 Nan Li1†
Nan Li1† Harvest F. Gu2*†
Harvest F. Gu2*†- 1Department of Endocrinology, Jiangsu Province Hospital of Traditional Chinese Medicine, The Affiliated Hospital of Nanjing University of Chinese Medicine, Nanjing, China
- 2Laboratory of Molecular Medicine, School of Basic Medicine and Clinical Pharmacy, China Pharmaceutical University, Nanjing, China
The SLC12A3 (Solute carrier family 12 member 3) gene encodes a sodium-chloride cotransporter and mediates Na+ and Cl− reabsorption in the distal convoluted tubule of kidneys. An experimental study has previously showed that with knockdown of zebrafish ortholog, slc12a3 led to structural abnormality of kidney pronephric distal duct at 1-cell stage, suggesting that SLC12A3 may have genetic effects in renal disorders. Many clinical reports have demonstrated that the function-loss mutations in the SLC12A3 gene, mainly including Thr60Met, Asp486Asn, Gly741Arg, Leu859Pro, Arg861Cys, Arg913Gln, Arg928Cys and Cys994Tyr, play the pathogenic effects in Gitelman syndrome. This kidney disease is inherited as an autosomal recessive trait. In addition, several population genetic association studies have indicated that the single nucleotide variant Arg913Gln in the SLC12A3 gene is associated with diabetic kidney disease in type 2 diabetes subjects. In this review, we first summarized bioinformatics of the SLC12A3 gene and its genetic variation. We then described the different genetic and biological effects of SLC12A3 in Gitelman syndrome and diabetic kidney disease. We also discussed about further genetic and biological analyses of SLC12A3 as pharmacokinetic targets of diuretics.
Introduction
As the urine-producing organs, kidneys are vital in maintaining normal body functions because they are responsible for maintaining the balance of water, electrolytes, and the homeostasis of the internal environment through their filtration and reabsorption (Wallace, 1998). The function of kidneys is related with age. In general speaking, kidneys reach full functionality after the age of 5 years and suffer a slow and progressive decline in their regulatory range from the age of 20 years. Features of renal aging include several functional alterations, such as reduction of glomerular filtration rate, Na+ reabsorption, K+ secretion, vitamin D3 synthesis, titratable acid excretion, responsiveness to hormones, and regulatory flexibility (Gekle, 2017; Hommos et al., 2017). For the whole lifetime, a human being’s survival depends upon the crucial functions and processes performed by the kidneys (Jourde-Chiche et al., 2019). Renal disorders such as Gitelman syndrome (GS, presented in the patients after 6 years old), diabetic kidney disease (DKD, the patients are adults) may strike anyone at any age and at any time (Chevalier, 2019; Akhtar et al., 2020). GS is characterized by hypokalemia, hypomagnesaemia and metabolic alkalosis (Knoers and Levtchenko, 2008; Blanchard et al., 2017; Fujimura et al., 2018). DKD is associated with increased matrix expansion that manifests morphologically as a diffuse or nodular expansion of the mesangium and diffuse thickening of the glomerular and tubular basement membranes (Reidy et al., 2014; Pugliese et al., 2019).
A mutation is a change that occurs in DNA sequence either during DNA replication or as the result of exposure to environmental factors such as smoking, sunlight and radiation. A missense mutation in which a base change or substitution results in a codon that causes insertion of a different amino acid into the growing polypeptide chain, giving rise to an altered protein (Abramowics and Gos, 2018). Accumulating evidence has demonstrated that the mutations in the solute carrier family 12 member 3 (SLC12A3) gene, mainly including Thr60Met, Ala313Val AAsp486Asn, Gly741Arg, Arg861Cys, Leu859Pro, Cys994Tyr, Arg913Gln and Arg928Cys, cause GS (Knoers and Levtchenko, 2008; Vargas-Poussou et al., 2011; Blanchard et al., 2017; Fujimura et al., 2018; Zeng et al., 2019). Furthermore, the single nucleotide variant (SNV) Arg913Gln in the SLC12A3 gene is found to be associated with DKD in type 2 diabetes (T2D) subjects (Tanaka et al., 2003; Nishiyama et al., 2005; Abu Seman et al., 2014; De la Cruz-Cano et al., 2019). In this review, we first summarized bioinformatics of SLC12A3 and its genetic variation. We then described the different genetic and biological effects of the SLC12A3 gene mutations in GS and DKD. Finally, we discuss about further genetic and biological studies of SLC12A3, and its relationship with other renal transporters in kidneys.
Bioinformatics and Biological Function of SLC12A3
The kidneys are complex organs, and each human kidney has the averaged 900,000 nephrons. The nephron is basic functional and structural unit of kidneys. Structurally, the nephron consists of the glomerulus (capillaries and podocytes) located within the Bowman’s capsule and the renal tubules, including the proximal tubule, the Loop of Henle, and the distal tubule. Functionally, the nephron plays a role in the filtration and reabsorption of water and electrolytes and the secretion of wastes (Oxburgh, 2018; Kanzaki et al., 2020; Vallon and Thomson, 2020). SLC12A3 functions as a Na-Cl cotransporter and services for salt homeostasis by mediating Na-Cl transport along the renal distal convoluted tubule (DCT). Evidence from in vivo experimental studies has demonstrated that slc12a3 is expressed predominantly in DCT cells in rodents (Costanzo, 1985; Ellison, 2003; Pizzonia et al., 1991; Gesek and Friedman, 1992). A schematic diagram shows that SLC12A3, as a transmembrane protein, passes through the epithelial cell membrane of renal distal convoluted tubules 12 times and physiologically functions ti transport Na and Cl ions from extra- to intra-cellular luminal sides (Figures 1A,B). In many developed countries, a high-salt diet has become an important risk factor for high blood pressure and cardiovascular diseases (CVD), and the average daily consumption has usually exceeded twice the recommended dose (5–6 g/day) (Zhao et al., 2015). SLC12A3 is sensitive to thiazide. Salt homeostasis can be affected pharmacologically by diuretic drugs (Glover et al., 2011). SLC12A3 is a good drug target, and the target mechanism is to cause salt wasting within the distal kidney nephron. By using thiazide to inhibit the SLC12A3 gene activity, a negative sodium balance, may be induced with great health benefit in preventing hypertension and CVD. Therefore, thiazide-type diuretics are among the most widely used agents in the management of hypertension and CVD by blocking SLC12A3 (Vormfelde et al., 2003; Glover et al., 2011).
