 Feng Zhang1Fuwei Li2Fujian Chen1Jinrong Huang1Qiong Luo1Xilong Du2Jiapeng Zhou2
Feng Zhang1Fuwei Li2Fujian Chen1Jinrong Huang1Qiong Luo1Xilong Du2Jiapeng Zhou2 Weiyue Gu2
Weiyue Gu2 Kaishou Xu3*
Kaishou Xu3*- 1Ganzhou Women and Children’s Health Care Hospital, Ganzhou, China
- 2Beijing Chigene Translational Medical Research Center Co. Ltd., Beijing, China
- 3Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangzhou, China
Developmental and epileptic encephalopathies (DEE) caused by heterozygous deleterious variants in Cut Like Homeobox2 (CUX2) is rare. To the best of our knowledge the only variant associated with a phenotype in this gene is the de novo missense variant c.1768G > A, p.Glu590Lys; however, further additional research is needed to characterize the relationship between disease and variants in this gene. In this study, we reported a patient from a non-consanguineous Chinese family presenting with epilepsy, developmental delay, and speech delay. Additionally, the patient responded well to levetiracetam, and at his last follow-up (5.5 years old), he had discontinued antiepileptic drug treatment and remained seizure-free for 6 months. To identify possible causative variants, trio-whole exome sequencing was performed. We identified a novel de novo missense CUX2 c.2834C > T, p. Thr945Met variant in the patient. Based on clinical and genetics information associated with the bioinformatics analyses, we hypothesized that this variant was the cause of the reported phenotype. AlphaFold and SWISS-MODEL homology modeling servers were used to predict the three-dimensional (3D) structure of CUX2 protein. Predictions based on the 3D-structure modeling indicated that the p.Thr945Met substitution was likely to alter the DNA-binding specificities and affect protein function. On the basis of clinical characteristics and genetic analysis, we presented one case diagnosed with DEE67. Our finding expanded the clinical and molecular spectrum of CUX2 variants.
Introduction
Developmental and epileptic encephalopathies (DEE) is characterized by onset in infancy/early childhood with refractory seizures, delayed psychomotor development or/and developmental regression. DEE is clinically and genetically heterogeneous, with over 50 genes known to be causative (Specchio and Curatolo, 2021). However, the genotype still has a limited effect on the phenotype. The Cut Like Homeobox 2 (CUX2) gene, which encodes a 1426 amino-acids transcription factor, plays an important role in the control of neuronal proliferation and differentiation in the brain (Quaggin et al., 1996). Recently, variants in CUX2 were found to be associated with developmental and epileptic encephalopathy 67 (DEE67, OMIM: 618141), an autosomal dominant disorder that typically manifests in infancy and is characterized by refractory seizures, global developmental delay with impaired motor and intellectual development, movement disorders, speech delay, and stereotypic or autistic behavior. To date, only 10 patients carrying recurrent de novo heterozygous Glu590-to-Lys (E590K) variant in the CUX2 gene have been reported (Barington et al., 2018; Chatron et al., 2018).
Here, we reported a 5.5 years old boy with epilepsy, developmental delay and speech delay. Trio (parents-proband) whole-exome sequence (WES) analysis revealed a novel de novo pathogenic missense variant c.2834C > T (p.Thr945Met) in the CUX2 gene. Our findings expand the genetic and phenotypic spectrum of DEE67 and contribute to our understanding of DEE on a genetic level.
Material and Methods
Patients
The proband was a 5.5-year-old boy, the third child of healthy non-consanguineous parents. He had two unaffected female siblings. The patient visited the hospital after experiencing seizures. A detailed clinical history, including a description of epilepsy, a detailed clinical, and a neurological examination was carried out. The study was approved by the Ethics Committee of Ganzhou Women and Children’s Health Care Hospital.
Variant Analysis
After obtaining informed consent, samples were taken from the proband and his parents. The whole-exome capture was carried out (xGen Exome Research Panel v1.0, IDT, IA, United States) according to the manufacturer’s protocol. High-throughput sequencing was performed using Illumina NovaSeq 6000 series sequencer (PE150), and at least 99% of the target sequence was sequenced. The sequencing process was performed by the Chigene Translational Medicine Research Center Co., Ltd., 100875, Beijing.
