development of unifocal retinoblastoma. The number and type of somatic copy number alterations spontaneously acquired during retinoblastoma development suggest Rb1/Rbl2 deficient murine retinoblastomas are more like their human counterparts (Kooi et al., 2017). The finding that Rb1 loss is not sufficient for retinoblastoma in mice is unexpected but has inspired more careful analysis to uncover possible explanations. Donovan et al. (2006) found that the pattern of pocket protein paralogue expression is different in mice and humans. In humans, RB1 is the primary paralogue expressed throughout retinal development. In mice, however, embryonic retinal progenitor cells express p107 while postnatal retinal progenitor cells express pRb. Both pRb and p130 are expressed in postmitotic retinal cells. Further, there is regulatory compensation between paralogues during retinal development. If pRb expression is abrogated, p107 expression is upregulated. A complementary compensatory mechanism is observed if p107 expression is eliminated causing up regulation of pRb expression. This compensation is not apparent in humans as ablation of pRb in the human fetal retina failed to elicit up regulation of p107. Murine retinoblastoma thus requires loss of multiple pocket proteins because of functional redundancy and feedback regulation between paralogues. This is one of the first formal demonstrations that pocket protein paralogues have overlapping tumor suppressor activity.
Pocket Protein Tumor Suppressor Activity in the Lung
Pocket protein pathway gene alterations are common and predict poor outcomes in human lung cancer (Bhateja et al., 2019), thus effects of pocket protein gene deletions have been investigated in the lungs of genetically engineered mice. Deletion of Rb1 in surfactant protein C expressing alveolar type 2 cells causes lung hyperplasia and defects in epithelial differentiation, but not cancer (Simpson et al., 2009). As in the retina, pRb loss was associated with compensatory p107 expression while p130 expression remain unchanged. Deletion of both Rb1/Rbl1, but not Rb1/Rbl2, caused development of non-small cell lung cancers in mice after long latency. Widespread deletion of all three pocket protein genes across multiple cell types within the mouse lung induces relatively benign tumorlets that stain positive for neuroendocrine lineage markers (Lázaro et al., 2017). Moreover, these mice are susceptible to chemical carcinogenesis as exposure to DHPN causes development of low grade malignant neuroendocrine tumors while DHPN did not cause tumorigenesis in control mice. These observations suggest all three pocket proteins may influence the initiation of neuroendocrine cancers in the lung. Yet it is RB1 loss that is a nearly universal feature of human high grade neuroendocrine lung cancers like small cell lung cancer. The other pocket protein genes are rarely altered in these cancers. This emphasizes RB1’s unique tumor suppressor activity in the lung.
In human small cell lung cancer RB1 loss nearly always occurs in combination with genetic alterations in other known oncogenes and tumor suppressor genes, particularly TP53. Rb1 and Trp53 deletion in the mouse lung causes development of tumors highly reminiscent of human small cell lung cancer with relatively long latency (Meuwissen et al., 2003). The cell type in which these deletions are made has a significant impact on resulting cancer phenotype (Sutherland et al., 2011). Deletion of Rb1/Trp53 in pulmonary neuroendocrine cells efficiently induces small cell lung cancer. Loss of Rb1/Trp53 in alveolar type 2 cells can also drive development of small cell lung cancer, but with reduced penetrance compared to deletion in pulmonary neuroendocrine cells. Clara cells are largely resistant to effects of Rb1/Trp53 deletion. These observations indicate that effects of Rb1/Trp53 loss are dependent on cell type, efficiently triggering neoplastic transform in some cell types but not triggering neoplastic transformation in others.
The efficiency of transformation also depends on other pocket proteins. Deletion of Rb1/Trp53 plus Rbl2 greatly accelerates the genesis of small cell lung cancers, and the gene expression pattern of these tumors is similar to the human disease (Schaffer et al., 2010). More recently Ng et al. used CRISPR-Cas9 mediated gene mutation to directly compare effects of p107 and p130 loss on small cell lung cancer induced by Rb1/Trp53 deletion. The ablation of p107 or p130 in addition to Rb1/Trp53 accelerated the rate of tumor progression and decreased the median survival of mice. Interestingly, distinct tumor phenotypes are observed depending on the pocket protein orthologues deleted. Rb1/Trp53/Rbl1 ablated mice developed fewer but larger tumors compared to Rb1/Trp53/Rbl2 deleted mice. The Rbl1-deleted small cell lung cancers exhibited a higher rate of mediastinal lymph node metastasis and lymphocyte infiltration compared to Rbl2-ablated mice. Thus Rbl1 and Rbl2 have tumor suppressor activity in the context of small cell lung cancer, but it is only revealed in the absence of RB1.
Pocket proteins also influence lung cancers initiated by other oncogenic drivers. EGFR and KRAS are the most frequently mutated oncogenes in human lung adenocarcinoma. Mutation of these oncogenes also induce lung adenocarcinoma in mice (Fisher et al., 2001; Jackson et al., 2001; Ji et al., 2006; Politi et al., 2006). Foggetti et al. used a CRISPR-Cas9 based approach to screen the effects of select mutations on development of lung adenocarcinoma initiated by Egfr/Trp53 or Kras/Trp53 mutations (Foggetti et al., 2021). Loss of Rb1, but not Cdkn2A, accelerated lung cancer progression induced by Egfr/Trp53 mutation. Loss of either Rb1 or Cdkn2A accelerated lung cancer progression initiated by Kras/Trp53 mutation, consistent with the higher frequency of CDKN2A alteration in KRAS versus EGFR initiated human lung adenocarcinoma. Effects of other pocket proteins were not tested. Such observations highlight that the effects of pocket protein pathway genetic alterations also depend on genetic background.
Ho et al. (2009) compared the effects of Rb1 or Rbl2 loss on Kras initiated lung adenocarcinoma. The median life span of these mice was reduced by additional loss of either Rb1 (20 weeks) or Rbl2 (25 weeks) compared to oncogenic Kras alone (32 weeks). However, loss of the pocket protein paralogues accelerated tumor progression differently. Loss of Rb1 accelerated the early stages of tumor development as indicated by an increased number of adenomas and tumor burden in younger mice, and it increased tumor grade. In contrast, loss of Rbl2 led to larger tumors in older mice. Analogous to the retina, lung tumors lacking pRb exhibited compensatory upregulation of p130 and p107 expression. Up regulation of p107 expression was also observed when Rbl2 was deleted in Kras mutant lung cancer. This is one of the few examples were a pocket protein paralogue exhibits tumor suppressor activity in the presence of wild type Rb1, suggesting Rb1 cannot completely compensate for loss of Rbl2 in some contexts.
Pocket Protein Tumor Suppressor Activity in the Epidermis
The pocket protein pathway is frequently mutated in human skin cancers. CDKN2A, for example, is lost in 35% of squamous cell carcinomas. Thus, several studies have focused on pocket protein function in the epidermis. Paramio et al. (1998) find that basal and lower spinous cells of the skin express both pRb and p107 while upper spinous and granular cells express p130. Forced overexpression of pRb and p107 in the human keratinocyte cell line HaCaT induces expression of early differentiation markers, while overexpression of all the pocket proteins induces expression of later differentiation markers. Overexpression of individual pocket proteins inhibit HaCaT cell proliferation, each to a different extent. p107 expression has the strongest inhibitory effect on cell proliferation, followed by pRb and finally p130. In this biological context, all the pocket protein paralogues cooperate to regulate differentiation and proliferation in epidermal cells, cells that exhibit a high turnover rate in order to maintain skin barrier function.
