 Yajuan Gao
Yajuan Gao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 23 May 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.797124
This article is part of the Research Topic The Genetics of Human Mendelian Skin Disorders View all 11 articles
Background: Gap junctions formed by connexins are channels on cytoplasm functioning in ion recycling and homeostasis. Some members of connexin family including connexin 31 are significant components in human skin and cochlea. In clinic, mutations of connexin 31 have been revealed as the cause of a rare hereditary skin disease called erythrokeratodermia variabilis (EKV) and non-syndromic hearing loss (NSHL).
Objective: To determine the underlying genetic cause of EKV, ichthyosis and NSHL in three members of a Chinese pedigree and skin histologic characteristics of the EKV patient.
Methods: By performing whole exome sequencing (WES), Sanger sequencing and skin biopsy, we demonstrate a Chinese pedigree carrying a mutation of GJB3 with three patients separately diagnosed with EKV, ichthyosis and NSHL.
Results: The proband, a 6-year-old Chinese girl, presented with demarcated annular red-brown plaques and hyperkeratotic scaly patches on her trunk and limbs. Her mother has ichthyosis with hyperkeratosis and geographic tongue while her younger brother had NSHL since birth. Mutation analysis revealed all of them carried a heterozygous missense mutation c.293G>A of GJB3. Skin biopsy showed many grain cells with dyskeratosis in the granular layer. Acanthosis, papillomatosis, and a mild superficial perivascular lymphocytic infiltrate were observed.
Conclusion: A mutation of GJB3 associated with EKV, ichthyosis and NSHL is reported in this case. The daughter with EKV and the son with NSHL in this Chinese family inherited the mutation from their mother with ichthyosis. The variation of clinical features may involve with genetic, epigenetic and environmental factors.
Gap junctions are channels or hemichannels assembled by connexins mediating cell-cell or cell-environment communication. Ions and small molecules can pass through gap junctions and guide embryonic development or pathogenic processes. Connexin 31(Cx31) coded by GJB3 (NM_024009.3), is one important member of connexin family. Highly expressed in upper differentiating epidermis (Di et al., 2001) and cochlear (Richard et al., 2000), mutations of GJB3 can result in different diseases including erythrokeratodermia variabilis (EKV) and non-syndromic hearing loss (NSHL) ranging from profound congenital deafness to mild, progressive hearing loss in late childhood.
EKV is a rare autosomal dominant skin disease featuring transient red patches that change over hours and days, along with fixed localized or generalized keratotic plaques. The disease is mainly caused by mutations in the GJB3, GJB4, and GJA1 genes, all coding members of connexin (Cx) family (Ishida-Yamamoto, 2016). Clinical presentation of EKV associated with GJB3 mutation can be variable ranging from typical keratotic lesions (Ishida-Yamamoto, 2016) to grey-brown and verrucous hyperkeratosis up to 2 cm thick (Glatz et al., 2011).
NSHL is a type of hereditary hearing loss without defects in other body parts and can be categorized as autosomal dominant, autosomal recessive, X-linked or mitochondrial mutation-related disease. Mutation of some important genes have been identified as the cause of NSHL, including GJB2, GJB3, and GJB6, which are all members of connexin family and generally involve with autosomal recessive or dominant hearing loss (Meena and Ayub, 2017). The Cx31 mutations lead to both recessive and dominant NSHL and severity can vary widely, from late-onset moderate deafness affecting high frequencies to congenital deafness (Liu et al., 2000).
Herein, we report a Chinese family with a missense mutation of GJB3 associated with different clinical symptoms covering EKV, ichthyosis and NSHL.
The study cohort includes a pair of parents, their daughter and son in a Chinese pedigree. The proband was a 6-year-old girl with demarcated annular red-brown plaques of variable sizes and colors spreading over the extensor side of right lower limb (Figure 1A), the right side of her chest (Figure 1B) and lumbar region (Figure 1C). Hyperkeratotic scaly patches were present mainly on the right thigh and knee. These manifestations had presented 6 months earlier, initially appearing on the right lower leg. No involvement of hair or nails was observed and no hearing impairment was found. Her mother has ichthyosis with hyperkeratosis on her limbs and geographic tongue while the patient’s younger brother was diagnosed with NSHL during hearing screening since birth. The father is an unaffected individual. The pedigree is shown in Figure 2A. The study was approved by institutional review board of Chinese Academy of Medical Sciences. Written informed consent was obtained from all participants, or from legal guardians in the case of minors.
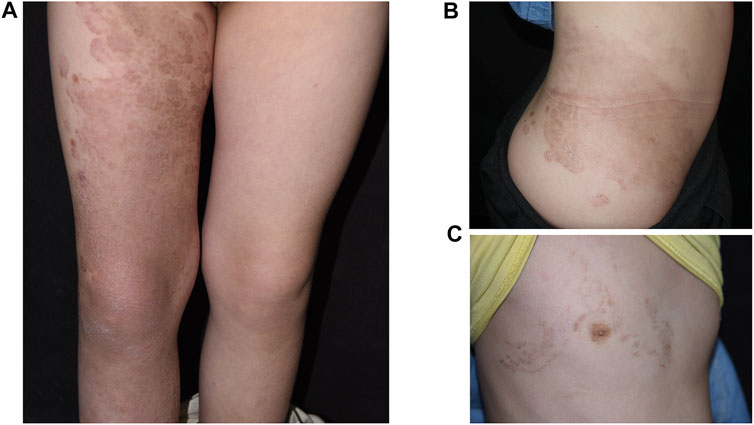
FIGURE 1. Clinical images showing demarcated, annular, red-brown plaques over the extensor side of right lower limb (A), lumbar region (B), and right side of the patient’s chest (C).
