 Zhi Cheng1,2
Zhi Cheng1,2 Fa Zou
Fa Zou Zhen-E Xu
Zhen-E Xu Chun Li
Chun Li Jingkun Miao
Jingkun Miao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 03 March 2022
Sec. Human and Medical Genomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.783799
This article is part of the Research Topic Inborn Errors of Metabolism: Mechanisms and Therapeutic Approaches View all 4 articles
Citrullinemia is a rare autosomal recessive disorder characterized by elevated concentrations of citrulline in the blood resulting from malfunction of the urea cycle. It is categorized into two types, types I and II, which are caused by argininosuccinate synthase 1 (ASS1), and citrin (SLC25A13) gene mutations, respectively. In this study, we performed genetic analysis on nine Chinese infants with citrullinemia using next-generation sequencing, which identified a novel mutation (p.Leu313Met) and a rare mutation (p.Thr323Ile, rs1250895424) of ASS1. We also found a novel splicing mutation of SLC25A13: c.1311 + 4_+7del. Functional analysis of the ASS1 missense mutations showed that both significantly impaired the enzyme activity of ASS1, with the p. Thr323Ile mutation clearly affecting the interaction between ASS1 and protein arginine methyltransferase 7 (PRMT7). These findings expand the mutational spectrum of ASS1 and SLC25A13, and further our understanding of the molecular genetic mechanism of citrullinemia in the Chinese population.
Citrullinemia is a rare autosomal recessive disorder characterized by elevated concentrations of citrulline in the blood from malfunctions of the urea cycle (Saheki et al., 1987). It is categorized into two types according to the molecular pathogenesis. Type I citrullinemia (CTLN1, OMIM# 2,15,700) is caused by argininosuccinate synthase 1 gene (ASS1) mutations, while type II citrullinemia is caused by citrin gene (SLC25A13) mutations (Woo et al., 2014).
Classic CTLN1 often presents early in the neonatal period in affected individuals with acute hyperammonemia and neurologic manifestations. If untreated, it can lead to life-threatening encephalopathy, metabolic coma, and death (Häberle et al., 2002). Late-onset forms of CTLN1 can also occur. These usually have milder phenotypes, including neurodisability, somnolence, and chronic intermittent hyperammonemia during childhood and adulthood (Häberle et al., 2003).
Type II citrullinemia has two main clinical phenotypes: neonatal intrahepatic cholestatic hepatitis caused by citrin deficiency (NICCD; OMIM# 6,05,814) and adult-onset type II citrullinemia (CTLN2; OMIM# 6,03,471) (Saheki and Kobayashi, 2002). NICCD is clinically characterized by intrahepatic cholestasis and metabolic abnormalities including multiple aminoacidemia, galactosemia, hypoglycemia, and hypoproteinemia. Most patients improve spontaneously without medical treatment before 1 year of age. However, some develop severe CTLN2 one or more decades later (Liu et al., 2014). Patients with CTLN2 suffer from various neuropsychological symptoms including disorientation, delirium, seizures, and coma because of hyperammonemia. Death from brain edema occurs in some cases (Tabata et al., 2008).
ASS1 is located at chromosome 9q24.11–9q23.12 and contains 16 exons. It encodes the argininosuccinate synthetase enzyme, which catalyzes the synthesis of argininosuccinate from citrulline, and aspartate. It is mainly expressed in the periportal hepatocytes of the liver, but also in most other body tissues (Engel et al., 2009). At least 153 ASS1 CTLN1 disease-causing mutations have been reported, of which most are missense mutations distributed within exons 3–15 (Diez-Fernandez et al., 2017).
SLC25A13 is located at chromosome 7q21.3. It encodes citrin, which functions as a calcium (Ca2+)-stimulated aspartate-glutamate carrier. Citrin is expressed in many tissues but most abundantly in the liver, and is localized to the mitochondrial inner membrane (Iijima et al., 2001). The first SLC25A13 disease-causing mutation was identified in a Japanese family with CTLN2 (Kobayashi et al., 1999). Later, some NICCD patients were also shown to carry homozygous and compound heterozygous mutations of SLC25A13 (Yamaguchi et al., 2002). To date, more than 110 pathogenic mutations of SLC25A13 have been reported, of which most are point mutations or short insertions/deletions (InDels). These were mainly identified in east Asian populations, including Japanese, Korean, and Chinese (Zhang et al., 2017).