Lara et al. (2008a), Lara et al. (2008b) and Ruiz et al. (2004) have extended the work in vivo using genetically engineered mice. Mice lacking pRb in the epidermis develop hyperplasia and hyperkeratosis due to abnormal proliferation and differentiation. However spontaneous tumorigenesis does not occur. Additional deletion of one or both p107 alleles exacerbates this phenotype. Complete loss of Rb1/Rbl1 causes mice to die shortly after birth, before tumorigenesis can be observed. To circumvent this issue, skin grafts from dual Rb1/Rbl1-ablated pups have been transplanted into immunodeficient mice. Papillomatous lesions develop spontaneously from these grafts and subsequently progress to squamous cell carcinomas. Skin grafts lacking pRb alone do not develop tumors. The effects of pocket protein loss in the context of Hras oncogenic mutation have also been tested. Primary keratinocytes expressing oncogenic mutant Hras and lacking both Rb1 and Rbl1 develop into large, poorly differentiated tumors upon transplantation into mouse hosts. Mutant Hras expressing keratinocytes lacking Rb1 alone develop tumors with delayed kinetics and a more differentiated phenotype. Molecular analyses has revealed reduced p53-dependent apoptosis in primary keratinocytes lacking both Rb1 and Rbl1, analogous to findings in mouse skin lacking both these pocket protein paralogues (Costa et al., 2012). This effect may contribute to the more aggressive skin tumors developing in mice lacking both paralogues. Thus both Rb1 and Rbl1 have detectable tumor suppressor activity in the skin.
Pocket Protein Tumor Suppressor Activity in the Intestine
Like the skin, the intestinal epithelium is a dynamic tissue exhibiting high cell turnover. Homeostasis of the intestinal epithelium is maintained by a population of constantly proliferating stem cells that give rise to progeny that progressively differentiate into enterocytes and other support cells as they migrate from the intestinal crypts towards the tips of the villi. Given stereotypical and spatially defined cell cycling within this tissue, it is attractive for assessing coordination between differentiation and the cell cycle. Normally, pRb and p130 expression is uniform across both the crypt and villi while p107 is most strongly expressed in proliferating crypt cells (Haigis et al., 2006; Guo et al., 2009). However, pRb is hyperphosphorylated in crypt cells while it is hypophosphorylated in villus cells. Mice lacking pRb in the intestine develop focal hyperplasia, increased proliferation in the crypt, and ectopic cell cycle re-entry within villus enterocytes, as long as the efficiency of gene deletion is sufficiently high. Differentiation is also impaired in the absence of Rb1 based on expression of differentiation markers. As in other tissues, the expression of p107 increases in both crypts and villi upon pRb loss. However this compensation is not sufficient to prevent intestinal hyperplasia. Deletion of pairs of pocket protein paralogues exacerbate the proliferative and differentiation phenotypes, with loss of Rb1 plus either of the other paralogues giving the most severe phenotype. These observations suggest Rb1 can more completely compensate for Rbbl1 and/or Rbl2 loss than either of these paralogues can compensate for Rb1 loss. As in other tissues noted above, therefore, this evidence suggests Rb1 function in linking cell cycle exit and differentiation in the intestine is relatively unique compared to other pocket protein family members.
Pocket Protein Tumor Suppressor Activity in Hematopoietic Stem Cells
Maintenance of quiescence in hematopoietic stem cells is necessary to maintain homeostasis in the bone marrow and prevent the formation of blood cancers such as leukemias and lymphomas. A number of studies have demonstrated that Rb1 deletion causes defects in hematopoiesis including defective maturation of erythrocytes (Macleod, 2008). However, many of these effects appear to be non-cell autonomous, confounding interpretation of observed phenotypes and the direct role of pRb. One exception to this is the documented effects of Rb1 loss on hematopoietic stem and progenitor cells under stress conditions where a cell intrinsic effect is observed (Daria et al., 2008). In contrast, loss of Rbl2 does not yield detectable hematopoietic phenotypes (Cobrinik et al., 1996) while Rbl1 loss causes mild myeloid hyperplasia that is only evident on the Balb/c genetic background (LeCouter et al., 1998). To explore this further, all three pocket protein paralogues have been deleted in hematopoietic cells of the mouse (Viatour et al., 2008; Kim et al., 2017). These triple knockout mice show reduced viability accompanied by myeloproliferation and myeloid cell infiltration in the kidney, liver, lungs, and spleen. Hematopoietic progenitor cells in the bone marrow of these mice showed increased proliferation and a shift towards the myeloid lineage. This is clearly a cell intrinsic defect as transplantation of triple knockout hematopoietic stem cells into wild type mice yield the same phenotype. Interestingly, mice retaining one wild-type allele of Rbl1 did not show any of the myeloid or hematopoietic progenitor cell phenotypes. Thus a single Rbl1 allele can fully compensate for loss of other pocket protein genes to maintain normal hematopoietic stem cell function and homeostasis. These findings highlight the important contributions that all pocket proteins make to normal hematopoiesis and may explain why upstream components of the core pocket protein pathway like cyclins, CDKs, and CDKIs are more commonly altered in leukemias and lymphoma than other cancers since this would functionally compromise all pocket protein paralogues simultaneously.
Pocket Proteins, Pluripotency, and Cancer Lineage Plasticity
While the canonical function of the pocket protein pathway is to regulate the cell division cycle, it has long been recognized that another hallmark feature of the family is its influence on cellular differentiation and development (Calo et al., 2010; Chinnam and Goodrich, 2011; Viatour and Sage, 2011; Julian and Blais, 2015; Dyson, 2016). Loss of pocket protein activity not only deregulates the cell cycle, but also compromises cellular differentiation. These two cellular processes are intimately linked, and the pocket protein pathway helps maintain this link during normal tissue homeostasis. Pocket protein pathway disruption is expected to sever this link, deregulating both differentiation and the cell cycle in emergent neoplastic cells. While loss of pocket protein cell cycle regulatory activity has long been recognized as a major cancer driver, loss of pocket protein mediated functions in cellular differentiation is an increasingly appreciated aspect of pocket protein tumor suppression.
One line of research inspiring this growing appreciation is the discovery of facultative tissue stem cells and experimentally induced pluripotent stem cells. This work has made clear that cellular differentiation is not irreversible and that normal cells can dedifferentiate into a stem or progenitor phenotype. RB1 is a barrier to this reversibility as loss of pRb increases the efficiency of induced stem cell reprogramming while pRb overexpression reduces reprogramming efficiency (Kareta et al., 2015). In this context, RB1 status does not alter cell cycle control. Instead pRb directly regulates the expression of pluripotency inducing transcription factors by binding to the relevant genes in complex with E2Fs, recruiting chromatin regulatory complexes like EZH2/PRC2 (Chicas et al., 2010; Kareta et al., 2015), and repressing RNA transcription. Indeed, RB1 loss can replace the requirement for SOX2 during induced stem cell reprogramming. These effects are not replicated by manipulation of the other pocket protein paralogues, highlighting a potentially unique aspect of pRb function. However, complete loss of all pocket protein paralogues is lethal for embryonic stem cells (Conklin et al., 2012), suggesting some level of pocket protein activity is needed to maintain the viability of pluripotent cells. This pRb-mediated effect on pluripotency is relevant to cancer as stem cell reprogramming factors like SOX2 are required for tumorigenesis initiated by RB1 loss (Li et al., 2012; Vilas et al., 2015). RBL2 has also been implicated in the direct regulation of SOX2 expression, but not RBL1 (Li et al., 2012; Vilas et al., 2015). This suggests RB1 uniquely regulates other aspects of pluripotency transcriptional networks to account for observed differences in the effects of pocket protein paralogues on induced stem cell reprogramming.
A second line of research emphasizing the importance of pocket protein mediated regulation of differentiation has been in area of cancer lineage plasticity. Cancer lineage plasticity describes the ability of cancer cells to reprogram their lineage state in order to adapt to selective pressures like therapy (Le Magnen et al., 2018; Quintanal-Villalonga et al., 2020). This mechanism of acquired therapeutic resistance has become more apparent in recent years as increasingly potent molecularly targeted therapies have been more widely deployed in the clinic (Sequist et al., 2011; Bluemn et al., 2017). The two key examples discussed above include prostate adenocarcinoma and EGFR mutant non-small cell lung cancer, two diseases where standard of care involves highly effective therapies targeting androgen receptor or EGFR signaling, respectively. In both diseases, a significant fraction of patients will relapse through therapy in association with an altered lineage state, including aggressive neuroendocrine variants. These lineage variants typically lack expression of the therapeutic target and are no longer dependent on it for growth and survival. This lineage transformation is associated with loss of RB1 and TP53 (Niederst et al., 2015; Ku et al., 2017; Mu et al., 2017), but does not directly involve changes in the cell cycle. Rather it involves epigenetic reprogramming promoted by factors reminiscent of those involved in stem cell reprogramming, like SOX2 and EZH2/PRC2. RB1/TP53 loss is not sufficient for neuroendocrine lineage transformation as many RB1/TP53 deficient cancers do not exhibit an obvious lineage state change. Even without a lineage state change, RB1/TP53 loss confers resistance to a wide range of cancer therapies. Perhaps this is because signals from many molecules targeted by cancer therapy converge on pocket protein pathway activity and depend on activities like pocket protein cell cycle control for therapeutic effectiveness (Knudsen et al., 2019; Nyquist et al., 2020). It is unclear whether loss of other pocket protein paralogues will phenocopy loss of RB1 in the context of cancer lineage plasticity and therapeutic resistance as systematic comparisons are lacking in the published literature.