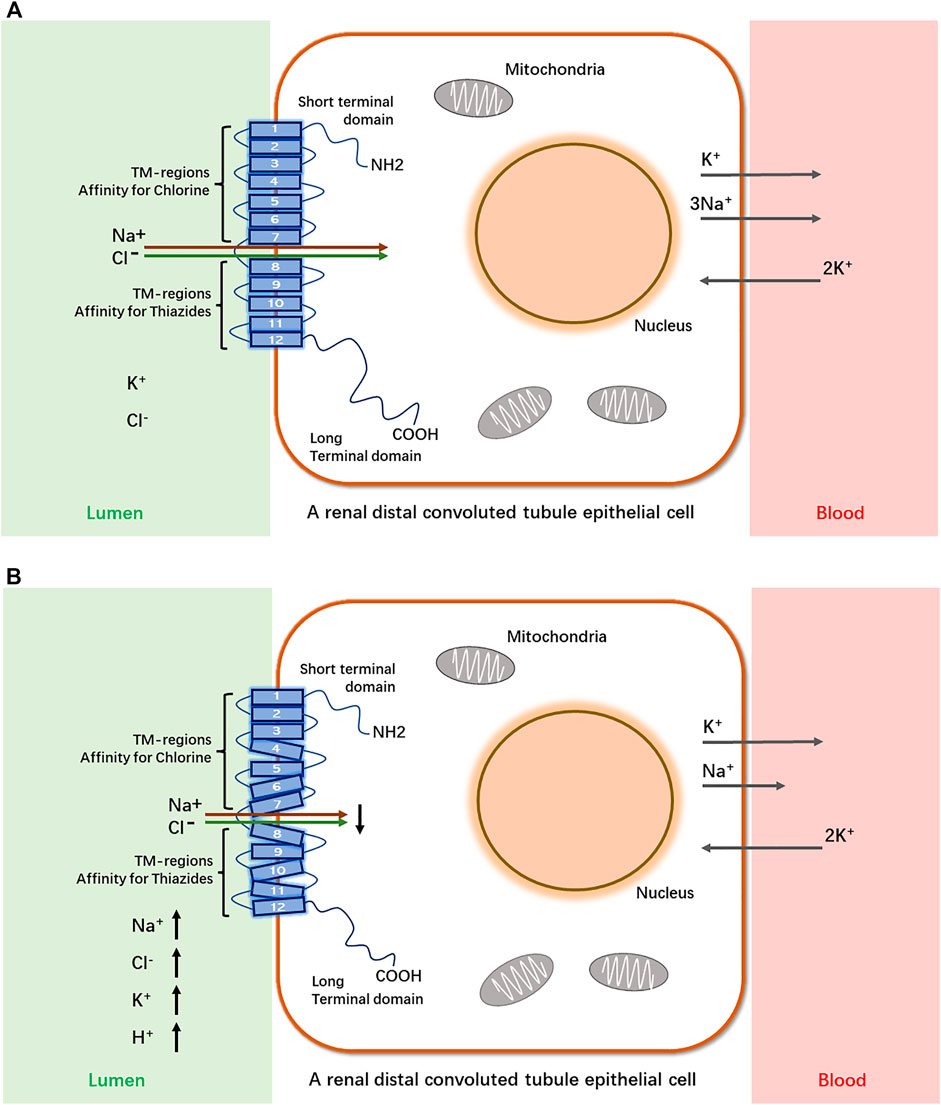
FIGURE 1. A schematic diagram of SLC12A3 as a transmembrane protein to transport Na+ and Cl− from extra- to intra-cellular luminal sides in a renal distal convoluted tubule epithelial cell and its dysfunction in Gitelman syndrome. SLC12A3 is a protein composed of cytoplasmic topological domains and helical transmembrane regions and flanked by a short amino-terminal domain (NH2) and a long carboxyterminal domain (COOH). (A) In heathy subjects, most of the Na+ and Cl− in distal convoluted tubule (DCT) of kidneys is reabsorbed by this protein. (B) In individuals with Gitelman syndrome, the dysfunction of SLC12A3 by inactivating mutations results in decreased reabsorption of Na+ and Cl− in DCT. As a heterotrimer, sodium channel epithelial 1 subunits alpha, beta, and gamma (SCNN1A, B and G) in the connecting tubule (CNT) and cortical collecting duct (CCD) function with co-reabsorption of Na+. While SLC12A3 is dysfunctional, SCNN1 (also known as ENaC) activity is compensatory increased, resulting in a greater tubular secretion of K+ and other ions (Mg2+and Na+) (Colussi et al., 1997).
We have searched for bioinformatics of the SLC12A3 gene mainly from GeneCards (https://www.genecards.org/cgi-bin/carddisp.pl?gene=SLC12A3) (Stelzer et al., 2016), and data are summarized in Table 1. Briefly, the SLC12A3 gene is localized in human chromosome 16q13. It should be emphasized that the SLC12A3 gene is 50,644 bp in length and consists of 26 exons. The first exon is relatively small and has a small 5′-untranslated region (UTR), while the last exon is the largest and has a large 3′-UTR. MicroRNAs (miRNAs) are endogenous RNAs and approximately 23 nt. They play important roles for gene-regulation to repress the gene activity by preferentially interacting with complementary sequence motifs in 3′-UTR of target mRNAs (Lai, 2002; Bartel, 2009). According to the information from genomic databases and literature searching, there are a total of 20 microRNAs that can regulate the SLC12A3 gene function (Table 1). Up to date, however, there is no report concerning the regulation of these miRNAs with the SLC12A3 gene expression in DKD except that Zhu Y et al. have reported an interaction between has-miRNA-6863 and SLC12A3 that potentially contribute to CVD (Zhu et al., 2019). In the promoter region of the SLC12A3 gene, there are several binding sites for transcription factors such as ATF6, E2F, GR (alpha and beta), ROR (alpha), TBP and USF1 but no CpG island exists. The SLC12A3 gene is predominantly and highly expressed in kidneys, while mRNA expression levels of this gene in adrenal gland, spleen, small intestine, and other tissues are very low. Data are adopted from GTEx, Illumina, BioGPS, and SAGE for SLC12A3 mRNA expression in normal human tissues (https://www.genecards.org/cgi-bin/carddisp.pl?gene=SLC12A3) and represented in Figure 2. In addition, the ortholog analysis has stated that the similarity of SLC12A3 in mRNA sequences between human and mouse is 87.08%. The slc12a3 mRNA expression levels are found to be over-expressed in kidneys of db/db mice from 6, 12, and 26 weeks at the age compared with the control mice at the same ages, suggesting that SLC12A3 may play an important role not only in the kidney cloacal development but also in progress of DKD (Abu Seman et al., 2014).