There are currently no well-recognized clinical/biochemical diagnostic criteria for either type of citrullinemia, yet molecular genetic analysis is critical for the diagnosis of patients. In this study, we performed genetic analysis of Chinese infants with citrullinemia using next-generation sequencing (NGS). We also carried out a functional investigation of ASS1 mutations identified in this study to better understand the genetic mechanism of this disease in the Chinese population.
From June 2014 to December 2020, nine infants with citrullinemia were enrolled in this study. The patients were diagnosed based on clinical findings and biochemical characterization. Clinical and biochemical data were recorded. Whole blood samples were collected for genetic analysis. A cohort of 100 healthy men was studied as a control group.
Genomic DNA was extracted from the whole blood of all recruited subjects using DNA isolation kits (Tiangen, Beijing, China). A total of 3 µg genomic DNA of affected infants was used to prepare indexed Illumina libraries according to the manufacturer’s protocol (Illumina, San Diego, CA, United States). Coding exons and flanking regions of 165 genes reported to be mutated in disorders of amino acid, organic acid, and fatty acid metabolism were selected and captured using the Agilent SureSelect Target Enrichment System (Agilent, Santa Clara, CA, United States). A list of the targeted genes is provided in Supplementary Table S1. The enriched libraries were sequenced on an Illumina HiSeq 2500 sequencer.
After sequencing, low-quality reads and adaptor sequences were filtered out using the Solexa QA package and the cutadapt program (https://cutadapt.readthedocs.org/), respectively (Cox et al., 2010). Clean reads were aligned to the human reference genome (hg19) using the SOAPaligner program (Li et al., 2009), which was also used to identify single nucleotide polymorphisms (SNPs). To detect InDels, reads were realigned to the reference genome using the Burrows-Wheeler alignment tool, and InDels were identified with the Genome Analysis Toolkit (Li and Durbin, 2009; DePristo et al., 2011). The impact of non-synonymous mutations was assessed in silico using Polyphen2 and SIFT (Kumar et al., 2009; Adzhubei et al., 2010), while the effect of splice site mutation was predicted by MutationTaster (Schwarz et al., 2014). The novelty of the mutations was confirmed by searching in dbSNP (http://www.ncbi.nlm.nih.gov/snp/), the 1,000 Genomes Project (https://www.internationalgenome.org/), and the HGMD Professional (http://www.hgmd.cf.ac.uk/ac/index.php) databases. The novel mutations were also confirmed by Sanger sequencing. Conservation analysis was performed using CLC Main Workbench Software.
The open reading frame (ORF) of human ASS1 was amplified by PCR from cDNA and inserted into the Bgl II- and BamH I-digested pEGFP-N1 vector. p. Leu313Met and p. Thr323Ile mutations were introduced into wild-type (WT) expression plasmids by PCR-based site-directed mutagenesis. Then, the ORFs of WT and mutant ASS1 were amplified and inserted into the Nde I- and Xba I-digested pCMV5-FLAG vector to create FLAG-tagged expression plasmids. The ORF of human PRMT7 was also PCR-amplified and inserted into Nde I and Xba I sites of the pCMV5-FLAG vector to create expression plasmids. All plasmids were verified by sequencing. The sequences of primers used in plasmid construction are shown in Supplementary Table S2.
Human embryonic kidney cells (HEK 293) were purchased from Shanghai cell bank (Chinese Academy of Sciences) and were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin in a humidified incubator containing 5% CO2 at 37°C. Transient transfection was carried out using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, United States) according to the manufacturer’s instructions.
ASS1 immunoprecipitation was performed as described previously with minor modifications (Herrera Sanchez et al., 2017; Miyamoto et al., 2017). Briefly, FLAG-tagged expression plasmids of ASS1 (WT or mutant) and empty vector were transfected into HEK 293 cells. Two days after transfection, cells were collected and lysed in NP-40 lysis buffer (Beyotime, Shanghai, China). The supernatants were collected after centrifugation at 10,000 × g for 15 min at 4°C and incubated with anti-FLAG M2 Beads (Sigma-Aldrich, Shanghai, China) at 4°C for 4 h. The beads were then washed three times with lysis buffer. Bound proteins were eluted by adding 100 μg/ml 3 × FLAG peptide, and eluted proteins were used for ASS1 activity assays.