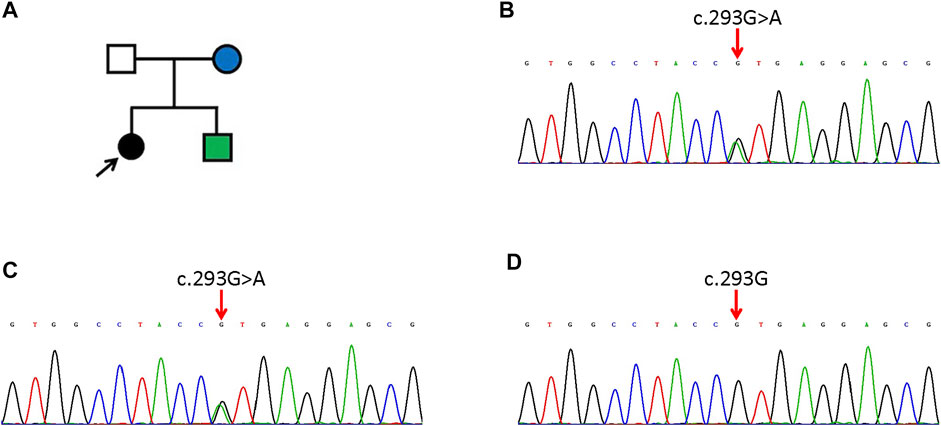
FIGURE 2. The pedigree of the family (A) and WES results of the proband (B), her mother (C) and her father (D). (A) The arrow indicates the proband. Black means EKV. The green one has NSHL and the blue one has ichthyosis. (B,C) The proband and her mother have a heterogenous missense mutation of c. 293G>A in GJB3. (D) The proband’s father has no mutation (c. 293G) in GJB3.
The genomic DNA was extracted from peripheral blood samples of all four participants using the QIAamp DNA Blood Mini Kit (QIAGEN, Hilden, Germany), according to the standard protocol and quantified by NanoDROP 2000 Spectrophotometer (Thermo Scientific; Waltham, MA, United States).
Whole exome sequence (WES) was conducted in the proband and her mother in Novogene company (Beijing, China) by using Illumina Novaseq plat, and the average sequencing depth is 100X. Sanger sequencing was performed in the proband’s brother for hot spot variants in NSHL-related genes (GJB2, GJB3, SLC26A4, and MT-RNR1). Raw sequence results were aligned to the human reference genome (GRCh37/hg38) annotated to get the candidate variants. Then the candidate variants were validated by Sanger sequencing to confirm the results of WES. And the primers were designed using primer3 Input for the suspected disease-causing genes.
The strategies of WES data filtering are as follows: 1) Variants with minor allele frequency (MAF)>0.01 were excluded, which were screened in normal population variant databases, including 1000G, ESP6500siv2 and gnomAD. 2) Variants in exons or alternative splicing regions were retained. 3) Synonymous mutations variants were removed, which were not located in highly conserved regions and would not affect splicing according to the same prediction software; and small non-frameshift insertion or deletion variants in the repeat regions were eliminated. 4) Variants that matched one of the following conditions were included: a) Variants were predicted to be pathogenic by at least one of the following programs including SIFT, Polyphen, MutationTaster, CADD. b) Variants were predicted to affect splicing by dbscSNV. 5) The remaining data were filtered by inheritance patterns and cutaneous phenotypes.
A skin biopsy was taken from the proband’s right thigh and viewed under the microscope for histopathological examination after hematoxylin-eosin staining.
Genetic tests revealed a highly pathogenic heterozygous missense mutation of GJB3 in the daughter and mother (Figure 2B, C). Sanger sequencing confirmed the existence of the same mutation in the younger brother. This mutation (NM_024009.3; c.293G>A; p.R98H) resulted in a change from a highly alkaline arginine residue at codon 98 to a slightly alkaline histidine residue, between the second transmembrane helix and intracellular domain of Cx31. The mutation was not detected in the father or healthy controls (Figure 2D). A diagnosis of EKV was made for the proband.
Histopathological examination showed many grain cells with dyskeratosis in the granular layer. Acanthosis, papillomatosis, and a mild superficial perivascular lymphocytic infiltrate were observed (Figure 3).