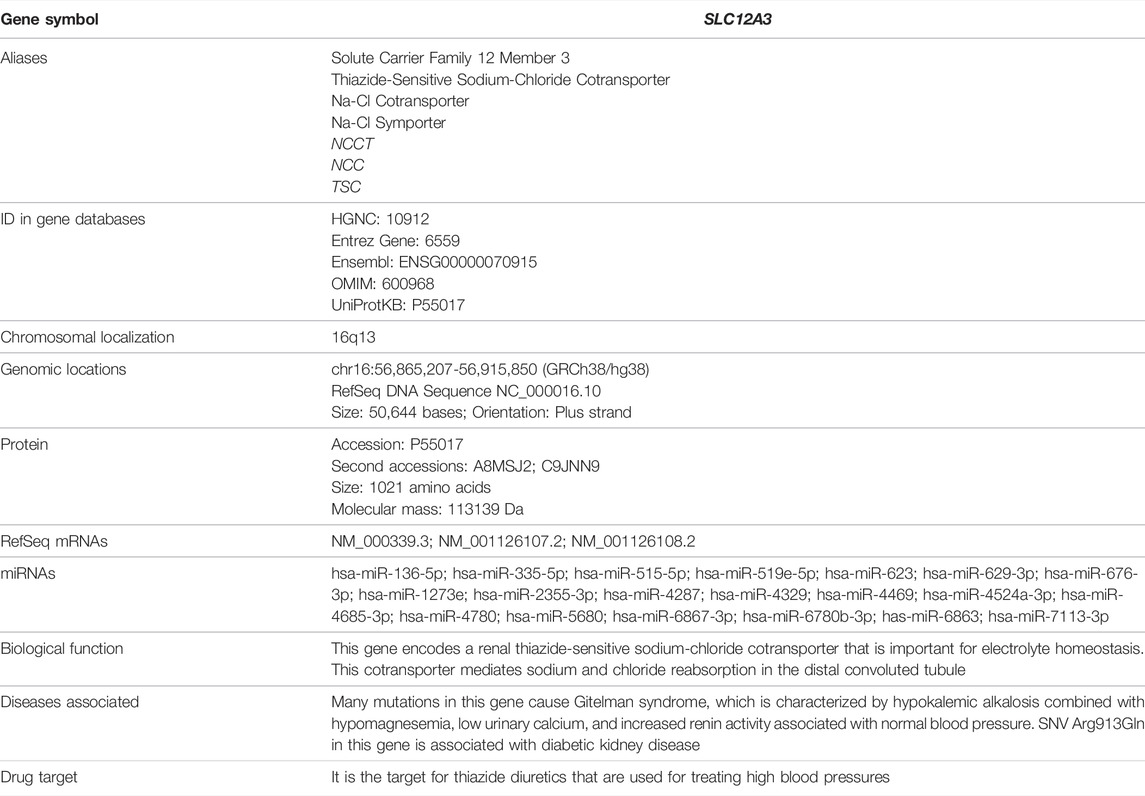
TABLE 1. Bioinformatics of the SLC12A3 gene.
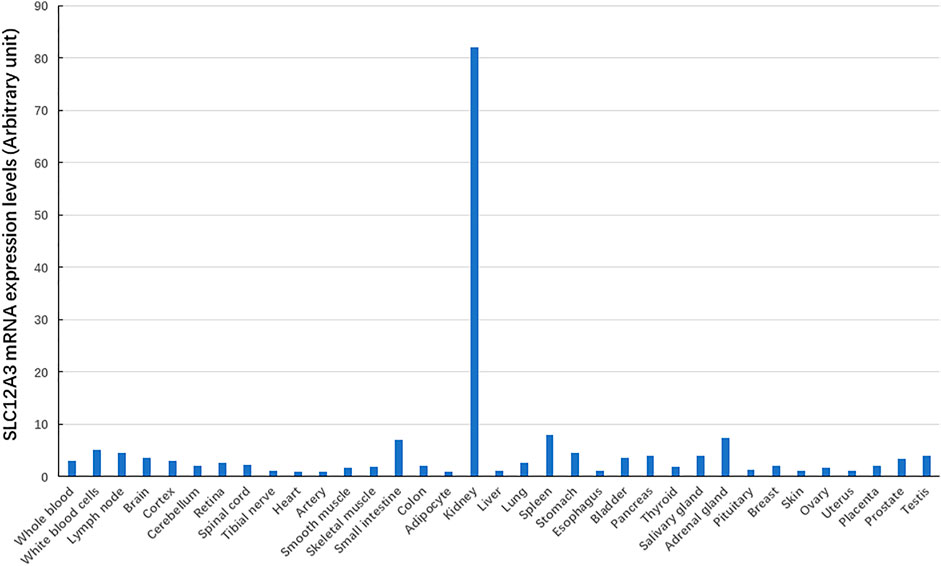
FIGURE 2. The SLC12A3 gene expression at mRNA levels in different tissues. The SLC12A3 gene expression at mRNA levels is predominant in kidneys, but low in adrenal gland, spleen, small intestine, and other tissues.
Zebrafish is a good alternative to mammalian models that can be used to apply powerful genetic experiments. In terms of kidney, zebrafish has proven itself to be the applicable and versatile experimental system, mainly due to the simplicity of its pronephros, which contains two nephrons that possess conserved structural and physiological aspects with mammalian nephrons (Morales and Wingert, 2017; Outtandy, 2019). The slc12a3 gene in zebrafish is conserved with 62% of amino acid identity compared with the human. We have previously applied a specific morpholino oligonucleotide-mediated antisense knockdown approach in zebrafish (Li, 2017) and found that the knockdown of zebrafish slc12a3 did not lead to global alteration of embryonic development compared to the wild-type embryos. Under the fluorescence microscopic analysis, pronephric duct epithelial structure defined by the signal (mCherry) in the cloacal portion was significantly altered at 4 dpf (Abu Seman et al., 2014). This has implicated that the SLC12A3 gene may have important genetic effects in renal diseases.
Genetic Effects of SLC12A3 in Gitelman Syndrome
Gene mutation refers to random alterations in DNA that occur in somatic and reproductive cells, often during replication and division. Effects of gene mutation can range from silent expression to self-destruction. In general, point mutations are classified as missense mutations (substitutions of bases can interfere with normal protein syntheses and functioning), nonsense mutations (occur when atypical base pairings produce a stop codon that may cause improper functioning or impede functioning altogether) and frameshift mutations (occur when a nucleotide pair is added or omitted in a gene sequence that shifts how codons are read and often result in different amino acids being added to the protein being synthesized). Furthermore, recent research has underlined the abundance and importance of splicing mutations in the etiology of inherited diseases. The splicing mutation may occur in both introns and exons and disrupt existing splice sites or splicing regulatory sequences (intronic and exonic splicing silencers and enhancers), create new ones, or activate the cryptic ones (Abramowics and Gos, 2018). Up to date, application of modern techniques has allowed us to identify the genetic variants in the SLC12A3 gene, which typically cause GS.