Conclusions and Speculations
Both human cancer genome sequencing data and experimental studies in genetically engineered mice formally demonstrate that all three pocket protein paralogues can function as tumor suppressors, but this data also reinforces the diversity of paralogue tumor suppressor activity and a dominant role for RB1 (Figure 5). Evidence for RB1 mediated tumor suppression is more readily detected across a wider spectrum of tissues and biological contexts while evidence for RBL1 and RBL2 tumor suppressor activity is more restricted. In many cases the tumor suppressor activity of RBL1 or RBL2 is only revealed in the absence of RB1. There is less evidence indicating loss of RBL1 or RBL2 potentiates cancer initiation or progression in contexts where RB1 remains intact. Nonetheless this conclusion is consistent with our understanding of pocket protein structure and function where they exhibit partially overlapping similarities, p107 and p130 being more similar to each other than they are to pRb. The greatest overlap between the pocket protein paralogues seems to converge on their cell cycle related functions, although even here there are differences in detail.
The body of data reviewed here also reveals that genetic disruption of pocket protein pathway function has non-uniform effects that differ depending on cancer type and the cancer’s genetic background. This is somewhat surprising given the widely held assumption that enforcing cell cycle control is the primary tumor suppressor function for the pathway. This potential paradox is particularly evident in cancers where genetic alteration of the pathway occurs late in cancer progression, well after the cell cycle is deregulated sufficiently to support development of the primary tumor. This apparent paradox can be resolved through identification of pocket protein mediated, context dependent, and non-canonical functions beyond its more universal role in the cell cycle. Rapidly accumulating experimental data has implicated the pocket protein pathway in multiple functions beyond canonical cell cycle control. A systematic review of all these functions is beyond the scope of this effort, but the topic has been addressed recently elsewhere (Dick et al., 2018; Knudsen et al., 2019). These non-canonical functions are likely to account for both the context dependent effects of pocket protein pathway disruption and the unique role for pRb in tumor suppression.
The cellular and molecular mechanisms mediating pRb’s unique tumor suppressor activity are not well understood. Nonetheless, we can speculate based on known differences between pocket protein paralogues from which these unique mechanisms may arise. The most fundamental differences are in their structure and biochemical activity. The pocket proteins all function as protein adaptors linking sequence specific DNA binding factors with protein complexes that modify chromatin. However, structural differences in their pocket domains specify preferential interactions with overlapping but distinct subsets of cellular proteins. Differences in the pocket protein interacting proteomes can potentially specify qualitatively distinct functions. RB1 has been implicated uniquely in some cancer relevant cellular functions, including pluripotency and cancer lineage plasticity. We propose that this pRb-specific function is a good candidate mechanism accounting for RB1’s dominant role in tumor suppression. RB1 not only derepresses pluripotency transcriptional networks, but loss of cell cycle control likely exacerbates epigenetic instability by destabilizing chromatin during cell cycle coupled disassembly and re-assembly. Resulting RB1 loss driven cancer lineage plasticity may facilitate adaptation of cancer cells to selective pressures experienced during metastatic dissemination and therapy, thereby driving cancer progression.
It is also clear that the pocket proteins are expressed in different cell types and in different stages of the cell cycle. This may also contribute to pRb’s unique tumor suppressor role if the cells where it normally functions are more likely to serve as the cells of cancer origin. RBL2 is expressed most highly in non-proliferative cells while RBL1 is expressed primarily in S phase. RB1 expression bridges these two and thus may be important in cell types transitioning between proliferative and non-proliferative states, like progenitor cells undergoing terminal differentiation. Or, pRb loss may have unique effects in pre-existing cancer cells. Loss of p107 or p130 may have little effect on pre-existing cancer cells while pRb loss may have more robust effects, such as increasing cancer lineage plasticity. Thus the unique role for pRb in cancer may be a function of both the unique molecular mechanisms it mediates and the unique cell types/states in which it carries out these functions.
An important caveat with the central conclusion of this review is that experimental studies systematically comparing the tumor suppressor activity of pocket protein paralogues are not abundant. Most studies focus on RB1 because of its already established importance in cancer, but this only further reinforces the bias. If we are to better understand the diversity of pocket protein tumor suppressor activity, the molecular mechanisms accounting for this diversity, and the clinical ramifications of these divergent mechanisms, future studies will need to address this bias by comparing pocket protein tumor suppressor activity systematically and directly.
Author Contributions
MF and DG developed the concept for the review, researched the scientific literature, analyzed publicly available data, and wrote the manuscript.
Funding
DG (CA234162, CA207757) and MF (CA234162-03S1) are supported by grants from the NIH and the Roswell Park Alliance Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We acknowledge helpful discussions with colleagues that have contributed to the thinking outlined in this review, particularly Roswell Park colleagues Pamela Hershberger, Erik Knudsen, Agnes Witkiewicz, and Dominic Smiraglia. We apologize to colleagues in the field whose important work we did not cite due to the scope of the topic addressed.
References
Abida, W., Cyrta, J., Heller, G., Prandi, D., Armenia, J., Coleman, I., et al. (2019). Genomic Correlates of Clinical Outcome in Advanced Prostate Cancer. Proc. Natl. Acad. Sci. U. S. A. 116 (23), 11428–11436. doi:10.1073/pnas.1902651116
Balanis, N. G., Sheu, K. M., Esedebe, F. N., Patel, S. J., Smith, B. A., Park, J. W., et al. (2019). Pan-cancer Convergence to a Small-Cell Neuroendocrine Phenotype that Shares Susceptibilities with Hematological Malignancies. Cancer cell 36, 17–34. doi:10.1016/j.ccell.2019.06.005
Ben-Porath, I., Weinberg, R. A., Ben-Porath, I., and Weinberg, R. A. (2005). The Signals and Pathways Activating Cellular Senescence. Int. J. Biochem. Cel Biol. 37, 961–976. doi:10.1016/j.biocel.2004.10.013
Berman, S. D., Calo, E., Landman, A. S., Danielian, P. S., Miller, E. S., West, J. C., et al. (2008). Metastatic Osteosarcoma Induced by Inactivation of Rb and P53 in the Osteoblast Lineage. Proc. Natl. Acad. Sci. 105, 11851–11856. doi:10.1073/pnas.0805462105
Bhateja, P., Chiu, M., Wildey, G., Lipka, M. B., Fu, P., Yang, M. C. L., et al. (2019). Retinoblastoma Mutation Predicts Poor Outcomes in Advanced Non Small Cell Lung Cancer. Cancer Med. 8, 1459–1466. doi:10.1002/cam4.2023
Black, E. P., Huang, E., Dressman, H., Rempel, R., Laakso, N., Asa, S. L., et al. (2003). Distinct Gene Expression Phenotypes of Cells Lacking Rb and Rb Family Members. Cancer Res. 63, 3716–3723.