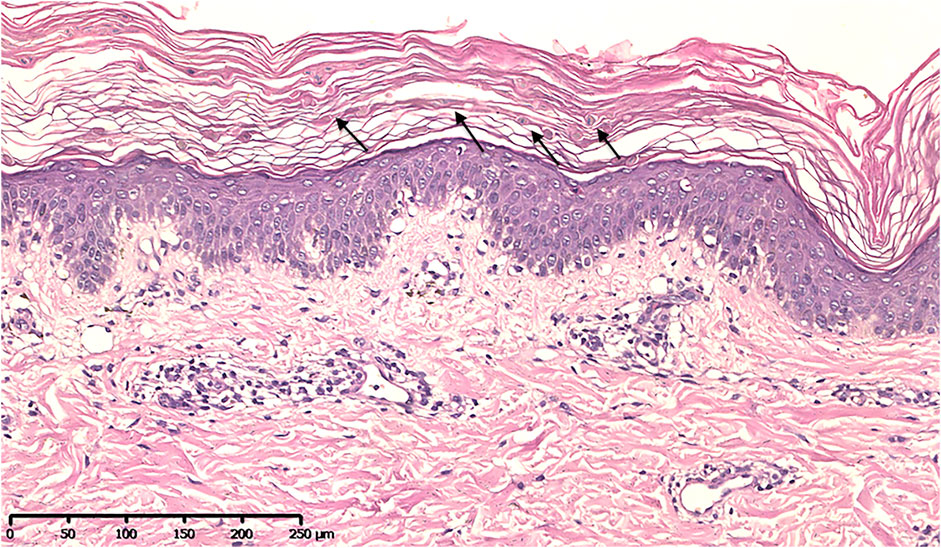
FIGURE 3. Histopathological image showing many grain cells with dyskeratosis in the granular layer, acanthosis, papillomatosis, and a mild superficial perivascular lymphocytic infiltrate (H&E).
Gap junctions are important for exchange of metabolites, ions and secondary messengers, especially in skin and cochlea. There are more than eight kinds of connexins expressed in skin epidermis, which contribute to its differentiation (Richard et al., 2000). Exchange of ions and small molecules helps maintain unique electrochemical environments which is important for cochlea normal function (Cohen-Salmon et al., 2002). GJB3 encodes Cx31 and is highly expressed in epidermis and cochlea, forming gap junctions (Scott and Kelsell, 2011), which is important in differentiation of keratinocytes and transfer of nerve pulses (Martinez et al., 2009). Gap junctions can be homomeric (consisting of one connexin type) or heteromeric (consisting of more than one connexin type) within the same cell (Kelly et al., 2015). Therefore, the connexons formed in epidermis and cochlea are intricate and delicate to guide the differentiation and maintain normal function.
EKV is a rare autosomal dominant skin disease associated with mutation of connexin genes, including GJB3, GJB4, and GJA1 (Ishida-Yamamoto, 2016). Several cases of autosomal recessive mutations of GJB3 causing EKV have also been reported (Gottfried et al., 2002; Terrinoni et al., 2004; Fuchs-Telem et al., 2011; Deng et al., 2019). Transient red patches and keratotic plaques are two prominent features of EKV. In this case, the patient with EKV and her mother both carry R98H mutation in Cx31 but the mother only shows the symptom of keratotic plaques and were diagnosed with ichthyosis. A severe case of EKV with grey-brown and verrucous hyperkeratosis up to 2 cm thick was reported caused by mutation of GJB3 (Glatz et al., 2011). Therefore, clinical symptoms of EKV may be diverse. Other genetic, epigenetic, and environmental factors are probably the explanation for variation of symptoms (Renner et al., 2008). Deep investigation is still needed. For the younger brother, no manifestation of skin is probably due to late onset characteristic of EKV or other factors related to genetics and environment.
Many kinds of connexins have been identified in cochlea and among them, Cx26 and Cx30 are predominant components while other types are limited (Wingard and Zhao, 2015). The mutations of Cx26 account for at least half of NSHL cases, while mutation of Cx31 is also a cause (Rabionet et al., 2000). Clinical symptoms of hearing loss resulted by GJB3 mutations range from congenital hearing loss since birth to late-onset hearing loss during childhood (Wingard and Zhao, 2015). Most NSHL cases related to Cx31 mutation are autosomal recessive while a few autosomal dominant cases were also reported (Liu et al., 2012; Oh et al., 2013). However, no case carrying the Cx31 mutation with both EKV and NSHL was reported but a pedigree with both Cx26 and Cx31 mutation presented hearing loss and palmoplantar keratoderma (Kelsell et al., 2000). Therefore, one possible explanation is that other connexin protein may make up the function loss of Cx31 in skin or cochlea while more studies are still required. In this family, three people harbor the same mutation but only the son has NSHL, which is probably due to partial penetrance. In earlier reports, female carriers with GJB3 dominant mutations in two deafness families have subclinical deafness or normal hearing while male carriers have NSHL (Xia et al., 1998), which indicates partial penetrance involving sex may be the reason of different symptoms of carriers.