GS is an autosomal recessive salt-losing renal tubulopathy disorder and characterized by hypokalemia, hypomagnesemia, hypocalciuria and secondary aldosteronism (Simon et al., 1996; Lemmink et al., 1998). The major clinical characters of GS are represented in Table 2 as below. GS may be the most frequent inherited renal tubular disorders. The prevalence is estimated at ∼25 per million and accordingly, the prevalence of heterozygotes is ∼1% in Caucasian populations (Vargas-Poussou et al., 2011). The prevalence of heterozygotes in Chinese populations is ∼3%, relatively higher than what in Caucasians (Mastroianni et al., 1996; Lin et al., 2004; Hsu et al., 2009; Takeuchi et al., 2015; Zeng et al., 2019). Accumulating reports have demonstrated that many genetic variants of the SLC12A3 gene, including missense mutations, insertion or deletion, and others such as single nucleotide variants (SNV) or repeated sequences, cause the loss of function of this gene and are responsible for the phenotypes in GS. In general speaking, point mutations in exon are categorized into missense, silent, or nonsense mutations, while missense alterations can induce the exclusion of an individual exon in various diseases. Hsu YJ et al. have analyzed DNA samples of 500 unrelated Chinese children by using PCR and restriction fragment length polymorphism approach and found that 15 mutations in the SLC12A3 gene are associated with GS. The overall incidence of positive heterozygous mutations in the SLC12A3 gene is 2.9%. There is no significant difference in systolic or diastolic blood pressure, biochemical profiles, or urine pH between children with heterozygous SLC12A3 mutations and non-affected controls (Hsu et al., 2009). Takeuchi Y et al. have used a bioinformatics program to analyze 88 missense mutations in the SLC12A3 gene and identified several mutations that may induce exon skipping in the gene (Takeuchi et al., 2015). Furthermore, Vargas-Poussou R et al. have reported that most of GS patients (70%) carry two mutations of the SLC12A3 gene based upon sequencing analysis of genomic DNA from a large cohort of 448 unrelated patients suspected of having GS (Vargas-Poussou et al., 2011). Recently, Zeng Y et al. have reported 90 mutations in the SLC12A3 gene based upon literature searching about Chinese patients with GS in the PubMed database and analyzing 8 GS Chinese patients (Zeng et al., 2019). Considering there is an overlap of the reported mutations in the SLC12A3 gene among GS patients, in this review, we have summarized and represent a list of all reported SLC12A3 genetic variants up to October 2021 in Table 3. There are a total of 150 genetic variants in the SLC12A3 gene. Of them, 86% are missense mutations. Other variants are ins/del, SNV and CNV. In Caucasians GS patients, 5 most frequent missense mutations in the SLC12A3 gene include Ala313Val, Gly741Arg, Arg861Cys, Leu859Pro and Cys994Tyr (blue letters in Figure 3A). Among Chinese GS patients, the most common missense mutations are Thr60Met, Asp486Asn, Arg913Gln and Arg928Cys (brown letters in Figure 3A). SLC12A3 protein (ID: P55017 in UniProtKB) is structured with six cytoplasmic topological domains and 11 helical trans-membranes (Figure 3B and https://alphafold.ebi.ac.uk/entry/P55017). The dysfunction of SLC12A3 in DCT, caused by SLC12A3 gene mutations, lead to GS, because the amino acid changes caused by these mutations result in the changes in the tertiary structure of the protein, and the function of the protein is reduced (Figure 3C). So far, our understanding of the changes in the functional characteristics of the most frequent SLC12A3 mutations in GS is still limited. The main reason is that such experimental research has a certain degree of difficulty. First, different frequent mutations of different populations have indicated location and ancestral diversity of SLC12A3 gene mutation (Maki et al., 2004; Ma et al., 2016). Second, different gene mutations can cause different changes in protein structure and function (Pandurangan and Blundell, 2020). In recent years, however, a few studies have integrated the protein configurations with the function of SLC12A3 mutations in vitro and in vivo and implicated that different mutated SLC12A3 may result in a mutation-triggered reduction in SLC12A3 protein expression, a reduction in the abundance at the plasma membrane, an impairment of protein glycosylation, and/or a disruption of phosphorylation (Yang et al., 2013; Valdez-Flores et al., 2016; Jiang et al., 2021). For instance, Thr60Met is one of most frequent SLC12A3 mutations in GS. Allele Thr60, as an important phosphorylation site, is very important for the membrane expression of SLC12A3 and phosphorylation of the adjacent Thr46 and Thr55 sites (Yang et al., 2013; Jiang et al., 2021). Furthermore, sodium channel epithelial 1 subunits alpha, beta, and gamma (SCNN1A, B and G) in the connecting tubule (CNT) and cortical collecting duct (CCD) function with co-reabsorption of Na+. When SLC12A3 is dysfunctional, SCNN1 activity is compensatory increased, resulting in a greater tubular secretion of K+ and other ions (such as Mg2+and Na+) (Hanukoglu and Hanukoglu, 2016) (Figure 1B).
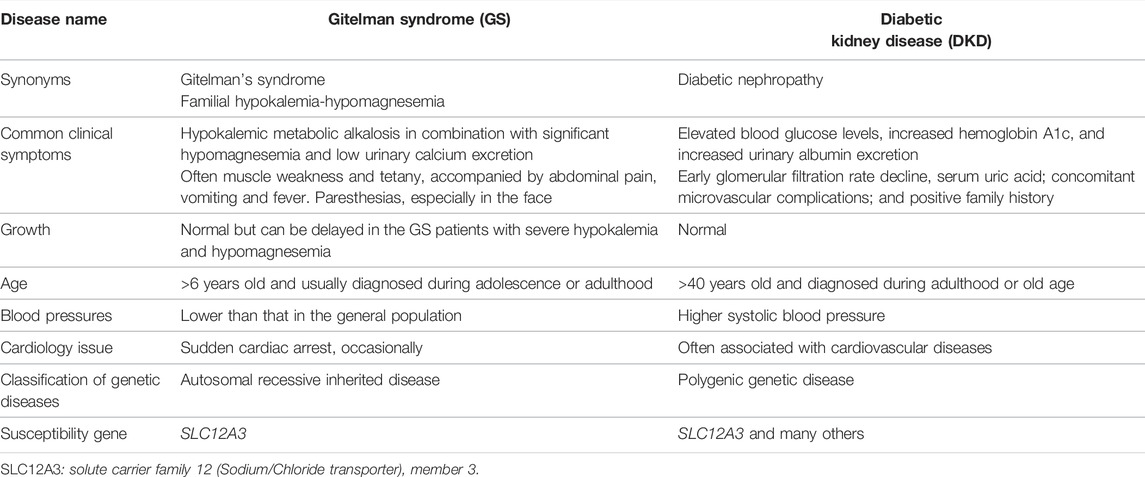
TABLE 2. Clinical characteristics and genetic disease classification of Gitelman syndrome and diabetic kidney disease.
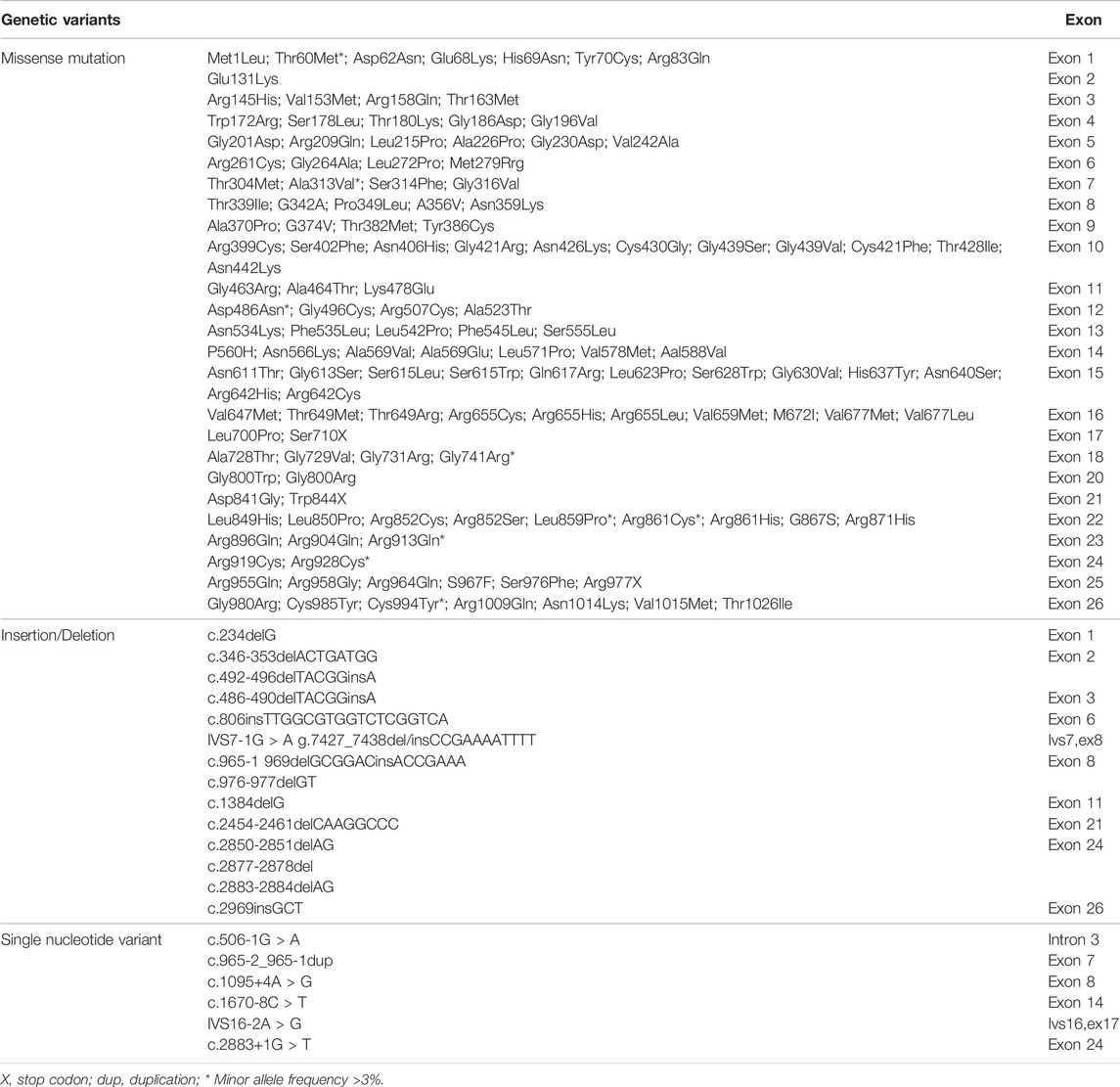
TABLE 3. Genetic variation in the SLC12A3 gene in Gitelman syndrome.