After eliminating adapters and low-quality reads, clean data were obtained and aligned to the Human genome (Hg19/GRC37) using the Burrows-Wheeler Alignment (BWA). The Genome Analysis Toolkit (GATK) was used to identify single nucleotide variants (SNVs) and insertion/deletion (InDel) variants. The Exome Aggregation Consortium ExAC was used to determine the allele frequencies of variants. Pathogenicity of nonsynonymous variants was predicted using the SIFT, Provean, REVEL, varianttaster, PolyPhen, and CADD tools. SNVs/indels were classified according to the standards and guidelines of the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) (Richards et al., 2015).
Variants were confirmed by Polymerase Chain Reaction (PCR) analysis, followed by Sanger sequencing. CUX2 was amplified using the following primers: forward primer (5′-CAGCCTGGTACAAGTCCCAAC-3′) and reverse primer (5′-CAGACTTATCCGCTGGTCCC-3′). DNA sequencing was performed using the 3730xl DNA Analyzer (Applied Biosystems, United States). The rare variant identified has been submitted to the ClinVar database (Accession number: SCV001946791) (https://www.ncbi.nlm.nih.gov/clinvar/variation/1296981).
Multiple protein sequence alignments were performed using MEGA X, and the modeling of the 3D structure was analyzed and visualized using Swiss-PdbViewer v4.1.
Results
Clinical Features
The proband (II:1) had the onset of febrile seizure at the age of 2 years with unconsciousness, up-rolling of the eyes, facial twitching, limb stretch, and no visible limb shaking; 6 months later, a second febrile seizure occurred, and 3 months later a third febrile seizure occurred. Each seizure lasted 3 min and was followed by spontaneous remission. At 2 years and 9 months the medical team started levetiracetam (40 mg/kg/day) and patient did not present any other febrile seizure. After 2 years without clinical or electrographic seizures, the boy has discontinued levetiracetam and remained seizure-free until the age of five. At 2 years and 9 months, the electroencephalograph (EEG) revealed a somewhat slower background activity and a small number of bilateral spikes and spike-wave discharges in the central, parietal, and posterior temporal regions during sleep, but subsequent magnetic resonance imaging (1.5T MRI) was normal. EEG performed at the age of 5 years without levetiracetam was normal. He was able to walk alone at 18 months. At the age of 2 years and 9 months, the patient was diagnosed with global developmental delay and he was not able to form three word sentences and just could pronounce few words with correct meaning. The results of the Gesell Development Diagnosis Scale (GDDS) revealed a total development quotient (DQ) of 56 (mild developmental delay). Then the patient has received home-based rehabilitation for 3 years. Now he is 5.5 years in kindergarten and the DQ score has been improved to 75 (borderline deficiency).
Variant Identification and Analysis
Trio-WES was performed and a heterozygous de novo missense variant (NM_015267.4: c.2834C > T, p.Thr945Met), in the CUX2 gene was identified in the proband. This c.2834C > T variant was not identified in the 1000 genome project or ExAC. Numerous computational tools, including PolyPhen (probably damaging, 1.0), Provean (deleterious, −4.16), SIFT (damaging, 0.0), mutationtaster (disease_causing, 1), and REVEL (deleterious, 0.582) suggested that the variant was deleterious and had a CADD score of 26.9. The de novo missense variant was confirmed by Sanger sequencing, the proband’s parents and healthy sisters did not carry the variant (Figures 1A,B). Based on these findings, the variant was classified as “Likely pathogenic” by ACMG/AMP guidelines (PS2, PM2, PP2, and PP3) (Richards et al., 2015; Harrison et al., 2019).