How the mutation in Cx31 affects cell function is believed to be related to where the mutation site lies (Sugiura et al., 2015). The structure of Cx31 mainly contains four transmembrane domains (M1-4) linked by one intracellular loop (CL) and two extracellular loops (E1 and E2) with conserved cysteine residues while N- and C-termini (NT and CT) are lying inside the cell (Kelly et al., 2015; Figure 4). The E1 domain plays an important role in formation of the gap junction channel (Richard et al., 2000). The M2 domain is known for function in voltage gating (Rabionet et al., 2000). The extracellular domain E2 probably functions in interaction between different types of connexin and formation of heterotypic connexons (Sugiura et al., 2015). Mutations of GJB3 resulting in NSHL mainly locate in E2 domain, which may interfere the interaction between Cx31 and Cx26 and damage the function of heterotypic connexons on the membrane of cochlear cells (Sugiura et al., 2015). However, there is seemingly no relationship between the mutation locus and phenotypes of EKV patients. Most mutations related to EKV are autosomal dominant while a few recessive mutations were also found (shown in Figure 4). Interestingly, a compound heterozygous case with two recessive mutations in GJB3 presented a mutation lying in E2, which was the first pathologic mutation involved with EKV identified in this domain (Deng et al., 2019). This patient had no symptoms of hearing loss probably because this mutation in E2 domain is recessive. By systematically searching the PubMed, Embase and Web of Science, we summarized all the GJB3 mutations reported leading to EKV and phenotypes in each case (Table 1) and autosomal dominant GJB3 mutation related to NSHL (Figure 4; Richard et al., 1998; Xia et al., 1998; Wilgoss et al., 1999; Lopez-Bigas et al., 2000; Richard et al., 2000; Gottfried et al., 2002; Alexandrino et al., 2004; Terrinoni et al., 2004; Common et al., 2005; Feldmeyer et al., 2005; Morley et al., 2005; Yang et al., 2007; Renner et al., 2008; Li et al., 2010; Fuchs-Telem et al., 2011; Glatz et al., 2011; Scott and Kelsell, 2011; Wang et al., 2011; Liu et al., 2012; Torres et al., 2012; Wang et al., 2012; Ikeya et al., 2013; Oh et al., 2013; Otaguchi et al., 2014; Beck et al., 2015; Sugiura et al., 2015; Takeichi et al., 2016; Deng et al., 2018; Imura et al., 2020). In this case, the substitution of R98H lying in the border of M2 and CL, which are important in voltage and pH gating (Richard et al., 2000), is the first mutation found involving both EKV and NSHL. The exact mechanism behind needs more investigation.
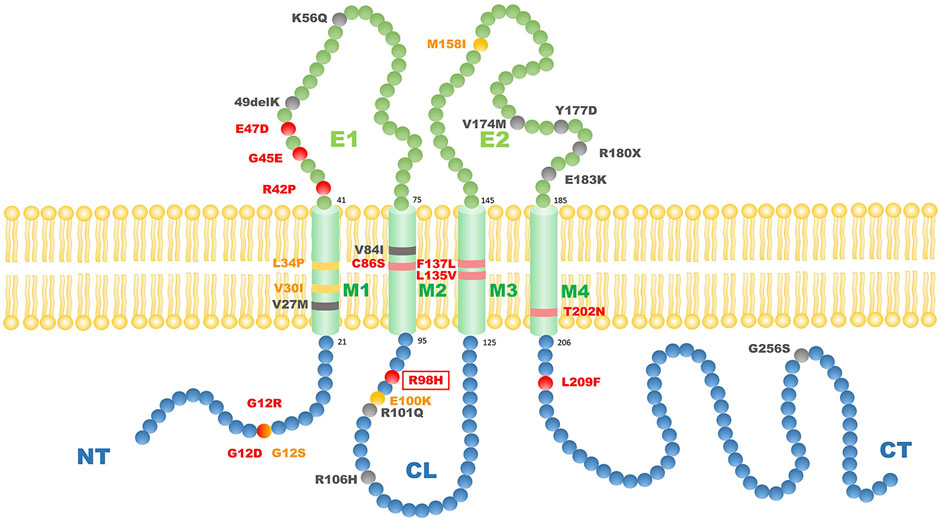
FIGURE 4. The scheme of reported GJB3 mutation related to EKV and autosomal dominant GJB3 point mutations related to NSHL. Red balls indicate autosomal dominant mutations with EKV phenotypes; yellow balls indicate autosomal recessive mutations with EKV phenotypes; black balls indicate common autosomal dominant GKB3 mutation related to NSHL. The red frame indicates the mutation we report in this case. M1–M4 refers to transmembrane domains. E1 and E2 refer to extracellular domains. CL refers to cytoplasmic loop. NT refers to cytoplasmic amino terminus. CT refers to cytoplasmic carboxy terminus.