For these assays, equal amounts of eluted proteins were resuspended in reaction buffer (20 mM Tris-HCl, pH 7.8, 2 mM ATP, 2 mM citrulline, 2 mM aspartate, 6 mM MgCl2, 20 mM KCl, and 0.1 U pyrophosphatase) to a final volume of 100 µl. Samples were incubated for 30 min at 37°C, then the reactions were stopped by the addition of 100 µl molybdate buffer (10 mM ascorbic acid, 2.5 mM ammonium molybdate, and 2% sulfuric acid). The accumulation of pyrophosphate was determined at 660 nm by spectrophotometry. Experiments were performed in triplicate and repeated three times.
Expression plasmid pCMV5-FLAG-PRMT7 was co-transfected with pEGFP-ASS1, pEGFP-ASS1-Leu313Met, pEGFP-ASS1-Thr323Ile, or pEGFP-N1 empty vector into HEK293 cells. Immunoprecipitation was carried out as described above. For western blotting, cell lysates and immunoprecipitates were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The membranes were blocked overnight with 5% (w/v) non-fat milk in Tris-buffered saline with 0.1% Tween 20, then probed with anti-green fluorescent protein (GFP) (Sungene, Tianjin, China), anti-β-actin (Sungene), or anti-FLAG M2 (Sigma-Aldrich, Shanghai, China) primary antibodies. They were then incubated with horseradish peroxidase-conjugated goat anti-mouse or goat anti-rabbit secondary antibodies and visualized by enhanced chemiluminescence (Sigma-Aldrich).
A total of nine patients (three females, six males) were included in this study. All had high plasma citrulline levels (>1,00 μmol/L), and most had elevated levels of blood arginine, methionine, and threonine. Five patients presented with hyperammonemia. Patient clinical and biochemical characteristics are summarized in Table 1.
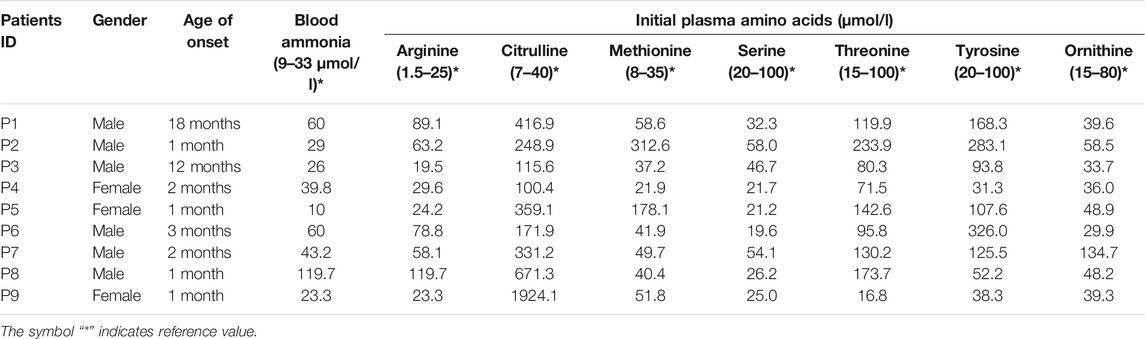
TABLE 1. Clinical and Biochemical characteristics of patients.
Six mutations (15 mutated alleles) of SLC25A13 were identified in the patients through NGS. The most common was c.851_854del (seven alleles, 47%), followed by c.1638_1660dup23 (four alleles, 27%). A novel splicing mutation of SLC25A13 was identified: c.1311 + 4_+7del (Supplementary Figure S1), which resulted in a deletion of “AGUA” at the 5′ splice site. This mutation was not observed in healthy controls, nor reported in dbSNP, 1,000 Genome Project, or the HGMD Professional databases. It was predicted to result in splice site changes and be disease-causing by MutationTaster.
Three ASS1 mutations were detected in the patients, of which a missense mutation was novel: c.937C > A (p.Leu313Met, shown in Supplementary Figure S2). The c.968C > T (p.Thr323Ile) mutation (rs1250895424, shown in Supplementary Figure S3) was identified by Trans-Omics for Precision Medicine (TOPMed) program previously. The minor allele frequency (MAF) is 0.000008. In the Genome Aggregation Database (gnomAD), the MAF of this mutation is 0.00002 in Asian population. So it was a rare mutation. Both mutations were not detected in healthy controls. And both are located at sites that are highly conserved among species (Figure 1). They were predicted to be pathogenic by Polyphen2 and SIFT (Supplementary Table S3). The mutations identified in this study are summarized in Table 2.