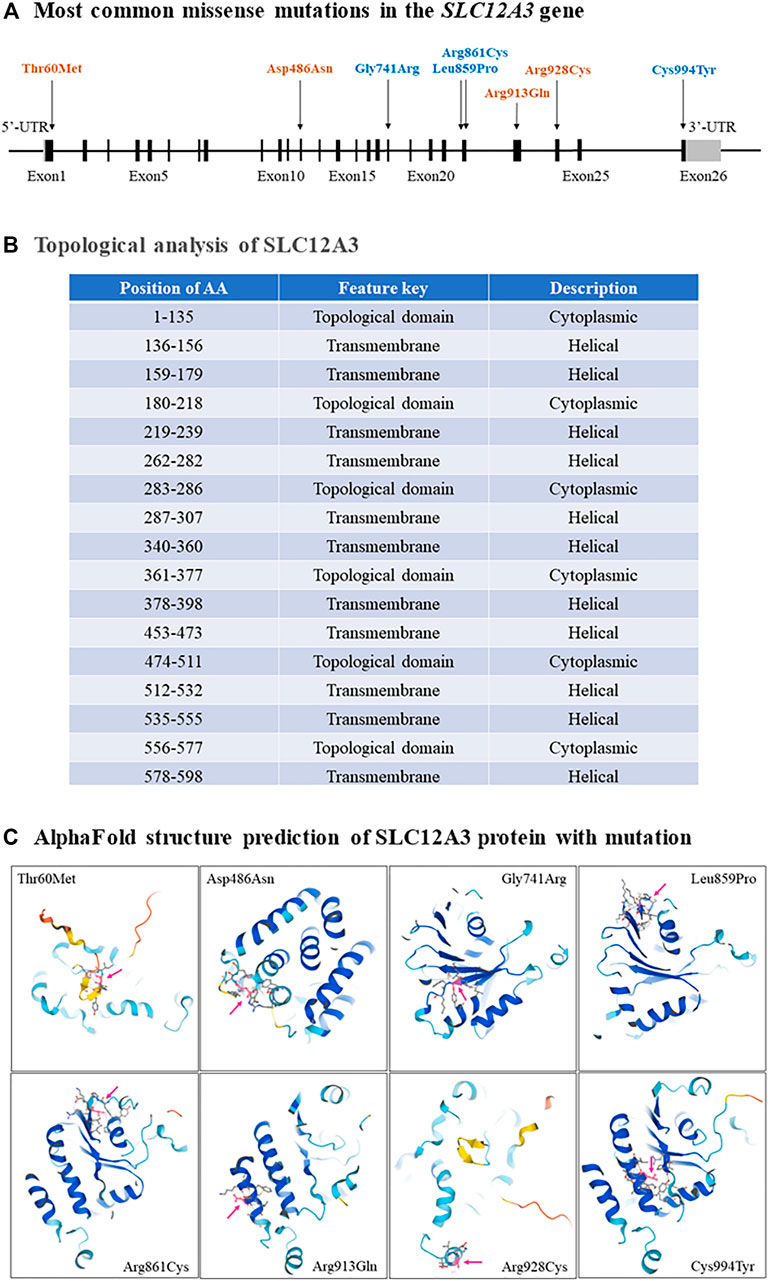
FIGURE 3. The most common missense mutations in the SLC12A3 gene. (A) A schematic diagram has shown that the most common missense mutations, including Thr60Met, Asp486Asn, Gly741Arg, Leu859Pro, Arg861Cys, Arg913Gln, Arg928Cys and Cys994Tyr, are located in exons 1, 12, 18, 22, 23, 24 and 26 of the SLC12A3 gene. (B) Topological analyses have indicated that all these missense mutations result in the changes of cytoplasmic topological domain but not in helical transmembrane. (C) The changes of SLC12A3 protein structure with each missense mutation are predicted respectively by using the AlphaFold (Jumper et al., 2021).
Genetic Effects of SLC12A3 in Diabetic Kidney Disease
DKD has a complex etiology due to synergistic interplay among genetic, epigenetic, and environmental factors. In recent years, researchers have undertaken genetic and epigenetic studies of DKD to better understand its molecular mechanisms (Gu, 2019). Tanaka et al. have previously performed a genome-wide analysis of gene-based single nucleotide variants (SNV) in a Japanese population and reported that SNV Arg913Gln in the SLC12A3 gene is associated with reduced risk to DKD in T2D (Tanaka et al., 2003). Another 10-years longitudinal study in the same population suggests that the 913Gln allele of this polymorphism may confer a protective effect in DKD (Nishiyama et al., 2005). After then, the association of Arg913Gln in the SLC12A3 gene with DKD has been confirmed with a genetic study in Malaysian population (Abu Seman et al., 2014). Db/db mice are characterized by hyperglycemia, obese and urinary albumin excretion enhancement and have been widely used as a genetic rodent model for study of T2D and DKD (Sharma et al., 2003; Wang et al., 2014). Abu Seman et al. have carried out the experiments to investigate the slc12a3 gene expression at both mRNA and protein levels in kidneys of db/db mice at the ages of 6, 12, and 26 weeks. Results demonstrate that slc12a3 expression levels at the stages of young (6 weeks old), diabetes (12 weeks old) and DKD conditions (26 weeks old) are higher than what in the control mice respectively. This implicates that SLC12A3 is a susceptibility gene to DKD (Abu Seman et al., 2014). Furthermore, genetic studies in Japanese and Malaysian subjects with T2D-DKD demonstrate that the mutant function-loss allele 913Glu has the protective genetic effects to T2D and DKD (Tanaka et al., 2003; Nishiyama et al., 2005; Abu Seman et al., 2014). However, the association between the SLC12A3 genetic polymorphisms and DKD is not detectable in a study with American Caucasians and possibly due to the limited statistical power in sample size (Ng et al., 2008). Furthermore, there is disagreement in the literature concerning the role of the Arg913Gln allele in diabetes and DKD, since it has been both as a predisposing and a protective allele. Two other studies in Korean and Chinese populations have considered the risk of allele 913Gln for DKD (Kim et al., 2006; Zhang et al., 2018). This disagreement is probably caused by misjudgment of the two alleles of SNV Arg913Gln in the SLC12A3 gene but unlikely due to the population specificity.