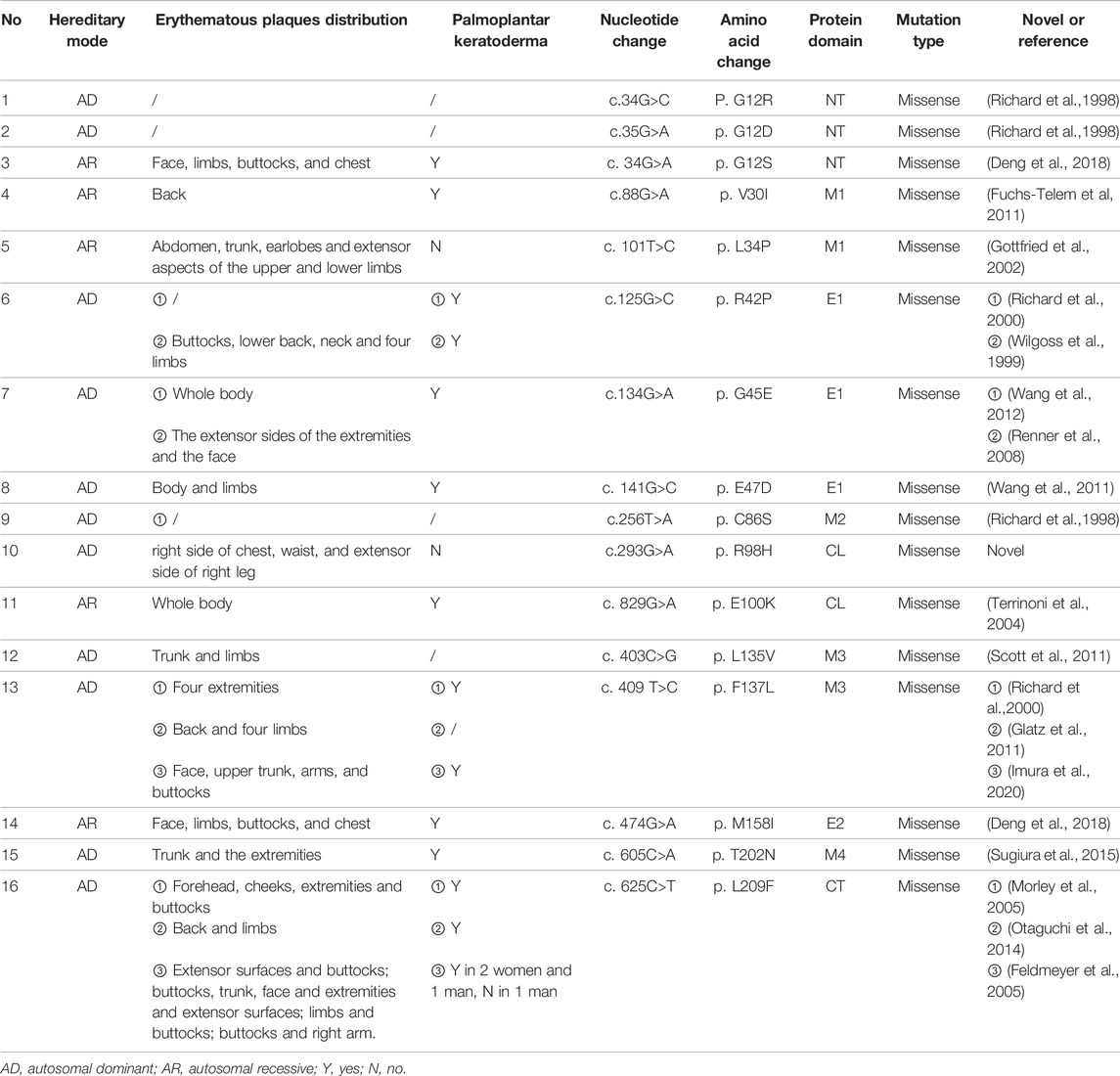
TABLE 1. Reported pathogenic mutations in GJB3 related to EKV and phenotypes.
Although the phenotypes of different pathologic mutations may be the same, the mechanisms behind them are likely different. In many vitro-studies, overexpression of Cx31 with the same mutation in cells may obtain different conclusions about pathogenic mechanisms possibly due to different experimental conditions. But overall, the viability of cells with EKV-related mutated Cx31 was decreased, while that of cells with NSHL-related Cx31 mutation was not (He et al., 2005; Tattersall et al., 2009; Easton et al., 2019). The mechanisms behind can be concluded into mainly two ways: 1) The mutated Cx31 protein accumulates in endoplasmic reticulum (ER) due to misfold, leading to ER stress response and finally cell death (Di et al., 2002; Tattersall et al., 2009; Chi et al., 2012). 2) Mutated Cx31 can be transferred to the cell membrane but only form dysfunctional gap junctions which may even interfere the normal function of plasma membrane (Rouan et al., 2003). However, a kind of rare mutation of Cx31 with G45E exhibits a new way to damage cells by inducing necrosis (Easton et al., 2019). Overexpression of Cx31G45E-GFP within HeLa cells and HaCaT cells led to expansion of the ER due to accumulation of mutated protein and finally cell necrosis rather than ER stress responses (Easton et al., 2019). Also, the interaction between mutated Cx31 and other wild-type connexins enables the accumulation of normal connexin in ER, which decreases the gap junctions on the cell membrane and interferes with normal function (Easton et al., 2019). The pathogenic mechanism of R98H in Cx31 needs experiments in vitro to identified.
In this case, we report a Chinese family with a mutation associated with EKV, ichthyosis and NSHL. The daughter with EKV and the son with NSHL in this Chinese family inherited the mutation from their mother with ichthyosis. The variation in clinical features may involve with genetic, epigenetic and environmental factors. One shortage of our research is that further experiments in vitro are needed to identify the possible pathogenic mechanism of this mutation. Our results indicate an important mutation site of Cx31 leading to EKV and NSHL with partial penetrance.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: GenBank database, accession number OL471368.
The studies involving human participants were reviewed and approved by Chinese Academy of Medical Sciences. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
TW participated in data receipt, clinical data collection and paper revision; YG participated in paper writing and literature review; QZ conducted gene mutation detection and verification and literature review. SZ and LY participated in case data collection; YPL and YHL were in charge of the research and revision of the paper.
This work was supported by grants from the Beijing Natural Science Foundation (Z210017 to TW), the Peking Union Medical College Hospital (grant number: ZC201903429 to YHL, ZC201911051 to TW), and the Center for Rare Diseases Research, Chinese Academy of Medical Sciences, Beijing, China.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Louise Adam, ELS(D), from Liwen Bianji, Edanz Editing China (www.liwenbianji.cn/ac) for editing the English text of a draft of this manuscript.