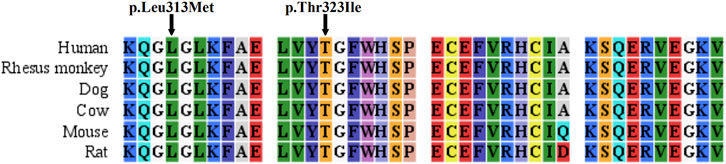
FIGURE 1. Sequence alignment of ASS1 proteins among species.
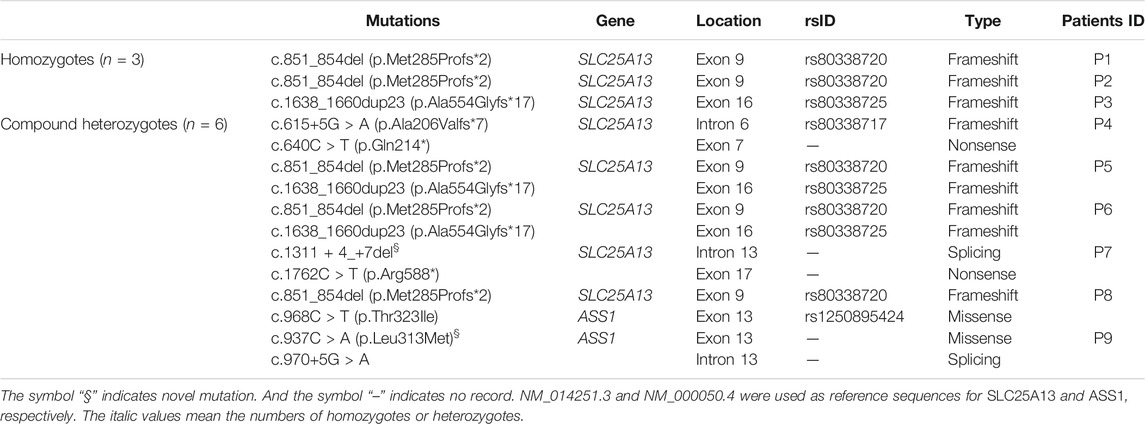
TABLE 2. Mutations detected in included infants.
Because splicing mutation can cause improper intron removal and alterations of the ORF, functional analyses were performed of the two ASS1 missense mutations. WT and mutant ASS1 proteins were over-expressed and purified from HEK293 cells. Assays of enzyme activity showed that both p. Leu313Met and p. Thr323Ile mutant proteins had significantly decreased activity compared with WT (Figure 2A).
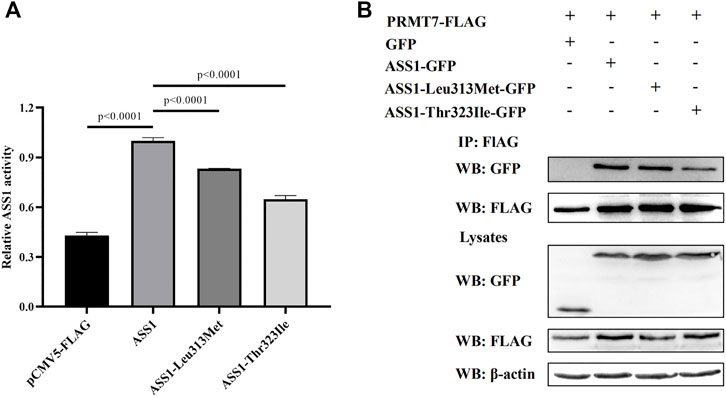
FIGURE 2. Functional analysis of ASS1 mutations. (A) Effect of p. Leu313Met and p. Thr323Ile mutations on ASS1 activity. The enzymatic activity of ASS1 immunoprecipitated from HEK293 cells expressing pCMV5-FLAG empty vector, ASS1-FLAG, ASS1-Leu313Met-FLAG, or ASS1-Thr323Ile-FALG was determined. Data are normalized to ASS1 wild-type protein (lane 2). Three independent experiments were performed. Statistical significance was determined by one-way ANOVA. (B) Effect of p. Leu313Met and p. Thr323Ile mutations on the ASS1–PRMT7 interaction. HEK293 cells co-expressing PRMT7-FLAG and GFP, or PRMT7-FLAG with ASS1-GFP, ASS1-Leu313Met-GFP, or ASS1-Thr323Ile-GFP were harvested. PRMT7-FLAG was immunoprecipitated using anti-FLAG M2 Beads. Immunoprecipitates were analyzed by western blotting using anti-GFP and anti-FLAG M2 antibodies. Total cell lysates were analyzed by western blotting using anti-GFP, anti-FLAG M2, and anti-β-actin antibodies.