Summary and Perspectives
Taking together, GS is a salt-losing tubulopathy characterized by hypokalemic alkalosis with hypomagnesemia and hypocalciuria. Up to date, more than 150 mutations in the SLC12A3 gene have been identified in GS patients. Most frequent missense mutations in the SLC12A3 gene include Thr60Met, Ala313Val, Gly741Arg, Arg861Cys, Leu859Pro Asp486Asn, Arg913Gln, Arg928Cys and Cys994Tyr. The mutant alleles of these missense mutation are function-loss-variants. One of them, i.e. Arg913Gln in the SLC12A3 gene is found to be associated with DKD as well. Based upon what we have described in this review, we suggest that in clinic, GS should be checked for adolescents or adults with hypokalemia, hypomagnesemia, or hypocalciuria with metabolic alkalosis, normal blood pressures. The gene detection method such as the direct sequencing of the SLC12A3 gene and identification of the specific mutations in the gene can be performed. The next-generation sequencing technology and multiple ligation probe amplification technology have been gradually applied for diagnosis of GS (Kim et al., 2016; Ishida and Gupta, 2021).
Author Contributions
LN contributed clinical data; HG provided with genetic information; Both prepared and revised the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors wish to thank for research grant supports from National Natural Science Foundation of China (82104751 NL) and China Pharmaceutical University (20180815 HG).
Abbreviation
ALPL, alkaline phosphatase, biomineralization associated; CCD, cortical collecting duct; CNT, connecting tubule; COL4A4, collagen type IV alpha 4 chain; CVD, cardiovascular diseases; DCT, distal convoluted tubule; DKD, diabetic kidney disease; GFR, glomerular filtration rate; GS, gitelman syndrome; MEFV, MEFV innate immuity regulator, pyrin; PCT, proximal convoluted tubule; S100A12, s100 calcium binding protein A12; SCNN1, sodium channel epithelial 1 subunit; SLC12A3, solute carrier family 12 member 3; SNV, single nucleotide variant; T2D, type 2 diabetes; UTR, untranslated region.
References
Abramowics, A., and Gos, M. (2018). Splicing Mutations in Human Genetic Disorders: Examples, Detection, and Confirmation. Journal of Applied Genetics 59 (3), 253–268. doi:10.1007/s13353-018-0444-7
Abu Seman, N., He, B., Ojala, J. R. M., Wan Mohamud, W. N., Östenson, C.-G., Brismar, K., et al. (2014). Genetic and Biological Effects of Sodium-Chloride Cotransporter (SLC12A3) in Diabetic Nephropathy. Am. J. Nephrol. 40 (5), 408–416. doi:10.1159/000368916
Akhtar, M., Taha, N. M., Nauman, A., Mujeeb, I. B., and Al-Nabet, A. D. M. H. (2020). Diabetic Kidney Disease: Past and Present. Adv. Anat. Pathol. 27 (2), 87–97. doi:10.1097/pap.0000000000000257
Bartel, D. P. (2009). MicroRNAs: Target Recognition and Regulatory Functions. Cell 136 (2), 215–233. doi:10.1016/j.cell.2009.01.002
Blanchard, A., Bockenhauer, D., Bolignano, D., Calò, L. A., Cosyns, E., Devuyst, O., et al. (2017). Gitelman Syndrome: Consensus and Guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 91 (1), 24–33. doi:10.1016/j.kint.2016.09.046
Chevalier, R. L. (2019). Evolution, Kidney Development, and Chronic Kidney Disease. Semin. Cell Developmental Biol. 91, 119–131. doi:10.1016/j.semcdb.2018.05.024
Colussi, G., Rombolà, G., Brunati, C., and De Ferrari, M. E. (1997). Abnormal Reabsorption of Na+/CI- by the Thiazide-Lnhibitable Transporter of the Distal Convoluted Tubule in Gitelman’s Syndrome. Am. J. Nephrol. 17 (2), 103–111. doi:10.1159/000169082
Costanzo, L. S. (1985). Localization of Diuretic Action in Microperfused Rat Distal Tubules: Ca and Na Transport. Am. J. Physiology-Renal Physiol. 248, F527–F535. doi:10.1152/ajprenal.1985.248.4.f527
De la Cruz-Cano, E., Jiménez-González, C. d. C., Morales-García, V., Pineda-Pérez, C., Tejas-Juárez, J. G., Rendón-Gandarilla, F. J., et al. (2019). Arg913Gln Variation of SLC12A3 Gene Is Associated with Diabetic Nephropathy in Type 2 Diabetes and Gitelman Syndrome: a Systematic Review. BMC Nephrol. 20 (1), 393. doi:10.1186/s12882-019-1590-9
Ellison, D. H. (2003). The Thiazide-Sensitive Na-Cl Cotransporter and Human Disease: Reemergence of an Old Player. Jasn 14 (2), 538–540. doi:10.1681/asn.v142538
Fujimura, J., Nozu, K., Yamamura, T., Minamikawa, S., Nakanishi, K., Horinouchi, T., et al. (2018). Clinical and Genetic Characteristics in Patients with Gitelman Syndrome. Kidney Int. Rep. 4 (1), 119–125. doi:10.1016/j.ekir.2018.09.015
Gekle, M. (2017). Kidney and Aging - A Narrative Review. Exp. Gerontol. 87 (Pt B), 153–155. doi:10.1016/j.exger.2016.03.013