Alexandrino, F., Oliveira, C. A., Reis, F. C., Maciel-Guerra, A. T., and Sartorato, E. L. (2004). Screening for Mutations in the GJB3 Gene in Brazilian Patients with Nonsyndromic Deafness. J. Appl. Genet. 45 (2), 249–254.
Beck, C., Pérez-Álvarez, J. C., Sigruener, A., Haubner, F., Seidler, T., Aslanidis, C., et al. (2015). Identification and Genotype/phenotype Correlation of Mutations in a Large German Cohort with Hearing Loss. Eur. Arch. Otorhinolaryngol. 272 (10), 2765–2776. doi:10.1007/s00405-014-3157-5
Chi, J., Li, L., Liu, M., Tan, J., Tang, C., Pan, Q., et al. (2012). Pathogenic Connexin-31 Forms Constitutively Active Hemichannels to Promote Necrotic Cell Death. PLoS One 7 (2), e32531. doi:10.1371/journal.pone.0032531
Cohen-Salmon, M., Ott, T., Michel, V., Hardelin, J.-P., Perfettini, I., Eybalin, M., et al. (2002). Targeted Ablation of Connexin26 in the Inner Ear Epithelial gap junction Network Causes Hearing Impairment and Cell Death. Curr. Biol. 12 (13), 1106–1111. doi:10.1016/s0960-9822(02)00904-1
Common, J. E. A., O'Toole, E. A., Leigh, I. M., Thomas, A., Griffiths, W. A. D., Venning, V., et al. (2005). Clinical and Genetic Heterogeneity of Erythrokeratoderma Variabilis. J. Invest. Dermatol. 125 (5), 920–927. doi:10.1111/j.0022-202X.2005.23919.x
Deng, Y., Wang, H., Mou, Y., Zeng, Q., and Xiong, X. (2019). Exome sequencing identifies novel compound heterozygous mutations in GJB3 gene that cause erythrokeratodermia variabilis et progressiva. Australas. J. Dermatol. 60 (1), e87–e89. doi:10.1111/ajd.12887
Di, W.-L., Rugg, E. L., Leigh, I. M., and Kelsell, D. P. (2001). Multiple Epidermal Connexins Are Expressed in Different Keratinocyte Subpopulations Including Connexin 31. J. Invest. Dermatol. 117 (4), 958–964. doi:10.1046/j.0022-202x.2001.01468.x
Di, W. L., Monypenny, J., Common, J. E., Kennedy, C. T., Holland, K. A., Leigh, I. M., et al. (2002). Defective Trafficking and Cell Death Is Characteristic of Skin Disease-Associated Connexin 31 Mutations. Hum. Mol. Genet. 11 (17), 2005–1h4. doi:10.1093/hmg/11.17.2005%J
Easton, J. A., Albuloushi, A. K., Kamps, M. A. F., Brouns, G. H. M. R., Broers, J. L. V., Coull, B. J., et al. (2019). A Rare Missense Mutation in GJB3 (Cx31G45E) Is Associated with a Unique Cellular Phenotype Resulting in Necrotic Cell Death. Exp. Dermatol. 28 (10), 1106–1113. doi:10.1111/exd.13542
Feldmeyer, L., Plantard, L., Mevorah, B., Huber, M., and Hohl, D. (2005). Novel Mutation of Connexin 31 Causing Erythrokeratoderma Variabilis. Br. J. Dermatol. 152 (5), 1072–1074. doi:10.1111/j.1365-2133.2005.06561.x
Fuchs-Telem, D., Pessach, Y., Mevorah, B., Shirazi, I., Sarig, O., and Sprecher, E. (2011). Erythrokeratoderma Variabilis Caused by a Recessive Mutation in GJB3. Clin. Exp. Dermatol. 36 (4), 406–411. doi:10.1111/j.1365-2230.2010.03986.x
Glatz, M., Steensel, M., Geel, M., Steijlen, P., and Wolf, P. (2011). An Unusual Missense Mutation in the GJB3 Gene Resulting in Severe Erythrokeratodermia Variabilis. Acta Derm Venerol 91 (6), 714–715. doi:10.2340/00015555-1135
Gottfried, I., Landau, M., Glaser, F., Di, W. L., Ophir, J., Mevorah, B., et al. (2002). A Mutation in GJB3 Is Associated with Recessive Erythrokeratodermia Variabilis (EKV) and Leads to Defective Trafficking of the Connexin 31 Protein. Hum. Mol. Genet. 11 (11), 1311–1316. doi:10.1093/hmg/11.11.1311%JHumanMolecularGenetics
He, L.-Q., Liu, Y., Cai, F., Tan, Z.-P., Pan, Q., Liang, D.-S., et al. (2005). Intracellular Distribution, Assembly and Effect of Disease-Associated Connexin 31 Mutants in HeLa Cells. Acta Biochim. Biophys. Sinica 37 (8), 547–554. doi:10.1111/j.1745-7270.2005.00080.x
Ikeya, S., Urano, S., Sakabe, J.-i., Ito, T., and Tokura, Y. (2013). Erythrokeratodermia Variabilis: First Japanese Case documentingGJB3mutation. J. Dermatol. 40 (5), 402–403. doi:10.1111/1346-8138.12101