Blakely, C. M., Watkins, T. B. K., Wu, W., Gini, B., Chabon, J. J., McCoach, C. E., et al. (2017). Evolution and Clinical Impact of Co-occurring Genetic Alterations in Advanced-Stage EGFR-Mutant Lung Cancers. Nat. Genet. 49, 1693–1704. doi:10.1038/ng.3990
Bluemn, E. G., Coleman, I. M., Lucas, J. M., Coleman, R. T., Hernandez-Lopez, S., Tharakan, R., et al. (2017). Androgen Receptor Pathway-independent Prostate Cancer Is Sustained through FGF Signaling. Cancer cell 32, 474–489. doi:10.1016/j.ccell.2017.09.003
Buchkovich, K., Duffy, L. A., and Harlow, E. (1989). The Retinoblastoma Protein Is Phosphorylated during Specific Phases of the Cell Cycle. Cell 58, 1097–1105. doi:10.1016/0092-8674(89)90508-4
Burke, J. R., Deshong, A. J., Pelton, J. G., and Rubin, S. M. (2010). Phosphorylation-induced Conformational Changes in the Retinoblastoma Protein Inhibit E2F Transactivation Domain Binding. J. Biol. Chem. 285, 16286–16293. doi:10.1074/jbc.m110.108167
Burke, J. R., Hura, G. L., and Rubin, S. M. (2012). Structures of Inactive Retinoblastoma Protein Reveal Multiple Mechanisms for Cell Cycle Control. Genes Dev. 26 (11), 1156–1166. doi:10.1101/gad.189837.112
Calo, E., Quintero-Estades, J. A., Danielian, P. S., Nedelcu, S., Berman, S. D., and Lees, J. A. (2010). Rb Regulates Fate Choice and Lineage Commitment In Vivo. Nature 466, 1110–1114. doi:10.1038/nature09264
Carr, S. M., Munro, S., Kessler, B., Oppermann, U., and La Thangue, N. B. (2011). Interplay between Lysine Methylation and Cdk Phosphorylation in Growth Control by the Retinoblastoma Protein. EMBO J. 30, 317–327. doi:10.1038/emboj.2010.311
Cerami, E., Gao, J., Dogrusoz, U., Gross, B. E., Sumer, S. O., Aksoy, B. A., et al. (2012). The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2, 401–404. doi:10.1158/2159-8290.cd-12-0095
Chan, H. M., Krstic-Demonacos, M., Smith, L., Demonacos, C., and Thangue, N. B. L. (2001). Acetylation Control of the Retinoblastoma Tumour-Suppressor Protein. Nat. Cel Biol 3, 667–674. doi:10.1038/35083062
Chen, D., Livne-bar, I., Vanderluit, J. L., Slack, R. S., Agochiya, M., and Bremner, R. (2004). Cell-specific Effects of RB or RB/p107 Loss on Retinal Development Implicate an Intrinsically Death-Resistant Cell-Of-Origin in Retinoblastoma. Cancer cell 5, 539–551. doi:10.1016/j.ccr.2004.05.025
Chen, P.-L., Scully, P., Shew, J.-Y., Wang, J. Y. J., and Lee, W.-H. (1989). Phosphorylation of the Retinoblastoma Gene Product Is Modulated during the Cell Cycle and Cellular Differentiation. Cell 58, 1193–1198. doi:10.1016/0092-8674(89)90517-5
Chen, W. S., Alshalalfa, M., Zhao, S. G., Liu, Y., Mahal, B. A., Quigley, D. A., et al. (2019). Novel RB1-Loss Transcriptomic Signature Is Associated with Poor Clinical Outcomes across Cancer Types. Clin. Cancer Res. 25 (14), 4290–4299. doi:10.1158/1078-0432.ccr-19-0404
Chicas, A., Wang, X., Zhang, C., McCurrach, M., Zhao, Z., Mert, O., et al. (2010). Dissecting the Unique Role of the Retinoblastoma Tumor Suppressor during Cellular Senescence. Cancer cell 17, 376–387. doi:10.1016/j.ccr.2010.01.023
Chinnam, M., and Goodrich, D. W. (2011). RB1, Development, and Cancer. Curr. Top. Dev. Biol. 94, 129–169. doi:10.1016/b978-0-12-380916-2.00005-x
Cho, H.-S., Hayami, S., Toyokawa, G., Maejima, K., Yamane, Y., Suzuki, T., et al. (2012). RB1 Methylation by SMYD2 Enhances Cell Cycle Progression through an Increase of RB1 Phosphorylation. Neoplasia 14, 476–IN8. doi:10.1593/neo.12656
Claudio, P. P., Howard, C. M., Baldi, A., De Luca, A., Fu, Y., Condorelli, G., et al. (1994). p130/pRb2 Has Growth Suppressive Properties Similar to yet Distinctive from Those of Retinoblastoma Family Members pRb and P107. Cancer Res. 54, 5556–5560.
Cobrinik, D., Lee, M. H., Hannon, G., Mulligan, G., Bronson, R. T., Dyson, N., et al. (1996). Shared Role of the pRB-Related P130 and P107 Proteins in Limb Development. Genes Dev. 10, 1633–1644. doi:10.1101/gad.10.13.1633
Cobrinik, D., Whyte, P., Peeper, D. S., Jacks, T., and Weinberg, R. A. (1993). Cell Cycle-specific Association of E2F with the P130 E1A-Binding Protein. Genes Dev. 7, 2392–2404. doi:10.1101/gad.7.12a.2392
Comings, D. E. (1973). A General Theory of Carcinogenesis. Proc. Natl. Acad. Sci. 70, 3324–3328. doi:10.1073/pnas.70.12.3324
Conklin, J. F., Baker, J., and Sage, J. (2012). The RB Family Is Required for the Self-Renewal and Survival of Human Embryonic Stem Cells. Nat. Commun. 3, 1244. doi:10.1038/ncomms2254
Connell-Crowley, L., Harper, J. W., and Goodrich, D. W. (1997). Cyclin D1/Cdk4 Regulates Retinoblastoma Protein-Mediated Cell Cycle Arrest by Site-specific Phosphorylation. MBoC 8, 287–301. doi:10.1091/mbc.8.2.287
Costa, C., Santos, M., Segrelles, C., Dueñas, M., Lara, M. F., Agirre, X., et al. (2012). A Novel Tumor Suppressor Network in Squamous Malignancies. Sci. Rep. 2, 828. doi:10.1038/srep00828
Dannenberg, J.-H., Schuijff, L., Dekker, M., van der Valk, M., and Riele, H. t. (2004). Tissue-specific Tumor Suppressor Activity of Retinoblastoma Gene Homologs P107 and P130. Genes Dev. 18, 2952–2962. doi:10.1101/gad.322004
Daria, D., Filippi, M.-D., Knudsen, E. S., Faccio, R., Li, Z., Kalfa, T., et al. (2008). The Retinoblastoma Tumor Suppressor Is a Critical Intrinsic Regulator for Hematopoietic Stem and Progenitor Cells under Stress. Blood 111, 1894–1902. doi:10.1182/blood-2007-02-071746
Dasgupta, P., Sun, J., Wang, S., Fusaro, G., Betts, V., Padmanabhan, J., et al. (2004). Disruption of the Rb-Raf-1 Interaction Inhibits Tumor Growth and Angiogenesis. Mol. Cel Biol 24, 9527–9541. doi:10.1128/mcb.24.21.9527-9541.2004
DeCaprio, J. A., Ludlow, J. W., Lynch, D., Furukawa, Y., Griffin, J., Piwnica-Worms, H., et al. (1989). The Product of the Retinoblastoma Susceptibility Gene Has Properties of a Cell Cycle Regulatory Element. Cell 58, 1085–1095. doi:10.1016/0092-8674(89)90507-2
Dick, F. A., Goodrich, D. W., Sage, J., and Dyson, N. J. (2018). Non-canonical Functions of the RB Protein in Cancer. Nat. Rev. Cancer 18, 442–451. doi:10.1038/s41568-018-0008-5
Donovan, S. L., Schweers, B., Martins, R., Johnson, D., and Dyer, M. A. (2006). Compensation by Tumor Suppressor Genes during Retinal Development in Mice and Humans. BMC Biol. 4, 14. doi:10.1186/1741-7007-4-14