Gesek, F. A., and Friedman, P. A. (1992). Mechanism of Calcium Transport Stimulated by Chlorothiazide in Mouse Distal Convoluted Tubule Cells. J. Clin. Invest. 90, 429–438. doi:10.1172/jci115878
Glover, M., Zuber, A. M., and O'Shaughnessy, K. M. (2011). Hypertension, Dietary Salt Intake, and the Role of the Thiazide-Sensitive Sodium Chloride Transporter NCCT. Cardiovasc. Ther. 29 (1), 68–76. doi:10.1111/j.1755-5922.2010.00180.x
Gu, H. F. (2019). Genetic and Epigenetic Studies in Diabetic Kidney Disease. Front. Genet. 10, 507. doi:10.3389/fgene.2019.00507
Hanukoglu, I., and Hanukoglu, A. (2016). Epithelial Sodium Channel (ENaC) Family: Phylogeny, Structure-Function, Tissue Distribution, and Associated Inherited Diseases. Gene 579 (2), 95–132. doi:10.1016/j.gene.2015.12.061
Hommos, M. S., Glassock, R. J., and Rule, A. D. (2017). Structural and Functional Changes in Human Kidneys with Healthy Aging. Jasn 28 (10), 2838–2844. doi:10.1681/asn.2017040421
Hsu, Y. J., Yang, S. S., Chu, N. F., Sytwu, H. K., Cheng, C. J., and Lin, S. H. (2009). Heterozygous Mutations of the Sodium Chloride Cotransporter in Chinese Children: Prevalence and Association with Blood Pressure. Nephrol. Dial. Transpl. 24 (4), 1170–1175. doi:10.1093/ndt/gfn619
Ishida, C., and Gupta, V. (2021). in StatPearls (Internet) (Treasure Island (FL): StatPearls Publishing).Genetics, Molecular Testing
Jiang, L., Peng, X., Zhao, B., Zhang, L., Xu, L., Li, X., et al. (2021). Frequent SLC12A3 Mutations in Chinese Gitelman Syndrome Patients: Structure and Function Disorder. Endocr. Connect 11 (1), e210262. doi:10.1530/EC-21-0262
Jourde-Chiche, N., Fakhouri, F., Dou, L., Bellien, J., Burtey, S., Frimat, M., et al. (2019). Endothelium Structure and Function in Kidney Health and Disease. Nat. Rev. Nephrol. 15 (2), 87–108. doi:10.1038/s41581-018-0098-z
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly Accurate Protein Structure Prediction with AlphaFold. Nature 596 (7873), 583–589. doi:10.1038/s41586-021-03819-2
Kanzaki, G., Tsuboi, N., Shimizu, A., and Yokoo, T. (2020). Human Nephron Number, Hypertension, and Renal Pathology. Anat. Rec. 303 (10), 2537–2543. doi:10.1002/ar.24302
Kim, J. H., Shin, H. D., Park, B. L., Moon, M. K., Cho, Y. M., Hwang, Y. H., et al. (2006). SLC12A3 (Solute Carrier Family 12 Member (Sodium/chloride) 3) Polymorphisms Are Associated with End-Stage Renal Disease in Diabetic Nephropathy. Diabetes 55, 843–848. doi:10.2337/diabetes.55.03.06.db05-1013
Kim, M. J., Cho, S. I., Chae, J.-H., Lim, B. C., Lee, J.-S., Lee, S. J., et al. (2016). Pitfalls of Multiple Ligation-dependent Probe Amplifications in Detecting DMD Exon Deletions or Duplications. J. Mol. Diagn. 18 (2), 253–259. doi:10.1016/j.jmoldx.2015.11.002
Knoers, N. V., and Levtchenko, E. N. (2008). Gitelman Syndrome. Orphanet J. Rare Dis. 3, 22. doi:10.1186/1750-1172-3-22
Lai, E. C. (2002). Micro RNAs Are Complementary to 3′ UTR Sequence Motifs that Mediate Negative post-transcriptional Regulation. Nat. Genet. 30 (4), 363–364. doi:10.1038/ng865
Lemmink, H. H., Knoers, N. V. A. M., Károlyi, L., van Dijk, H., Niaudet, P., Antignac, C., et al. (1998). Novel Mutations in the Thiazide-Sensitive NaCl Cotransporter Gene in Patients with Gitelman Syndrome with Predominant Localization to the C-Terminal Domain. Kidney Int. 54, 720–730. doi:10.1046/j.1523-1755.1998.00070.x
Li, Y.-F. (2017). End-Modifications on Morpholino Oligos. Methods Mol. Biol. 1565, 39–50. doi:10.1007/978-1-4939-6817-6_4
Lieber, M., Kallinich, T., Lohse, P., Klotsche, J., Holzinger, D., Foell, D., et al. (2015). Increased Serum Concentrations of Neutrophil-Derived Protein S100A12 in Heterozygous Carriers of MEFV Mutations. Clin. Exp. Rheumatol. 33 (6 Suppl. 94), S113–S116.
Lin, S.-H., Cheng, N.-L., Hsu, Y.-J., and Halperin, M. L. (2004). Intrafamilial Phenotype Variability in Patients with Gitelman Syndrome Having the Same Mutations in Their Thiazide-Sensitive Sodium/chloride Cotransporter. Am. J. Kidney Dis. 43, 304–312. doi:10.1053/j.ajkd.2003.10.018
Ma, J., Ren, H., Lin, L., Zhang, C., Wang, Z., Xie, J., et al. (2016). Genetic Features of Chinese Patients with Gitelman Syndrome: Sixteen Novel SLC12A3 Mutations Identified in a New Cohort. Am. J. Nephrol. 44 (2), 113–121. doi:10.1159/000447366
Maki, N., Komatsuda, A., Wakui, H., Ohtani, H., Kigawa, A., Aiba, N., et al. (2004). Four Novel Mutations in the Thiazide-Sensitive Na-Cl Co-transporter Gene in Japanese Patients with Gitelman's Syndrome. Nephrol. Dial. Transplant. 19 (7), 1761–1766. doi:10.1093/ndt/gfh239
Mastroianni, N., Bettinelli, A., Bianchetti, M., Colussi, G., De Fusco, M., Sereni, F., et al. (1996). Novel Molecular Variants of the Na-Cl Cotransporter Gene Are Responsible for Gitelman Syndrome. Am. J. Hum. Genet. 59, 1019–1026.