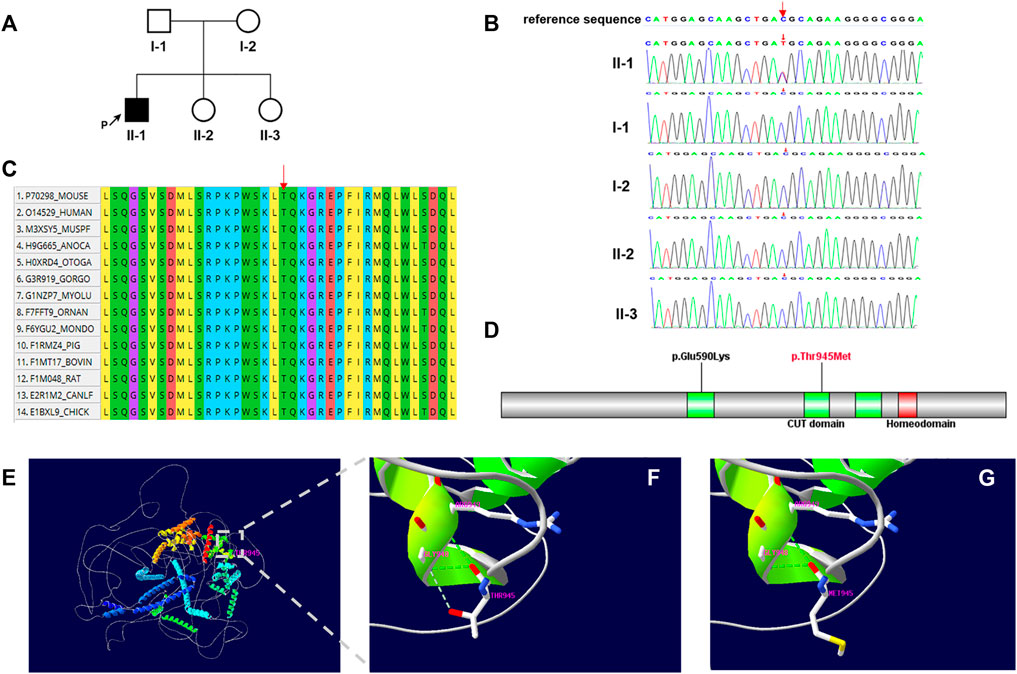
FIGURE 1. (A) Family pedigree. The black arrow indicates the proband. (B) Sanger sequencing of the patient and family members confirmed the de novo variant c.2834C > T. (C) Multiple-sequence alignment of CUX2 protein among species. Red arrow shows the Thr945 site. (D) Domain structure of CUX2 protein (CUT domain, green; Homeodomain, red). Localization of amino acid changes p.Glu590Lys and p.Thr945Met is indicated. (E–G) Structural model of the CUX2 protein. Ribbon representations show details of the second CUT domain for wild-type (p.Thr945) (F) and mutated (p.Thr945Met) (G) CUX2. Noted loss of the weaker hydrogen bond between Thr945 and Gly948. Green dashed lines: hydrogen bonds; grey dashed lines: weaker hydrogen bonds.
Multiple sequence alignment of the protein shows that the Thr-945 residue is highly conserved across species (Figure 1C). This variant locates in the second CUT domain of the human homeobox protein CUX2 (Cut-like2) (Figure 1D) and results in a threonine to methionine amino acid substitution. By substituting a polar and uncharged residue with a non-polar and hydrophobic amino acid, this variant may further affect the formation of a weaker hydrogen bond between Thr 945and Gly948 (Figures 1E–G).
Discussion
In this study, we performed trio-whole exome sequencing and characterized a novel missense variant c.2834C > T (p.Thr945Met) in CUX2 in a Chinese male patient with DEE. In previous studies, all known patients (ten unrelated probands with DEE) presented with a recurrent de novo missense variant, c.1768G > A (p.Glu590Lys) (Table 1) (Rauch et al., 2012; Allen et al., 2013; Geisheker et al., 2017; Barington et al., 2018; Chatron et al., 2018). The ten patients presented with a variety of epilepsy forms, including myoclonic seizures (3), absence seizures (2), focal, tonic, atonic, and generalized tonic-clonic. The age of onset ranged from 2 months to 9 years. EEG was performed in 9 of 10 patients. All participants had abnormal EEG findings, and 5 individuals presented with generalized spike-wave (GSW) or polyspike-wave (GPSW) patterns. Brain MRI was performed in 9 of 10 patients and was unremarkable in 6 of 9 patients. Cerebellar atrophy, hippocampal asymmetry, thin posterior corpus callosum were observed in some patients. In our case, the EEG revealed bi-parieto-temporal discharges while the MRI was normal. All patients had developmental delays, in addition to a variety of other variable features such as nonverbal (7), movement disorders (6), and autistic features (3). Our patient presented with febrile seizure when 2 years old. Besides, he has developmental delay.