Dyson, N. J. (2016). RB1: a Prototype Tumor Suppressor and an enigma. Genes Dev. 30, 1492–1502. doi:10.1101/gad.282145.116
Ewen, M. E., Xing, Y., Lawrence, J. B., and Livingston, D. M. (1991). Molecular Cloning, Chromosomal Mapping, and Expression of the cDNA for P107, a Retinoblastoma Gene Product-Related Protein. Cell 66, 1155–1164. doi:10.1016/0092-8674(91)90038-z
Fisher, G. H., Wellen, S. L., Klimstra, D., Lenczowski, J. M., Tichelaar, J. W., Lizak, M. J., et al. (2001). Induction and Apoptotic Regression of Lung Adenocarcinomas by Regulation of a K-Ras Transgene in the Presence and Absence of Tumor Suppressor Genes. Genes Dev. 15, 3249–3262. doi:10.1101/gad.947701
Foggetti, G., Li, C., Cai, H., Hellyer, J. A., Lin, W.-Y., Ayeni, D., et al. (2021). Genetic Determinants of EGFR-Driven Lung Cancer Growth and Therapeutic Response In Vivo. Cancer Discov. 11, 1736–1753. doi:10.1158/2159-8290.cd-20-1385
Friend, S. H., Bernards, R., Rogelj, S., Weinberg, R. A., Rapaport, J. M., Albert, D. M., et al. (1986). A Human DNA Segment with Properties of the Gene that Predisposes to Retinoblastoma and Osteosarcoma. Nature 323, 643–646. doi:10.1038/323643a0
Fung, Y.-K. T., Murphree, A. L., T'Ang, A., Qian, J., Hinrichs, S. H., and Benedict, W. F. (1987). Structural Evidence for the Authenticity of the Human Retinoblastoma Gene. Science 236, 1657–1661. doi:10.1126/science.2885916
Gao, J., Aksoy, B. A., Dogrusoz, U., Dresdner, G., Gross, B., Sumer, S. O., et al. (2013). Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 6, pl1. doi:10.1126/scisignal.2004088
Goodrich, D. W. (2006). The Retinoblastoma Tumor-Suppressor Gene, the Exception that Proves the Rule. Oncogene 25, 5233–5243. doi:10.1038/sj.onc.1209616
Goodrich, D. W., Wang, N. P., Qian, Y.-W., Lee, E. Y.-H. P., and Lee, W.-H. (1991). The Retinoblastoma Gene Product Regulates Progression through the G1 Phase of the Cell Cycle. Cell 67, 293–302. doi:10.1016/0092-8674(91)90181-w
Gubern, A., Joaquin, M., Marquès, M., Maseres, P., Garcia-Garcia, J., Amat, R., et al. (2016). The N-Terminal Phosphorylation of RB by P38 Bypasses its Inactivation by CDKs and Prevents Proliferation in Cancer Cells. Mol. Cel 64, 25–36. doi:10.1016/j.molcel.2016.08.015
Guiley, K. Z., Liban, T. J., Felthousen, J. G., Ramanan, P., Litovchick, L., and Rubin, S. M. (2015). Structural Mechanisms of DREAM Complex Assembly and Regulation. Genes Dev. 29, 961–974. doi:10.1101/gad.257568.114
Guo, J., Longshore, S., Nair, R., and Warner, B. W. (2009). Retinoblastoma Protein (pRb), but Not P107 or P130, Is Required for Maintenance of Enterocyte Quiescence and Differentiation in Small Intestine. J. Biol. Chem. 284, 134–140. doi:10.1074/jbc.m806133200
Guo, J., Sheng, G., and Warner, B. W. (2005). Epidermal Growth Factor-Induced Rapid Retinoblastoma Phosphorylation at Ser780 and Ser795 Is Mediated by ERK1/2 in Small Intestine Epithelial Cells. J. Biol. Chem. 280, 35992–35998. doi:10.1074/jbc.m504583200
Haigis, K., Sage, J., Glickman, J., Shafer, S., and Jacks, T. (2006). The Related Retinoblastoma (pRb) and P130 Proteins Cooperate to Regulate Homeostasis in the Intestinal Epithelium. J. Biol. Chem. 281, 638–647. doi:10.1074/jbc.m509053200
Hannon, G. J., Demetrick, D., and Beach, D. (1993). Isolation of the Rb-Related P130 through its Interaction with CDK2 and Cyclins. Genes Dev. 7, 2378–2391. doi:10.1101/gad.7.12a.2378
Hansen, K., Farkas, T., Lukas, J., Holm, K., Ronnstrand, L., and Bartek, J. (2001). Phosphorylation-dependent and -independent Functions of P130 Cooperate to Evoke a Sustained G1 Block. EMBO J. 20, 422–432. doi:10.1093/emboj/20.3.422
Hasan, M. M., Brocca, S., Sacco, E., Spinelli, M., Papaleo, E., Lambrughi, M., et al. (2013). A Comparative Study of Whi5 and Retinoblastoma Proteins: from Sequence and Structure Analysis to Intracellular Networks. Front. Physiol. 4, 315. doi:10.3389/fphys.2013.00315
Hassler, M., Singh, S., Yue, W. W., Luczynski, M., Lakbir, R., Sanchez-Sanchez, F., et al. (2007). Crystal Structure of the Retinoblastoma Protein N Domain Provides Insight into Tumor Suppression, Ligand Interaction, and Holoprotein Architecture. Mol. Cel 28, 371–385. doi:10.1016/j.molcel.2007.08.023
Hatakeyama, M., Brill, J. A., Fink, G. R., and Weinberg, R. A. (1994). Collaboration of G1 Cyclins in the Functional Inactivation of the Retinoblastoma Protein. Genes Dev. 8, 1759–1771. doi:10.1101/gad.8.15.1759
Hattori, T., Uchida, C., Takahashi, H., Yamamoto, N., Naito, M., and Taya, Y. (2014). Distinct and Site-specific Phosphorylation of the Retinoblastoma Protein at Serine 612 in Differentiated Cells. PLoS ONE 9, e86709. doi:10.1371/journal.pone.0086709
Hilgendorf, K. I., Leshchiner, E. S., Nedelcu, S., Maynard, M. A., Calo, E., Ianari, A., et al. (2013). The Retinoblastoma Protein Induces Apoptosis Directly at the Mitochondria. Genes Dev. 27, 1003–1015. doi:10.1101/gad.211326.112
Hirschi, A., Cecchini, M., Steinhardt, R. C., Schamber, M. R., Dick, F. A., and Rubin, S. M. (2010). An Overlapping Kinase and Phosphatase Docking Site Regulates Activity of the Retinoblastoma Protein. Nat. Struct. Mol. Biol. 17 (9), 1051–1057. doi:10.1038/nsmb.1868
Ho, V. M., Schaffer, B. E., Karnezis, A. N., Park, K. S., and Sage, J. (2009). The Retinoblastoma Gene Rb and its Family Member P130 Suppress Lung Adenocarcinoma Induced by Oncogenic K-Ras. Oncogene 28, 1393–1399. doi:10.1038/onc.2008.491
Hornbeck, P. V., Zhang, B., Murray, B., Kornhauser, J. M., Latham, V., and Skrzypek, E. (2015). PhosphoSitePlus, 2014: Mutations, PTMs and Recalibrations. Nucleic Acids Res. 43, D512–D520. doi:10.1093/nar/gku1267
Hou, S. T., Xie, X., Baggley, A., Park, D. S., Chen, G., and Walker, T. (2002). Activation of the Rb/E2F1 Pathway by the Nonproliferative P38 MAPK during Fas (APO1/CD95)-Mediated Neuronal Apoptosis. J. Biol. Chem. 277, 48764–48770. doi:10.1074/jbc.m206336200
Indovina, P., Marcelli, E., Casini, N., Rizzo, V., and Giordano, A. (2013). Emerging Roles of RB Family: New Defense Mechanisms against Tumor Progression. J. Cell. Physiol. 228, 525–535. doi:10.1002/jcp.24170
Inoue, Y., Kitagawa, M., and Taya, Y. (2007). Phosphorylation of pRB at Ser612 by Chk1/2 Leads to a Complex between pRB and E2F-1 after DNA Damage. EMBO J. 26, 2083–2093. doi:10.1038/sj.emboj.7601652