Morales, E. E., and Wingert, R. A. (2017). Zebrafish as a Model of Kidney Disease. Results Probl. Cell Differ 60, 55–75. doi:10.1007/978-3-319-51436-9_3
Ng, D. P. K., Nurbaya, S., Choo, S., Koh, D., Chia, K.-s., and Krolewski, A. S. (2008). Genetic Variation at the SLC12A3 Locus Is Unlikely to Explain Risk for Advanced Diabetic Nephropathy in Caucasians with Type 2 Diabetes. Nephrol. Dial. Transplant. 23, 2260–2264. doi:10.1093/ndt/gfm946
Nishiyama, K., Tanaka, Y., Nakajima, K., Mokubo, A., Atsumi, Y., Matsuoka, K., et al. (2005). Polymorphism of the Solute Carrier Family 12 (Sodium/chloride Transporters) Member 3, SLC12A3, Gene at Exon 23 (+78G/A: Arg913Gln) Is Associated with Elevation of Urinary Albumin Excretion in Japanese Patients with Type 2 Diabetes: a 10-year Longitudinal Study. Diabetologia 48, 1335–1338. doi:10.1007/s00125-005-1785-4
Outtandy, P., Russell, C., Kleta, R., and Bockenhauer, D. (2019). Zebrafish as a Model for Kidney Function and Disease. Pediatr. Nephrol. 34 (5), 751–762. doi:10.1007/s00467-018-3921-7
Oxburgh, L. (2018). Kidney Nephron Determination. Annu. Rev. Cell Dev. Biol. 34, 427–450. doi:10.1146/annurev-cellbio-100616-060647
Pandurangan, A. P., and Blundell, T. L. (2020). Prediction of Impacts of Mutations on Protein Structure and Interactions: SDM, a Statistical Approach, and mCSM, Using Machine Learning. Protein Sci. 29 (1), 247–257. doi:10.1002/pro.3774
Pizzonia, J. H., Gesek, F. A., Kennedy, S. M., Coutermarsh, B. A., Bacskai, B. J., and Friedman, P. A. (1991). Immunomagnetic Separation, Primary Culture, and Characterization of Cortical Thick Ascending Limb Plus Distal Convoluted Tubule Cells from Mouse Kidney. Vitro Cell Dev Biol - Anim. 27, 409–416. doi:10.1007/bf02630961
Pugliese, G., Penno, G., Natali, A., Barutta, F., Di Paolo, S., Reboldi, G., et al. (2019). Diabetic Kidney Disease: New Clinical and Therapeutic Issues. Joint Position Statement of the Italian Diabetes Society and the Italian Society of Nephrology on "The Natural History of Diabetic Kidney Disease and Treatment of Hyperglycemia in Patients with Type 2 Diabetes and Impaired Renal Function". Nutr. Metab. Cardiovasc. Dis. 29 (11), 1127–1150. doi:10.1016/j.numecd.2019.07.017
Reidy, K., Kang, H. M., Hostetter, T., and Susztak, K. (2014). Molecular Mechanisms of Diabetic Kidney Disease. J. Clin. Invest. 124 (6), 2333–2340. doi:10.1172/jci72271
Sharma, K., McCue, P., and Dunn, S. R. (2003). Diabetic Kidney Disease in Thedb/dbmouse. Am. J. Physiology-Renal Physiol. 284 (6), F1138–F1144. doi:10.1152/ajprenal.00315.2002
Simon, D. B., Nelson-Williams, C., Johnson Bia, M., Ellison, D., Karet, F. E., Morey Molina, A., et al. (1996). Gitelman's Variant of Barter's Syndrome, Inherited Hypokalaemic Alkalosis, Is Caused by Mutations in the Thiazide-Sensitive Na-Cl Cotransporter. Nat. Genet. 12 (1), 24–30. doi:10.1038/ng0196-24
Stelzer, Y., Wu, H., Song, Y., Shivalila, C. S., Markoulaki, S., Jaenisch, R., et al. (2016). Parent-of-Origin DNA Methylation Dynamics During Mouse Development. Cell Reports 16 (12), 3167–3180. doi:10.1016/j.celrep.2016.08.066
Taillandier, A., Domingues, C., Dufour, A., Debiais, F., Guggenbuhl, P., Roux, C., et al. (2018). Genetic Analysis of Adults Heterozygous for ALPL Mutations. J. Bone Miner Metab. 36 (6), 723–733. doi:10.1007/s00774-017-0888-6
Takeuchi, Y., Mishima, E., Shima, H., Akiyama, Y., Suzuki, C., Suzuki, T., et al. (2015). Exonic Mutations in the SLC12A3 Gene Cause Exon Skipping and Premature Termination in Gitelman Syndrome. Jasn 26 (2), 271–279. doi:10.1681/asn.2013091013
Tanaka, N., Babazono, T., Saito, S., Sekine, A., Tsunoda, T., Haneda, M., et al. (2003). Association of Solute Carrier Family 12 (Sodium/chloride) Member 3 with Diabetic Nephropathy, Identified by Genome-wide Analyses of Single Nucleotide Polymorphisms. Diabetes 52 (11), 2848–2853. doi:10.2337/diabetes.52.11.2848
Valdez-Flores, M. A., Vargas-Poussou, R., Verkaart, S., Tutakhel, O. A. Z., Valdez-Ortiz, A., Blanchard, A., et al. (2016). Functionomics of NCC Mutations in Gitelman Syndrome Using a Novel Mammalian Cell-Based Activity Assay. Am. J. Physiology-Renal Physiol. 311 (6), F1159–F1167. doi:10.1152/ajprenal.00124.2016
Vallon, V., and Thomson, S. C. (2020). The Tubular Hypothesis of Nephron Filtration and Diabetic Kidney Disease. Nat. Rev. Nephrol. 16 (6), 317–336. doi:10.1038/s41581-020-0256-y
Vargas-Poussou, R., Dahan, K., Kahila, D., Venisse, A., Riveira-Munoz, E., Debaix, H., et al. (2011). Spectrum of Mutations in Gitelman Syndrome. Jasn 22 (4), 693–703. doi:10.1681/asn.2010090907
Vormfelde, S. V., Burckhardt, G., Zirk, A., Wojnowski, L., and Brockmöller, J. (2003). Pharmacogenomics of Diuretic Drugs: Data on Rare Monogenic Disorders and on Polymorphisms and Requirements for Further Research. Pharmacogenomics 4 (6), 701–734. doi:10.1517/phgs.4.6.701.22817
Wallace, M. A. (1998). Anatomy and Physiology of the Kidney. AORN J. 68 (5), 800. doi:10.1016/s0001-2092(06)62377-6
Wang, B., Chandrasekera, P., and Pippin, J. (2014). Leptin- and Leptin Receptor-Deficient Rodent Models: Relevance for Human Type 2 Diabetes. Cdr 10 (2), 131–145. doi:10.2174/1573399810666140508121012
Yang, S.-S., Fang, Y.-W., Tseng, M.-H., Chu, P.-Y., Yu, I.-S., Wu, H.-C., et al. (2013). Phosphorylation Regulates NCC Stability and Transporter Activity In Vivo. Jasn 24 (10), 1587–1597. doi:10.1681/asn.2012070742
Zeng, Y., Li, P., Fang, S., Wu, C., Zhang, Y., Lin, X., et al. (2019). Genetic Analysis of SLC12A3 Gene in Chinese Patients with Gitelman Syndrome. Med. Sci. Monit. 25, 5942–5952. doi:10.12659/msm.916069
Zhang, R., Zhuang, L., Li, M., Zhang, J., Zhao, W., Ge, X., et al. (2018). Arg913Gln of SLC12A3 Gene Promotes Development and Progression of End-Stage Renal Disease in Chinese Type 2 Diabetes Mellitus. Mol. Cell Biochem 437 (1-2), 203–210. doi:10.1007/s11010-017-3120-z
Zhao, F., Zhang, P., Zhang, L., Niu, W., Gao, J., Lu, L., et al. (2015). Consumption and Sources of Dietary Salt in Family Members in Beijing. Nutrients 7 (4), 2719–2730. doi:10.3390/nu7042719
Keywords: diabetic kidney disease, genetic variant, gitelman syndrome, SLC12a3, sodium and chloride reabsorption
Citation: Li N and Gu HF (2022) Genetic and Biological Effects of SLC12A3, a Sodium-Chloride Cotransporter, in Gitelman Syndrome and Diabetic Kidney Disease. Front. Genet. 13:799224. doi: 10.3389/fgene.2022.799224
Received: 21 October 2021; Accepted: 12 January 2022;
Published: 03 May 2022.
Edited by:
Radha Dutt Singh, University of Calgary, CanadaReviewed by:
Nadia Akawi, United Arab Emirates University, United Arab EmiratesGiacomo Colussi, Niguarda Ca’ Granda Hospital, Italy
Birgitte Moenster Christensen, Aarhus University, Denmark
Copyright © 2022 Li and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Harvest F. Gu, ZmVuZy5ndUBjcHUuZWR1LmNu
†ORCID: Nan Li, orcid.org/0000000233412373; Harvest F. Gu, orcid.org/0000000345785449