PRMT7 was previously reported to interact with ASS1 (Verma et al., 2017), so we next investigated the effect of the mutations on this interaction. Co-immunoprecipitation experiments using FLAG-tagged PRMT7 and GFP-tagged ASS1 revealed similar expression levels of the two mutant proteins with that of WT ASS1 in HEK293 cells. A similar amount of p. Leu313Met mutant protein to WT ASS1 was co-immunoprecipitated by PRMT7. However, less p. Thr323Ile mutant protein was co-immunoprecipitated by PRMT7 compared with WT (Figure 2B), suggesting that the p. Thr323Ile mutant protein binds weakly to PRMT7.
Although citrullinemia types I and II are caused by mutations in different genes, they exhibit partial similarities in clinical phenotypes. In the present study, we performed genetic analysis of nine Chinese infants with citrullinemia, and identified homozygous or compound heterozygous mutations of ASS1 and SLC25A13 (Table 2). This demonstrated that genetic analysis can help determine the subgroup of citrullinemia besides clinical and biochemical examinations.
Our study identified a novel splicing mutation of SLC25A13, which leads to the deletion of “AGUA” at the 5′ splice site. This mutation is very likely to affect pre-mRNA splicing for three main reasons. First, during the splicing process, the 5′ splice site (CAG/GUAAGU sequence) and 3′ splice site (NYAG/G sequence) are recognized by spliceosome components (Ohno et al., 2018). While most common mutations affect +1 and +2 residues at the 5′ donor splice site and −1 and −2 residues at the 3′ acceptor splice site, any mutations in these canonical sequences could impair the interaction between pre-mRNA and the spliceosome, leading to abnormal pre-mRNA splicing (Anna and Monika, 2018). The OXCT1 c.1248+5G > A mutation, IKBKAP c.2204+6T > C mutation, and CDHR1 c.2040+5G > T mutation were previously found to cause exon skipping (Ibrahim et al., 2007; Hori et al., 2013; Stingl et al., 2017). Moreover, the ATF6 c.82+5G > T mutation led to intron retention (Kohl et al., 2015), while ASS1 mutations c.773+4A > C and c.970+5G > A were identified in patients with citrullinemia (Kobayashi et al., 1995; Lin et al., 2019). Second, the MutationTaster predicted that the mutation was disease-causing. Finally, high levels of plasma citrulline (331.2 μmol/l; normal: 7–40 μmol/l) were detected in the patient carrying this mutation. Considering that he also carried a nonsense mutation of SLC25A13 (p.Arg588*), it is likely that these compound heterozygous mutations caused the production of defective citrin protein.
We also identified a novel mutation (p.Leu313Met) and a rare mutation (p.Thr323Ile) of ASS1, which are both located at highly conserved sites and were absent from the 100 healthy controls. Polyphen2 and SIFT predicted them to be damaging, and functional analyses showed that they significantly impaired the enzyme activity of ASS1. Furthermore, PRMT7 encodes a protein arginine methyltransferase which catalyzes arginine methylation. A previous study showed that PRMT7 interacts with ASS1, and that several mutations associated with citrullinemia disrupt this interaction (Verma et al., 2017). In this study, we found that the p. Thr323Ile mutation also considerably decreased the binding of ASS1 to PRMT7. Therefore, we believe that these two mutations are pathogenic for citrullinemia.
Digenic inheritance refers to mutations in two distinct genes causing a genetic phenotype or disease. With increasing exome and genome sequence data being generated through NGS, the number of human diseases exhibiting digenic inheritance continues to grow (Gazzo et al., 2016; Deltas, 2018). It provides new insights into the genetics underlying many disorders classically considered monogenic (Schäffer, 2013). Distal renal tubular acidosis (dRTA) is just one example (Nagara et al., 2018). In the present study, we identified a patient (P8) harboring both a heterozygous mutation of ASS1 (p.Thr323Ile) and a heterozygous mutation of SLC25A13 (p.Met285Profs*2). The patient had high levels of plasma citrulline (671.3 μmol/l, normal: 7–40 μmol/l) and arginine (119.7 μmol/l, normal: 1.5–25 μmol/l). Together, these results might implicate a putative digenic inheritance mechanism in citrullinemia. A mutated allele of ASS1 may reduce the formation of argininosuccinate from citrulline and aspartate, while a mutated SLC25A13 allele is likely to further inhibit the reaction by limiting the supply of aspartate from mitochondria. Thus, the compound mutations would be predicted to lead to the upstream accumulation of citrulline. Further analyses are required to investigate the effects of these compound mutations on cell metabolism.