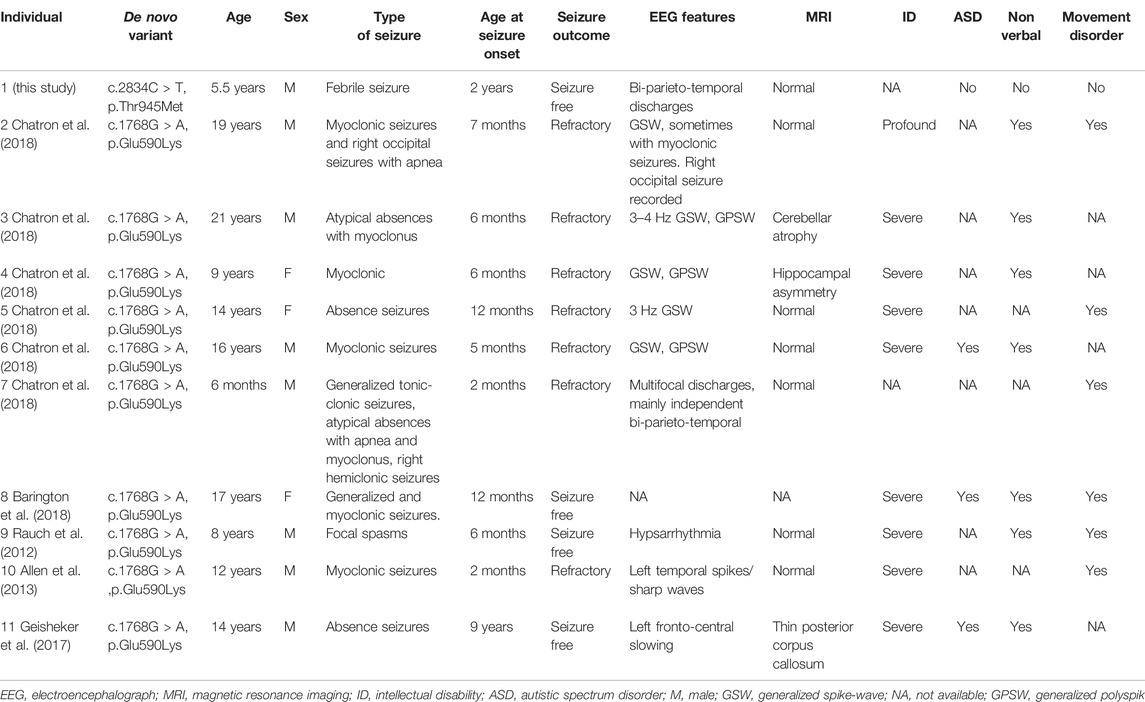
TABLE 1. Clinical features of individuals with the CUX2 variant.
The majority of patients with the p.Glu590Lys variant failed to respond to multiple antiepileptic drugs, including valproate, carbamazepine, clobazam, levetiracetam, and lamotrigine. Only three patients achieved seizure-free status while using valproate or a combination of valproate and lamotrigine. In our case, seizures were controlled after treatment with levetiracetam (Barington et al., 2018; Chatron et al., 2018). At the last follow-up, he had discontinued antiepileptic drug treatment (at age of 5 years) and remained seizure-free for 6 months. Taken together, our patient had a less severe phenotype than previously reported in patients carrying the p.Glu590Lys variant, although the relationship between the reported variant and the described phenotype requires more case reports to be established.
The CUX2 protein consists of three CUT domains and a homeodomain; which are important for DNA binding (Gingras et al., 2005). Additionally, the sequences of all four DNA-binding domains are highly conserved (Quaggin et al., 1996). The CUT domain 1 is located between amino acids 549 and 627, while the CUT domain 2 is located between amino acids 892 and 967 in CUX2. The previously reported variant p.Glu590Lys, which is located at the CUT domain 1, destabilized the effect in-silico analyses and interfered with DNA binding (Chatron et al., 2018). In this study, we modeled the 3D structure of the wild-type CUX2 protein (1–1486) and examined the potential functional impact of the p.Thr945Met variant (Figures 1E–G). Firstly, the variant alters the charge at residue 945 (polar to non-polar) (CUPSAT: ΔΔG = −6.21 kcal/mol, I-mutant3.0: ΔΔG = −0.37 kcal/mol), potentially disrupting the structure and affecting inter and intramolecular interactions (Chatron et al., 2018). Additionally, the Thr945Met variant affects a highly-conserved threonine residue in the CUT domain 2, which could result in structural changes and alter DNA-binding specificities (Gingras et al., 2005). However, additional molecular functional studies will be required to confirm the potential mechanism of pathogenic variants.