Imura, K., Ikeya, S., Ogata, T., and Tokura, Y. (2020). Erythrokeratodermia Variabilis et Progressiva with a Rare GJB3 Mutation. J. Dermatol. 47 (4), e111–e113. doi:10.1111/1346-8138.15206
Ishida-Yamamoto, A. (2016). Erythrokeratodermia variabilis et progressiva. J. Dermatol. 43 (3), 280–285. doi:10.1111/1346-8138.13220
Kelly, J. J., Simek, J., and Laird, D. W. (2015). Mechanisms Linking Connexin Mutations to Human Diseases. Cell Tissue Res 360 (3), 701–721. doi:10.1007/s00441-014-2024-4
Kelsell, D. P., Wilgoss, A. L., Richard, G., Stevens, H. P., Munro, C. S., and Leigh, I. M. (2000). Connexin Mutations Associated with Palmoplantar Keratoderma and Profound Deafness in a Single Family. Eur. J. Hum. Genet. 8 (2), 141–144. doi:10.1038/sj.ejhg.5200407
Li, Y. H., Jiang, H., Yang, L. J., Xu, H. X., Li, H., Li, H. W., et al. (2010). Study of mtDNA 12S rRNA A1555G, GJB2, GJB3 Gene Mutation in Uighur and Han People with Hereditary Nonsyndromic Hearing Loss in Xinjiang. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 45 (8), 645–651. doi:10.3760/cma.j.issn.1673-0860.2010.08.008
Liu, H., Liu, H., Fu, X.-A., Yu, Y.-X., Zhou, G.-Z., Lu, X.-M., et al. (2012). Mutation Analysis of GJB3 and GJB4 in Chinese Patients with Erythrokeratodermia Variabilis. J. Dermatol. 39 (4), 400–401. doi:10.1111/j.1346-8138.2011.01314.x
Liu, X. Z., Xia, X. J., Xu, L. R., Pandya, A., Liang, C. Y., Blanton, S. H., et al. (2000). Mutations in Connexin31 Underlie Recessive as Well as Dominant Non-syndromic Hearing Loss. Hum. Mol. Genet. 9 (1), 63–67. doi:10.1093/hmg/9.1.63%25JHumanMolecularGenetics
López-Bigas, N., Rabionet, R., Martínez, E., Banchs, I., Volpini, V., Vance, J. M., et al. (2000). Identification of Seven Novel SNPS (Five Nucleotide and Two Amino Acid Substitutions) in the Connexin31 (GJB3) Gene. Hum. Mutat. 15 (5), 481–482. doi:10.1002/(SICI)1098-1004(200005)15:5<481::AID-HUMU15>3.0.CO;2-7
Martínez, A. D., Acuña, R., Figueroa, V., Maripillan, J., and Nicholson, B. (2009). Gap-junction Channels Dysfunction in Deafness and Hearing Loss. Antioxid. Redox Signaling 11 (2), 309–322. doi:10.1089/ars.2008.2138
Meena, R., and Ayub, M. (2017). Genetics of Human Hereditary Hearing Impairment. J. Ayub Med. Coll. Abbottabad 29 (4), 671–676.
Morley, S. M., White, M. I., Rogers, M., Wasserman, D., Ratajczak, P., McLean, W. H. I., et al. (2005). A New, Recurrent Mutation of GJB3 (Cx31) in Erythrokeratodermia Variabilis. Br. J. Dermatol. 152 (6), 1143–1148. doi:10.1111/j.1365-2133.2005.06610.x
Oh, S.-K., Choi, S.-Y., Yu, S. H., Lee, K.-Y., Hong, J. H., Hur, S. W., et al. (2013). Evaluation of the Pathogenicity of GJB3 and GJB6 Variants Associated with Nonsyndromic Hearing Loss. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis. 1832 (1), 285–291. doi:10.1016/j.bbadis.2012.05.009
Otaguchi, R., Kawakami, T., Matsuoka, M., Kimura, S., Soma, Y., Matsuda, M., et al. (2014). A Sporadic Elder Case of Erythrokeratodermia Variabilis with a Single Base-Pair Transversion in GJB3 Gene Successfully Treated with Systemic Vitamin A Derivative. J. Dermatol. 41 (11), a–n. doi:10.1111/1346-8138.12628
Rabionet, R., Gasparini, P., and Estivill, X. (2000). Molecular Genetics of Hearing Impairment Due to Mutations in gap junction Genes Encoding Beta Connexins. Hum. Mutat. 16 (3), 190–202. doi:10.1002/1098-1004(200009)16:3<190::aid-humu2>3.0.co;2-i
Renner, R., Paasch, U., Simon, J., Froster, U., and Heinritz, W. (2008). A New Mutation in the GJB3 Gene in a Patient with Erythrokeratodermia Variabilis. J. Eur. Acad. Dermatol. Venerol 22 (6), 750–751. doi:10.1111/j.1468-3083.2007.02447.x
Richard, G., Brown, N., Smith, L. E., Terrinoni, A., Melino, G., Mackie, R. M., et al. (2000). The Spectrum of Mutations in Erythrokeratodermias - Novel and De Novo Mutations in GJB3. Hum. Genet. 106 (3), 321–329. doi:10.1007/s00439005104510.1007/s004390000258
Richard, G., Smith, L. E., Bailey, R. A., Itin, P., Hohl, D., Epstein, E. H., et al. (1998). Mutations in the Human Connexin Gene GJB3 Cause Erythrokeratodermia Variabilis. Nat. Genet. 20 (4), 366–369. doi:10.1038/3840