The clinical presentation of citrullinemia is very heterogeneous. In our study, patient P9 with ASS1 mutations displayed the most severe clinical symptoms. She had the highest plasma citrulline level (1924.1 μmol/l; normal: 7–40 μmol/l), and died at 1 year of age. This indicates that ASS1 mutations might cause more severe clinical manifestations than SLC25A13 mutations. Consistent with this, patient P8 carrying compound heterozygous mutations of ASS1 and SLC25A13 also showed more severe clinical manifestations than patients only harboring SLC25A13 mutations. Most mutations of SLC25A13 identified in this study led to truncated proteins. However, no firm genotype–phenotype correlations could be observed in patients carrying SLC25A13 mutations.
The present study had some limitations. First, we did not perform functional analysis of the c.1311 + 4_+7del mutation to support its predicted effect on SLC25A13 splicing. Second, because of limited sample sources, genetic analysis of the patients’ parents was not carried out to determine if the identified novel mutations were de novo; additionally, the segregation test for identified mutations could not be performed.
In summary, we carried out genetic analysis and functional investigation in Chinese infants with citrullinemia. We identified a novel mutation of ASS1 and a novel mutation of SLC25A13. These findings expand the mutational spectrum of ASS1 and SLC25A13, and improve our understanding of the molecular genetic mechanism of citrullinemia in the Chinese population.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the Ethics Committee of the Children’s Hospital affiliated to Chongqing Medical University (No. 2021078) and Chongqing Health Center for Women and Children (No. 2020024). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
JM conceived the study. Z-EX, CL, and HL were responsible for recruiting patients and collection of blood samples. ZC performed the experimental work. XH and FZ participated in the experimental work. JM and ZC drafted the manuscript, tables, and figures. All authors read and approved the final manuscript.
This study was supported by Chongqing Health Center for Women and Children: (No. 2019YJMS08), the Fundamental Research Funds for Non-profit Public Scientific Research Institutions of Chongqing (No. 2016CSTC-jbky-01711).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.783799/full#supplementary-material
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 7, 248–249. doi:10.1038/nmeth0410-248
Anna, A., and Monika, G. (2018). Splicing Mutations in Human Genetic Disorders: Examples, Detection, and Confirmation. J. Appl. Genet. 59, 253–268. doi:10.1007/s13353-018-0444-7
Cox, M. P., Peterson, D. A., and Biggs, P. J. (2010). SolexaQA: At-A-Glance Quality Assessment of Illumina Second-Generation Sequencing Data. BMC Bioinformatics 11, 485. doi:10.1186/1471-2105-11-485
Deltas, C. (2018). Digenic Inheritance and Genetic Modifiers. Clin. Genet. 93, 429–438. doi:10.1111/cge.13150
DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., et al. (2011). A Framework for Variation Discovery and Genotyping Using Next-Generation DNA Sequencing Data. Nat. Genet. 43, 491–498. doi:10.1038/ng.806
Diez-Fernandez, C., Rüfenacht, V., and Häberle, J. (2017). Mutations in the Human Argininosuccinate Synthetase (ASS1) Gene, Impact on Patients, Common Changes, and Structural Considerations. Hum. Mutat. 38, 471–484. doi:10.1002/humu.23184
Engel, K., Höhne, W., and Häberle, J. (2009). Mutations and Polymorphisms in the Human Argininosuccinate Synthetase (ASS1) Gene. Hum. Mutat. 30, 300–307. doi:10.1002/humu.20847