Jacks, T., Fazeli, A., Schmitt, E. M., Bronson, R. T., Goodell, M. A., and Weinberg, R. A. (1992). Effects of an Rb Mutation in the Mouse. Nature 359, 295–300. doi:10.1038/359295a0
Jackson, E. L., Willis, N., Mercer, K., Bronson, R. T., Crowley, D., Montoya, R., et al. (2001). Analysis of Lung Tumor Initiation and Progression Using Conditional Expression of Oncogenic K-Ras. Genes Dev. 15, 3243–3248. doi:10.1101/gad.943001
Ji, H., Li, D., Chen, L., Shimamura, T., Kobayashi, S., McNamara, K., et al. (2006). The Impact of Human EGFR Kinase Domain Mutations on Lung Tumorigenesis and In Vivo Sensitivity to EGFR-Targeted Therapies. Cancer cell 9, 485–495. doi:10.1016/j.ccr.2006.04.022
Jiang, J., Gu, Y., Liu, J., Wu, R., Fu, L., Zhao, J., et al. (2016). Coexistence of p16/CDKN2A Homozygous Deletions and Activating EGFR Mutations in Lung Adenocarcinoma Patients Signifies a Poor Response to EGFR-TKIs. Lung Cancer 102, 101–107. doi:10.1016/j.lungcan.2016.10.015
Julian, L. M., and Blais, A. (2015). Transcriptional Control of Stem Cell Fate by E2Fs and Pocket Proteins. Front. Genet. 6, 161. doi:10.3389/fgene.2015.00161
Kareta, M. S., Gorges, L. L., Hafeez, S., Benayoun, B. A., Marro, S., Zmoos, A.-F., et al. (2015). Inhibition of Pluripotency Networks by the Rb Tumor Suppressor Restricts Reprogramming and Tumorigenesis. Cell stem cell 16, 39–50. doi:10.1016/j.stem.2014.10.019
Kim, E., Cheng, Y., Bolton-Gillespie, E., Cai, X., Ma, C., Tarangelo, A., et al. (2017). Rb Family Proteins Enforce the Homeostasis of Quiescent Hematopoietic Stem Cells by Repressing Socs3 Expression. J. Exp. Med. 214, 1901–1912. doi:10.1084/jem.20160719
Kim, H.-Y., Ahn, B. Y., and Cho, Y. (2001). Structural Basis for the Inactivation of Retinoblastoma Tumor Suppressor by SV40 Large T Antigen. EMBO J. 20, 295–304. doi:10.1093/emboj/20.1.295
Kim, H.-Y., and Cho, Y. (1997). Structural Similarity between the Pocket Region of Retinoblastoma Tumour Suppressor and the Cyclin-Box. Nat. Struct. Mol. Biol. 4, 390–395. doi:10.1038/nsb0597-390
Kim, K. Y., Wang, D.-H., Campbell, M., Huerta, S. B., Shevchenko, B., Izumiya, C., et al. (2015). PRMT4-mediated Arginine Methylation Negatively Regulates Retinoblastoma Tumor Suppressor Protein and Promotes E2F-1 Dissociation. Mol. Cell. Biol. 35, 238–248. doi:10.1128/mcb.00945-14
Knudsen, E. S., Nambiar, R., Rosario, S. R., Smiraglia, D. J., Goodrich, D. W., and Witkiewicz, A. K. (2020). Pan-cancer Molecular Analysis of the RB Tumor Suppressor Protein Pathway. Commun. Biol. 3, 158.
Knudsen, E. S., Pruitt, S. C., Hershberger, P. A., Witkiewicz, A. K., and Goodrich, D. W. (2019). Cell Cycle and beyond: Exploiting New RB1 Controlled Mechanisms for Cancer Therapy. Trends Cancer 5, 308–324. doi:10.1016/j.trecan.2019.03.005
Knudson, A. G. (1971). Mutation and Cancer: Statistical Study of Retinoblastoma. Proc. Natl. Acad. Sci. 68, 820–823. doi:10.1073/pnas.68.4.820
Kooi, I. E., van Mil, S. E., MacPherson, D., Mol, B. M., Moll, A. C., Meijers-Heijboer, H., et al. (2017). Genomic Landscape of Retinoblastoma inRb−/−p130−/−mice Resembles Human Retinoblastoma. Genes Chromosomes Cancer 56, 231–242. doi:10.1002/gcc.22429
Ku, S. Y., Rosario, S., Wang, Y., Mu, P., Seshadri, M., Goodrich, Z. W., et al. (2017). Rb1 and Trp53 Cooperate to Suppress Prostate Cancer Lineage Plasticity, Metastasis, and Antiandrogen Resistance. Science 355, 78–83. doi:10.1126/science.aah4199
Lara, M. F., García-Escudero, R., Ruiz, S., Santos, M., Moral, M., Martínez-Cruz, A. B., et al. (2008). Gene Profiling Approaches Help to Define the Specific Functions of Retinoblastoma Family in Epidermis. Mol. Carcinog. 47, 209–221. doi:10.1002/mc.20376
Lara, M. F., Santos, M., Ruiz, S., Segrelles, C., Moral, M., Martínez-Cruz, A. B., et al. (2008). p107 Acts as a Tumor Suppressor in pRb-Deficient Epidermis. Mol. Carcinog. 47, 105–113. doi:10.1002/mc.20367
Laudato, S., Aparicio, A., and Giancotti, F. G. (2019). Clonal Evolution and Epithelial Plasticity in the Emergence of AR-Independent Prostate Carcinoma. Trends Cancer 5, 440–455. doi:10.1016/j.trecan.2019.05.008
Lázaro, S., Pérez-Crespo, M., Enguita, A. B., Hernández, P., Martínez-Palacio, J., Oteo, M., et al. (2017). Ablating All Three Retinoblastoma Family Members in Mouse Lung Leads to Neuroendocrine Tumor Formation. Oncotarget 8, 4373–4386. doi:10.18632/oncotarget.13875
Le Magnen, C., Shen, M. M., and Abate-Shen, C. (2018). Lineage Plasticity in Cancer Progression and Treatment. Annu. Rev. Cancer Biol. 2, 271–289. doi:10.1146/annurev-cancerbio-030617-050224
LeCouter, J. E., Kablar, B., Hardy, W. R., Ying, C., Megeney, L. A., May, L. L., et al. (1998). Strain-dependent Myeloid Hyperplasia, Growth Deficiency, and Accelerated Cell Cycle in Mice Lacking the Rb-Related P107 Gene. Mol. Cel Biol 18, 7455–7465. doi:10.1128/mcb.18.12.7455
Ledl, A., Schmidt, D., and Müller, S. (2005). Viral Oncoproteins E1A and E7 and Cellular LxCxE Proteins Repress SUMO Modification of the Retinoblastoma Tumor Suppressor. Oncogene 24, 3810–3818. doi:10.1038/sj.onc.1208539
Leduc, C., Claverie, P., Eymin, B., Col, E., Khochbin, S., Brambilla, E., et al. (2006). p14ARF Promotes RB Accumulation through Inhibition of its Tip60-dependent Acetylation. Oncogene 25, 4147–4154. doi:10.1038/sj.onc.1209446
Lee, C., Chang, J. H., Lee, H. S., and Cho, Y. (2002). Structural Basis for the Recognition of the E2F Transactivation Domain by the Retinoblastoma Tumor Suppressor. Genes Dev. 16, 3199–3212. doi:10.1101/gad.1046102
Lee, J.-O., Russo, A. A., and Pavletich, N. P. (1998). Structure of the Retinoblastoma Tumour-Suppressor Pocket Domain Bound to a Peptide from HPV E7. Nature 391, 859–865. doi:10.1038/36038
Lee, J. K., Lee, J., Kim, S., Kim, S., Youk, J., Park, S., et al. (2017). Clonal History and Genetic Predictors of Transformation into Small-Cell Carcinomas from Lung Adenocarcinomas. J. Clin. Oncol. 35 (26), 3065–3074. doi:10.1200/jco.2016.71.9096
Lee, W.-H., Bookstein, R., Hong, F., Young, L.-J., Shew, J.-Y., and Lee, E. Y.-H. P. (1987). Human Retinoblastoma Susceptibility Gene: Cloning, Identification, and Sequence. Science 235, 1394–1399. doi:10.1126/science.3823889
Leng, X., Noble, M., Adams, P. D., Qin, J., and Harper, J. W. (2002). Reversal of Growth Suppression by P107 via Direct Phosphorylation by Cyclin D1/cyclin-dependent Kinase 4. Mol. Cel Biol 22, 2242–2254. doi:10.1128/mcb.22.7.2242-2254.2002