In summary, patients with de novo CUX2 variants have developmental and epileptic encephalopathies characterized by developmental delay, speech delay, movement disorders, and autistic behavior. Our findings have further expanded the clinical and molecular spectrum of DEE67.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, SCV001946791.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Ganzhou Women and Children’s Health Care Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
FZ and KX wrote the manuscript. FL, XD, JZ, and WG were responsible for exome sequencing and genetic analysis. FZ, FC, JH, and QL managed the patient and provided clinical information. WG and KX critically reviewed the manuscript. All the authors read and approved the final manuscript.
Funding
This study was supported by the Science and Technology Plan Program of the Health Commission of Jiangxi Province (No. 20204662).
Conflict of Interest
Authors FL, XD, JZ, and WG were employed by Beijing Chigene Translational Medical Research Center Co. Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are incredibly thankful to the participants in our study.
References
Allen, A. S., Berkovic, S. F., Cossette, P., Delanty, N., Dlugos, D., Eichler, E. E., et al. (2013). De Novo mutations in Epileptic Encephalopathies. Nature 501 (7466), 217–221. doi:10.1038/nature12439
Barington, M., Risom, L., Ek, J., Uldall, P., and Ostergaard, E. (2018). A Recurrent De Novo CUX2 Missense Variant Associated with Intellectual Disability, Seizures, and Autism Spectrum Disorder. Eur. J. Hum. Genet. 26 (9), 1388–1391. doi:10.1038/s41431-018-0184-5
Chatron, N., Møller, R. S., Champaigne, N. L., Schneider, A. L., Kuechler, A., Labalme, A., et al. (2018). The Epilepsy Phenotypic Spectrum Associated with a Recurrent CUX2 Variant. Ann. Neurol. 83 (5), 926–934. doi:10.1002/ana.25222
Geisheker, M. R., Heymann, G., Wang, T., Coe, B. P., Turner, T. N., Stessman, H. A. F., et al. (2017). Hotspots of Missense Mutation Identify Neurodevelopmental Disorder Genes and Functional Domains. Nat. Neurosci. 20 (8), 1043–1051. doi:10.1038/nn.4589
Gingras, H., Cases, O., Krasilnikova, M., Bérubé, G., and Nepveu, A. (2005). Biochemical Characterization of the Mammalian Cux2 Protein. Gene 344, 273–285. doi:10.1016/j.gene.2004.11.008
Harrison, S. M., Biesecker, L. G., and Rehm, H. L. (2019). Overview of Specifications to the ACMG/AMP Variant Interpretation Guidelines. Curr. Protoc. Hum. Genet. 103 (1), 93. doi:10.1002/cphg.93
Quaggin, S. E., Heuvel, G. B. V., Golden, K., Bodmer, R., and Igarashi, P. (1996). Primary Structure, Neural-specific Expression, and Chromosomal Localization of , a Second Murine Homeobox Gene Related to. J. Biol. Chem. 271 (37), 22624–22634. doi:10.1074/jbc.271.37.22624
Rauch, A., Wieczorek, D., Graf, E., Wieland, T., Endele, S., Schwarzmayr, T., et al. (2012). Range of Genetic Mutations Associated with Severe Non-syndromic Sporadic Intellectual Disability: an Exome Sequencing Study. Lancet 380 (9854), 1674–1682. doi:10.1016/s0140-6736(12)61480-9
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Keywords: trio-whole exome sequencing, developmental and epileptic encephalopathy 67, CUX2, levetiracetam treatment, CUX2 clinical phenotype
Citation: Zhang F, Li F, Chen F, Huang J, Luo Q, Du X, Zhou J, Gu W and Xu K (2022) Novel Variant Expands the Clinical Spectrum of CUX2-Associated Developmental and Epileptic Encephalopathies. Front. Genet. 13:808181. doi: 10.3389/fgene.2022.808181
Received: 03 November 2021; Accepted: 14 June 2022;
Published: 01 July 2022.
Edited by:
María L. Couce, Complejo Hospitalario Universitario de Santiago, SpainReviewed by:
Carlotta Stipa, IRCCS Institute of Neurological Sciences of Bologna (ISNB), ItalyCybel Mehawej, Lebanese American University, Lebanon
Isabella Peixoto De Barcelos, Children’s Hospital of Philadelphia, United States
Copyright © 2022 Zhang, Li, Chen, Huang, Luo, Du, Zhou, Gu and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kaishou Xu, eGtzeWlAMTI2LmNvbQ==