Gazzo, A. M., Daneels, D., Cilia, E., Bonduelle, M., Abramowicz, M., Van Dooren, S., et al. (2016). DIDA: A Curated and Annotated Digenic Diseases Database. Nucleic Acids Res. 44, D900–D907. doi:10.1093/nar/gkv1068
Häberle, J., Pauli, S., Linnebank, M., Kleijer, W., Bakker, H., Wanders, R., et al. (2002). Structure of the Human Argininosuccinate Synthetase Gene and an Improved System for Molecular Diagnostics in Patients with Classical and Mild Citrullinemia. Hum. Genet. 110, 327–333. doi:10.1007/s00439-002-0686-6
Häberle, J., Pauli, S., Schmidt, E., Schulze-Eilfing, B., Berning, C., and Koch, H. G. (2003). Mild Citrullinemia in Caucasians Is an Allelic Variant of Argininosuccinate Synthetase Deficiency (Citrullinemia Type 1). Mol. Genet. Metab. 80, 302–306. doi:10.1016/j.ymgme.2003.08.002
Herrera Sanchez, M. B., Previdi, S., Bruno, S., Fonsato, V., Deregibus, M. C., Kholia, S., et al. (2017). Extracellular Vesicles from Human Liver Stem Cells Restore Argininosuccinate Synthase Deficiency. Stem Cel. Res. Ther. 8, 176. doi:10.1186/s13287-017-0628-9
Hori, T., Fukao, T., Murase, K., Sakaguchi, N., Harding, C. O., and Kondo, N. (2013). Molecular Basis of Two-Exon Skipping (Exons 12 and 13) by c.1248+5g>a in OXCT1 Gene: Study on Intermediates of OXCT1 Transcripts in Fibroblasts. Hum. Mutat. 34, 473–480. doi:10.1002/humu.22258
Ibrahim, E. C., Hims, M. M., Shomron, N., Burge, C. B., Slaugenhaupt, S. A., and Reed, R. (2007). Weak Definition of IKBKAP Exon 20 Leads to Aberrant Splicing in Familial Dysautonomia. Hum. Mutat. 28, 41–53. doi:10.1002/humu.20401
Iijima, M., Jalil, A., Begum, L., Yasuda, T., Yamaguchi, N., Xian Li, M., et al. (2001). Pathogenesis of Adult-Onset Type II Citrullinemia Caused by Deficiency of Citrin, a Mitochondrial Solute Carrier Protein: Tissue and Subcellular Localization of Citrin. Adv. Enzyme Regul. 41, 325–342. doi:10.1016/s0065-2571(00)00022-4
Kobayashi, K., Kakinoki, H., Fukushige, T., Shaheen, N., Terazono, H., and Saheki, T. (1995). Nature and Frequency of Mutations in the Argininosuccinate Synthetase Gene that Cause Classical Citrullinemia. Hum. Genet. 96, 454–463. doi:10.1007/BF00191806
Kobayashi, K., Sinasac, D. S., Iijima, M., Boright, A. P., Begum, L., Lee, J. R., et al. (1999). The Gene Mutated in Adult-Onset Type II Citrullinaemia Encodes a Putative Mitochondrial Carrier Protein. Nat. Genet. 22, 159–163. doi:10.1038/9667
Kohl, S., Zobor, D., Chiang, W.-C., Weisschuh, N., Staller, J., Menendez, I. G., et al. (2015). Mutations in the Unfolded Protein Response Regulator ATF6 Cause the Cone Dysfunction Disorder Achromatopsia. Nat. Genet. 47, 757–765. doi:10.1038/ng.3319
Kumar, P., Henikoff, S., and Ng, P. C. (2009). Predicting the Effects of Coding Non-synonymous Variants on Protein Function Using the SIFT Algorithm. Nat. Protoc. 4, 1073–1081. doi:10.1038/nprot.2009.86
Li, H., and Durbin, R. (2009). Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 25, 1754–1760. doi:10.1093/bioinformatics/btp324
Li, R., Yu, C., Li, Y., Lam, T.-W., Yiu, S.-M., Kristiansen, K., et al. (2009). SOAP2: an Improved Ultrafast Tool for Short Read Alignment. Bioinformatics 25, 1966–1967. doi:10.1093/bioinformatics/btp336
Lin, Y., Gao, H., Lu, B., Zhou, S., Zheng, T., Lin, W., et al. (2019). Citrullinemia Type I Is Associated with a Novel Splicing Variant, c.773 + 4A > C, in ASS1: a Case Report and Literature Review. BMC Med. Genet. 20, 110. doi:10.1186/s12881-019-0836-5
Liu, G., Wei, X., Chen, R., Zhou, H., Li, X., Sun, Y., et al. (2014). A Novel Mutation of the SLC25A13 Gene in a Chinese Patient with Citrin Deficiency Detected by Target Next-Generation Sequencing. Gene 533, 547–553. doi:10.1016/j.gene.2013.10.021