Li, H., Collado, M., Villasante, A., Matheu, A., Lynch, C. J., Cañamero, M., et al. (2012). p27Kip1 Directly Represses Sox2 during Embryonic Stem Cell Differentiation. Cell stem cell 11, 845–852. doi:10.1016/j.stem.2012.09.014
Liban, T. J., Medina, E. M., Tripathi, S., Sengupta, S., Henry, R. W., Buchler, N. E., et al. (2017). Conservation and Divergence of C-Terminal Domain Structure in the Retinoblastoma Protein Family. Proc. Natl. Acad. Sci. USA 114, 4942–4947. doi:10.1073/pnas.1619170114
Litovchick, L., Sadasivam, S., Florens, L., Zhu, X., Swanson, S. K., Velmurugan, S., et al. (2007). Evolutionarily Conserved Multisubunit RBL2/p130 and E2F4 Protein Complex Represses Human Cell Cycle-dependent Genes in Quiescence. Mol. Cel 26, 539–551. doi:10.1016/j.molcel.2007.04.015
Liu, S.-Y., Bao, H., Wang, Q., Mao, W.-M., Chen, Y., Tong, X., et al. (2021). Genomic Signatures Define Three Subtypes of EGFR-Mutant Stage II-III Non-small-cell Lung Cancer with Distinct Adjuvant Therapy Outcomes. Nat. Commun. 12, 6450. doi:10.1038/s41467-021-26806-7
Liu, X., and Marmorstein, R. (2007). Structure of the Retinoblastoma Protein Bound to Adenovirus E1A Reveals the Molecular Basis for Viral Oncoprotein Inactivation of a Tumor Suppressor. Genes Dev. 21, 2711–2716. doi:10.1101/gad.1590607
Macleod, K. F. (2008). The Role of the RB Tumour Suppressor Pathway in Oxidative Stress Responses in the Haematopoietic System. Nat. Rev. Cancer 8, 769–781. doi:10.1038/nrc2504
MacPherson, D., Sage, J., Kim, T., Ho, D., McLaughlin, M. E., and Jacks, T. (2004). Cell Type-specific Effects of Rb Deletion in the Murine Retina. Genes Dev. 18, 1681–1694. doi:10.1101/gad.1203304
Marcoux, N., Gettinger, S. N., O’Kane, G., Arbour, K. C., Neal, J. W., Husain, H., et al. (2019). EGFR-mutant Adenocarcinomas that Transform to Small-Cell Lung Cancer and Other Neuroendocrine Carcinomas: Clinical Outcomes. Jco 37, 278–285. doi:10.1200/jco.18.01585
Markham, D., Munro, S., Soloway, J., O'Connor, D. P., and La Thangue, N. B. (2006). DNA‐damage‐responsive Acetylation of pRb Regulates Binding to E2F‐1. EMBO Rep. 7, 192–198. doi:10.1038/sj.embor.7400591
McNair, C., Xu, K., Mandigo, A. C., Benelli, M., Leiby, B., Rodrigues, D., et al. (2018). Differential Impact of RB Status on E2F1 Reprogramming in Human Cancer. J. Clin. Invest. 128, 341–358. doi:10.1172/JCI93566
Meng, F., Qian, J., Yue, H., Li, X., and Xue, K. (2016). SUMOylation of Rb Enhances its Binding with CDK2 and Phosphorylation at Early G1 Phase. Cell Cycle 15, 1724–1732. doi:10.1080/15384101.2016.1182267
Meuwissen, R., Linn, S. C., Linnoila, R. I., Zevenhoven, J., Mooi, W. J., and Berns, A. (2003). Induction of Small Cell Lung Cancer by Somatic Inactivation of Both Trp53 and Rb1 in a Conditional Mouse Model. Cancer cell 4, 181–189. doi:10.1016/s1535-6108(03)00220-4
Miwa, S., Uchida, C., Kitagawa, K., Hattori, T., Oda, T., Sugimura, H., et al. (2006). Mdm2-mediated pRB Downregulation Is Involved in Carcinogenesis in a P53-independent Manner. Biochem. Biophysical Res. Commun. 340, 54–61. doi:10.1016/j.bbrc.2005.11.148
Morris, E. J., and Dyson, N. J. (2001). Retinoblastoma Protein Partners. Adv. Cancer Res. 82, 1–54. doi:10.1016/s0065-230x(01)82001-7
Mu, P., Zhang, Z., Benelli, M., Karthaus, W. R., Hoover, E., Chen, C.-C., et al. (2017). SOX2 Promotes Lineage Plasticity and Antiandrogen Resistance in TP53 - and RB1 -deficient Prostate Cancer. Science 355, 84–88. doi:10.1126/science.aah4307
Munro, S., Khaire, N., Inche, A., Carr, S., and La Thangue, N. B. (2010). Lysine Methylation Regulates the pRb Tumour Suppressor Protein. Oncogene 29, 2357–2367. doi:10.1038/onc.2009.511
Nair, J. S., Ho, A. L., Tse, A. N., Coward, J., Cheema, H., Ambrosini, G., et al. (2009). Aurora B Kinase Regulates the Postmitotic Endoreduplication Checkpoint via Phosphorylation of the Retinoblastoma Protein at Serine 780. MBoC 20, 2218–2228. doi:10.1091/mbc.e08-08-0885
Narasimha, A. M., Kaulich, M., Shapiro, G. S., Choi, Y. J., Sicinski, P., and Dowdy, S. F. (2014). Cyclin D Activates the Rb Tumor Suppressor by Mono-Phosphorylation. Elife 3, e02872. doi:10.7554/elife.02872
Nath, N., Wang, S., Betts, V., Knudsen, E., and Chellappan, S. (2003). Apoptotic and Mitogenic Stimuli Inactivate Rb by Differential Utilization of P38 and Cyclin-dependent Kinases. Oncogene 22, 5986–5994. doi:10.1038/sj.onc.1206843
Nguyen, D. X., Baglia, L. A., Huang, S.-M., Baker, C. M., and McCance, D. J. (2004). Acetylation Regulates the Differentiation-specific Functions of the Retinoblastoma Protein. EMBO J. 23, 1609–1618. doi:10.1038/sj.emboj.7600176
Niederst, M. J., Sequist, L. V., Poirier, J. T., Mermel, C. H., Lockerman, E. L., Garcia, A. R., et al. (2015). RB Loss in Resistant EGFR Mutant Lung Adenocarcinomas that Transform to Small-Cell Lung Cancer. Nat. Commun. 6, 6377. doi:10.1038/ncomms7377
Nyquist, M. D., Corella, A., Coleman, I., De Sarkar, N., Kaipainen, A., Ha, G., et al. (2020). Combined TP53 and RB1 Loss Promotes Prostate Cancer Resistance to a Spectrum of Therapeutics and Confers Vulnerability to Replication Stress. Cel Rep. 31, 107669. doi:10.1016/j.celrep.2020.107669
Paramio, J. M., Laín, S., Segrelles, C., Lane, E. B., and Jorcano, J. L. (1998). Differential Expression and Functionally Co-operative Roles for the Retinoblastoma Family of Proteins in Epidermal Differentiation. Oncogene 17, 949–957. doi:10.1038/sj.onc.1202031
Pilkinton, M., Sandoval, R., and Colamonici, O. R. (2007). Mammalian Mip/LIN-9 Interacts with Either the P107, p130/E2F4 Repressor Complex or B-Myb in a Cell Cycle-phase-dependent Context Distinct from the Drosophila dREAM Complex. Oncogene 26, 7535–7543. doi:10.1038/sj.onc.1210562
Politi, K., Zakowski, M. F., Fan, P.-D., Schonfeld, E. A., Pao, W., and Varmus, H. E. (2006). Lung Adenocarcinomas Induced in Mice by Mutant EGF Receptors Foundin Human Lung Cancers Respondto a Tyrosine Kinase Inhibitor Orto Down-Regulation of the Receptors. Genes Dev. 20, 1496–1510. doi:10.1101/gad.1417406
Priestley, P., Baber, J., Lolkema, M. P., Steeghs, N., de Bruijn, E., Shale, C., et al. (2019). Pan-cancer Whole-Genome Analyses of Metastatic Solid Tumours. Nature 575, 210–216. doi:10.1038/s41586-019-1689-y