Rouan, F., Lo, C. W., Fertala, A., Wahl, M., Jost, M., Rodeck, U., et al. (2003). Divergent Effects of Two Sequence Variants ofGJB3(G12D and R32W) on the Function of Connexin 31In Vitro. Exp. Dermatol. 12 (2), 191–197. doi:10.1034/j.1600-0625.2003.120210.x
Scott, C. A., and Kelsell, D. P. (2011). Key Functions for gap Junctions in Skin and Hearing. Biochem. J. 438 (2), 245–254. doi:10.1042/BJ20110278
Sugiura, K., Arima, M., Matsunaga, K., and Akiyama, M. (2015). The novelGJB3mutation p.Thr202Asn in the M4 Transmembrane Domain Underlies Erythrokeratodermia Variabilis. Br. J. Dermatol. 173 (1), 309–311. doi:10.1111/bjd.13641
Takeichi, T., Sugiura, K., Hsu, C., Nomura, T., Takama, H., Simpson, M., et al. (2016). Erythrokeratoderma Variabilis Caused by p.Gly45Glu in Connexin 31: Importance of the First Extracellular Loop Glycine Residue for Gap Junction Function. Acta Derm Venerol 96 (4), 557–559. doi:10.2340/00015555-2307
Tattersall, D., Scott, C. A., Gray, C., Zicha, D., and Kelsell, D. P. (2009). EKV Mutant Connexin 31 Associated Cell Death Is Mediated by ER Stress. Hum. Mol. Genet. 18 (24), 4734–4745. doi:10.1093/hmg/ddp436%25JHumanMolecularGenetics
Terrinoni, A., Leta, A., Pedicelli, C., Candi, E., Ranalli, M., Puddu, P., et al. (2004). A Novel Recessive Connexin 31 (GJB3) Mutation in a Case of Erythrokeratodermia Variabilis. J. Invest. Dermatol. 122 (3), 837–839. doi:10.1111/j.0022-202X.2004.22311.x
Torres, T., Velho, G., Sanches, M., and Selores, M. (2012). A Case of Erythrokeratodermia Variabilis with Connexin 31 Gene Mutation (Cx31F137L). Int. J. Dermatol. 51 (4), 494–496. doi:10.1111/j.1365-4632.2010.04640.x
Wang, W., Liu, L. H., Chen, G., Gao, M., Zhu, J., Zhou, F. S., et al. (2012). A Missense Mutation in theGJB3gene Responsible for Erythrokeratodermia Variabilis in a Chinese Family. Clin. Exp. Dermatol. 37 (8), 919–921. doi:10.1111/j.1365-2230.2012.04406.x
Wang, Z.-X., Lu, W.-S., Li, H., Lin, D., Zhou, F.-S., Sun, L.-D., et al. (2011). A Novel GJB3 (Cx31) Missense Mutation in a Chinese Patient with Erythrokeratodermia Variabilis. J. Eur. Acad. Dermatol. Venereol. 25 (1), 113–115. doi:10.1111/j.1468-3083.2010.03691.x
Wingard, J. C., and Zhao, H.-B. (2015). Cellular and Deafness Mechanisms Underlying Connexin Mutation-Induced Hearing Loss €" A Common Hereditary Deafness. Front. Cel. Neurosci. 9, 202. doi:10.3389/fncel.2015.00202
Wilgoss, A., Leigh, I. M., Barnes, M. R., Dopping-Hepenstal, P., Eady, R. A., Walter, J. M., et al. (1999). Identification of a Novel Mutation R42P in the Gap Junction Protein Beta-3 Associated With Autosomal Dominant Erythrokeratoderma Variabilis. J. Invest Dermatol. 113 (6), 1119–1122. doi:10.1046/j.1523-1747.1999.00792.x
Xia., J.-h., Liu, C.-y., Tang, B.-s., Pan, Q., Huang, L., Dai, H.-p., et al. (1998). Mutations in the Gene Encoding gap junction Protein β-3 Associated with Autosomal Dominant Hearing Impairment. Nat. Genet. 20 (4), 370–373. doi:10.1038/3845
Keywords: connexin gene, GJB3, erythrokeratodermia variabilis, ichthyosis, nonsyndromic hearing loss
Citation: Gao Y, Zhang Q, Zhang S, Yang L, Liu Y, Liu Y and Wang T (2022) A Connexin Gene (GJB3) Mutation in a Chinese Family With Erythrokeratodermia Variabilis, Ichthyosis and Nonsyndromic Hearing Loss: Case Report and Mutations Update. Front. Genet. 13:797124. doi: 10.3389/fgene.2022.797124
Received: 20 November 2021; Accepted: 28 March 2022;
Published: 23 May 2022.
Edited by:
Jia Zhang, Xinhua Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, ChinaReviewed by:
Muhammad Ansar, Quaid-i-Azam University, PakistanCopyright © 2022 Gao, Zhang, Zhang, Yang, Liu, Liu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaping Liu, ypliu@ibms.pumc.edu.cn; Yuehua Liu, yuehualiu63@163.com; Tao Wang, wangtaopumch@126.com
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.