Miyamoto, T., Lo, P. H. Y., Saichi, N., Ueda, K., Hirata, M., Tanikawa, C., et al. (2017). Argininosuccinate Synthase 1 Is an Intrinsic Akt Repressor Transactivated by P53. Sci. Adv. 3, e1603204. doi:10.1126/sciadv.1603204
Nagara, M., Papagregoriou, G., Ben Abdallah, R., Landoulsi, Z., Bouyacoub, Y., Elouej, S., et al. (2018). Distal Renal Tubular Acidosis in a Libyan Patient: Evidence for Digenic Inheritance. Eur. J. Med. Genet. 61, 1–7. doi:10.1016/j.ejmg.2017.10.002
Ohno, K., Takeda, J. I., and Masuda, A. (2018). Rules and Tools to Predict the Splicing Effects of Exonic and Intronic Mutations. Wiley. Interdiscip. Rev. RNA. 9, e1451. doi:10.1002/wrna.1451
Saheki, T., and Kobayashi, K. (2002). Mitochondrial Aspartate Glutamate Carrier (Citrin) Deficiency as the Cause of Adult-Onset Type II Citrullinemia (CTLN2) and Idiopathic Neonatal Hepatitis (NICCD). J. Hum. Genet. 47, 333–341. doi:10.1007/s100380200046
Saheki, T., Kobayashi, K., Ichiki, H., Matuo, S., Tatsuno, M., Imamura, Y., et al. (1987). Molecular Basis of Enzyme Abnormalities in Urea Cycle Disorders. Enzyme 38, 227–232. doi:10.1159/000469209
Schäffer, A. A. (2013). Digenic Inheritance in Medical Genetics. J. Med. Genet. 50, 641–652. doi:10.1136/jmedgenet-2013-101713
Schwarz, J. M., Cooper, D. N., Schuelke, M., and Seelow, D. (2014). MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 11, 361–362. doi:10.1038/nmeth.2890
Stingl, K., Mayer, A. K., Llavona, P., Mulahasanovic, L., Rudolph, G., Jacobson, S. G., et al. (2017). CDHR1 Mutations in Retinal Dystrophies. Sci. Rep. 7, 6992. doi:10.1038/s41598-017-07117-8
Tabata, A., Sheng, J.-S., Ushikai, M., Song, Y.-Z., Gao, H.-Z., Lu, Y.-B., et al. (2008). Identification of 13 Novel Mutations Including a Retrotransposal Insertion in SLC25A13 Gene and Frequency of 30 Mutations Found in Patients with Citrin Deficiency. J. Hum. Genet. 53, 534–545. doi:10.1007/s10038-008-0282-2
Verma, M., Charles, R. C. M., Chakrapani, B., Coumar, M. S., Govindaraju, G., Rajavelu, A., et al. (2017). PRMT7 Interacts with ASS1 and Citrullinemia Mutations Disrupt the Interaction. J. Mol. Biol. 429, 2278–2289. doi:10.1016/j.jmb.2017.05.026
Woo, H. I., Park, H.-D., and Lee, Y.-W. (2014). Molecular Genetics of Citrullinemia Types I and II. Clin. Chim. Acta 431, 1–8. doi:10.1016/j.cca.2014.01.032
Yamaguchi, N., Kobayashi, K., Yasuda, T., Nishi, I., Iijima, M., Nakagawa, M., et al. (2002). Screening of SLC25A13 Mutations in Early and Late Onset Patients with Citrin Deficiency and in the Japanese Population: Identification of Two Novel Mutations and Establishment of Multiple DNA Diagnosis Methods for Nine Mutations. Hum. Mutat. 19, 122–130. doi:10.1002/humu.10022
Zhang, Z.-H., Lin, W.-X., Zheng, Q.-Q., Guo, L., and Song, Y.-Z. (2017). Molecular Diagnosis of Citrin Deficiency in an Infant with Intrahepatic Cholestasis: Identification of a 21.7kb Gross Deletion that Completely Silences the Transcriptional and Translational Expression of the Affected SLC25A13 Allele. Oncotarget 8, 87182–87193. doi:10.18632/oncotarget.19901
Keywords: citrullinemia, genetic analysis, mutation, ASS1, SLC25A13
Citation: Cheng Z, He X, Zou F, Xu Z-E, Li C, Liu H and Miao J (2022) Identification of Novel Mutations in Chinese Infants With Citrullinemia. Front. Genet. 13:783799. doi: 10.3389/fgene.2022.783799
Received: 27 September 2021; Accepted: 15 February 2022;
Published: 03 March 2022.
Edited by:
Chen Huei Leo, Singapore University of Technology and Design, SingaporeCopyright © 2022 Cheng, He, Zou, Xu, Li, Liu and Miao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingkun Miao, amVubmFtaWFvOTlAc2luYS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.