Quintanal-Villalonga, Á., Chan, J. M., Yu, H. A., Pe’er, D., Sawyers, C. L., Sen, T., et al. (2020). Lineage Plasticity in Cancer: a Shared Pathway of Therapeutic Resistance. Nat. Rev. Clin. Oncol. 17, 360–371. doi:10.1038/s41571-020-0340-z
Robinson, D. R., Wu, Y.-M., Lonigro, R. J., Vats, P., Cobain, E., Everett, J., et al. (2017). Integrative Clinical Genomics of Metastatic Cancer. Nature 548, 297–303. doi:10.1038/nature23306
Rubin, S. M., Gall, A.-L., Zheng, N., and Pavletich, N. P. (2005). Structure of the Rb C-Terminal Domain Bound to E2F1-DP1: A Mechanism for Phosphorylation-Induced E2F Release. Cell 123, 1093–1106. doi:10.1016/j.cell.2005.09.044
Ruiz, S., Santos, M., Segrelles, C., Leis, H., Jorcano, J. L., Berns, A., et al. (2004). Unique and Overlapping Functions of pRb and P107 in the Control of Proliferation and Differentiation in Epidermis. Development 131, 2737–2748. doi:10.1242/dev.01148
Saddic, L. A., West, L. E., Aslanian, A., Yates, J. R., Rubin, S. M., Gozani, O., et al. (2010). Methylation of the Retinoblastoma Tumor Suppressor by SMYD2. J. Biol. Chem. 285, 37733–37740. doi:10.1074/jbc.m110.137612
Sanidas, I., Morris, R., Fella, K. A., Rumde, P. H., Boukhali, M., Tai, E. C., et al. (2019). A Code of Mono-Phosphorylation Modulates the Function of RB. Mol. Cel 73, 985–1000. doi:10.1016/j.molcel.2019.01.004
Schade, A. E., Oser, M. G., Nicholson, H. E., and DeCaprio, J. A. (2019). Cyclin D-CDK4 Relieves Cooperative Repression of Proliferation and Cell Cycle Gene Expression by DREAM and RB. Oncogene 38, 4962–4976. doi:10.1038/s41388-019-0767-9
Schaffer, B. E., Park, K.-S., Yiu, G., Conklin, J. F., Lin, C., Burkhart, D. L., et al. (2010). Loss of P130 Accelerates Tumor Development in a Mouse Model for Human Small-Cell Lung Carcinoma. Cancer Res. 70, 3877–3883. doi:10.1158/0008-5472.can-09-4228
Schonfeld, S. J., Kleinerman, R. A., Abramson, D. H., Seddon, J. M., Tucker, M. A., and Morton, L. M. (2021). Long-term Risk of Subsequent Cancer Incidence Among Hereditary and Nonhereditary Retinoblastoma Survivors. Br. J. Cancer 124, 1312–1319. doi:10.1038/s41416-020-01248-y
Sequist, L. V., Waltman, B. A., Dias-Santagata, D., Digumarthy, S., Turke, A. B., Fidias, P., et al. (2011). Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci. Transl Med. 3, 75ra26. doi:10.1126/scitranslmed.3002003
Simpson, D. S., Mason-Richie, N. A., Gettler, C. A., and Wikenheiser-Brokamp, K. A. (2009). Retinoblastoma Family Proteins Have Distinct Functions in Pulmonary Epithelial Cells In Vivo Critical for Suppressing Cell Growth and Tumorigenesis. Cancer Res. 69, 8733–8741. doi:10.1158/0008-5472.can-09-1359
Skoulidis, F., and Heymach, J. V. (2019). Co-occurring Genomic Alterations in Non-small-cell Lung Cancer Biology and Therapy. Nat. Rev. Cancer 19, 495–509. doi:10.1038/s41568-019-0179-8
Sutherland, K. D., Proost, N., Brouns, I., Adriaensen, D., Song, J.-Y., and Berns, A. (2011). Cell of Origin of Small Cell Lung Cancer: Inactivation of Trp53 and Rb1 in Distinct Cell Types of Adult Mouse Lung. Cancer cell 19, 754–764. doi:10.1016/j.ccr.2011.04.019
Tedesco, D., Lukas, J., and Reed, S. I. (2002). The pRb-Related Protein P130 Is Regulated by Phosphorylation-dependent Proteolysis via the Protein-Ubiquitin Ligase SCFSkp2. Genes Dev. 16, 2946–2957. doi:10.1101/gad.1011202
Topacio, B. R., Zatulovskiy, E., Cristea, S., Xie, S., Tambo, C. S., Rubin, S. M., et al. (2019). Cyclin D-Cdk4,6 Drives Cell-Cycle Progression via the Retinoblastoma Protein's C-Terminal Helix. Mol. Cel 74, 758–770. doi:10.1016/j.molcel.2019.03.020
Ventura, E., Pentimalli, F., and Giordano, A. (2018). RBL2/p130: a Direct AKT Substrate and Mediator of AKT Inhibition-Induced Apoptosis. Oncoscience 5, 278–280. doi:10.18632/oncoscience.467
Viatour, P., and Sage, J. (2011). Newly Identified Aspects of Tumor Suppression by RB. Dis. Model. Mech. 4, 581–585. doi:10.1242/dmm.008060
Viatour, P., Somervaille, T. C., Venkatasubrahmanyam, S., Kogan, S., McLaughlin, M. E., Weissman, I. L., et al. (2008). Hematopoietic Stem Cell Quiescence Is Maintained by Compound Contributions of the Retinoblastoma Gene Family. Cell stem cell 3, 416–428. doi:10.1016/j.stem.2008.07.009
Vilas, J. M., Ferreirós, A., Carneiro, C., Morey, L., Silva-Álvarez, S. D., Fernandes, T., et al. (2015). Transcriptional Regulation of Sox2 by the Retinoblastoma Family of Pocket Proteins. Oncotarget 6, 2992–3002. doi:10.18632/oncotarget.2996
Weinberg, R. A. (1995). The Retinoblastoma Protein and Cell Cycle Control. Cell 81, 323–330. doi:10.1016/0092-8674(95)90385-2
Wirt, S. E., and Sage, J. (2010). p107 in the Public Eye: an Rb Understudy and More. Cell Div 5, 9. doi:10.1186/1747-1028-5-9
Xiao, B., Spencer, J., Clements, A., Ali-Khan, N., Mittnacht, S., Broceno, C., et al. (2003). Crystal Structure of the Retinoblastoma Tumor Suppressor Protein Bound to E2F and the Molecular Basis of its Regulation. Proc. Natl. Acad. Sci. 100, 2363–2368. doi:10.1073/pnas.0436813100
Zhang, J., Benavente, C. A., McEvoy, J., Flores-Otero, J., Ding, L., Chen, X., et al. (2012). A Novel Retinoblastoma Therapy from Genomic and Epigenetic Analyses. Nature 481, 329–334. doi:10.1038/nature10733
Zhang, J., Schweers, B., and Dyer, M. A. (2004). The First Knockout Mouse Model of Retinoblastoma. Cell Cycle 3, 952–959. doi:10.4161/cc.3.7.1002
Zhou, Z., Flesken-Nikitin, A., Corney, D. C., Wang, W., Goodrich, D. W., Roy-Burman, P., et al. (2006). Synergy of P53 and Rb Deficiency in a Conditional Mouse Model for Metastatic Prostate Cancer. Cancer Res. 66, 7889–7898. doi:10.1158/0008-5472.can-06-0486
Keywords: tumor suppressor gene, pocket protein, retinoblastoma, cell cycle, cellular differentiation, therapeutic resistance
Citation: Flores M and Goodrich DW (2022) Retinoblastoma Protein Paralogs and Tumor Suppression. Front. Genet. 13:818719. doi: 10.3389/fgene.2022.818719
Received: 19 November 2021; Accepted: 25 February 2022;
Published: 18 March 2022.
Edited by:
Sandhya Payankaulam, Michigan State University, United StatesReviewed by:
Lisa Marie Julian, Simon Fraser University, CanadaDavid N. Arnosti, Michigan State University, United States
Copyright © 2022 Flores and Goodrich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David W. Goodrich, ZGF2aWQuZ29vZHJpY2hAcm9zd2VsbHBhcmsub3Jn