 Sitao Li
Sitao Li Congcong Shi
Congcong Shi Yao Cai
Yao Cai Xia Gu1
Xia Gu1 Fei Ma
Fei Ma Hu Hao
Hu Hao- 1Department of Pediatrics, The Sixth Affiliated Hospital, Sun Yat sen University, Guangzhou, China
- 2Inborn Errors of Metabolism Laboratory, The Sixth Affiliated Hospital, Sun Yat sen University, Guangzhou, China
- 3Department of Child Care, The Maternity and Child Health Care Hospital of Tianhe District, Guangzhou, China
Isolated methylmalonic acidemia (MMA) is an inherited organic acid metabolic disorder in an autosomal recessive manner, caused by mutations in the methylmalonyl coenzyme A mutase gene, and the isolated MMA patients often suffer from multi-organ damage. The present study aimed to profile the differential proteome of serum between isolated MAA patients and healthy control. The in vivo proteome of isolated MAA patients and healthy subjects was detected by an isobaric tag for relative and absolute quantitation (iTRAQ). A total of 94 differentially expressed proteins (DEPs) were identified between MMA patients and healthy control, including 58 upregulated and 36 downregulated DEPs in MMA patients. Among them, the most significantly upregulated proteins were CRP and immunoglobulins, and the top five most significantly downregulated proteins were all different types of immunoglobulins in MMA patients. GO analysis showed that these DEPs were mainly enriched in immune-related function and membrane protein-related function. KEGG revealed that these DEPs were mainly enriched in lysosome and cholesterol metabolism pathways. Also, these DEPs were predicted to contribute to lipid metabolic diseases. We addressed the proteomes of isolated MMA patients and identified DEPs. Our study expands our current understanding of MMA, and the DEPs could be valuable for designing alternative therapies to alleviate MMA symptoms.
Introduction
Methylmalonic acidemia (MMA) is an inherited organic acid metabolic disorder in an autosomal recessive manner. According to the biochemical manifestation, MMA can be classified into two common types: isolated MMA and MMA combined with homocysteinemia (Manoli and Venditti, 2005). Isolated MMA is mostly caused by mutations in the methylmalonyl coenzyme A mutase (MCM) gene (MUT) (Han et al., 2017), and a few cases have been shown to be the result of other gene mutations, such as MMAA, MCEE, and others (He et al., 2020). MMA usually resulted in various clinical symptoms, including severe metabolic acidosis, thrombocytopenia, hyperammonemia, ketosis and ketonuria, developmental delay, neutropenia, and hyperglycinemia (Manoli and Venditti, 2005). Clinical diagnosis of MMA relies upon specialized metabolic testing. Definitive diagnosis of MMA relies on the analysis of organic acids in plasma and/or urine by gas-liquid chromatography and mass spectrometry; the concentration of methylmalonic acid is greatly increased in the plasma, urine, and cerebrospinal fluid of affected individuals (Manoli et al., 1993). Based on complex and comprehensive clinical diagnosis, the abnormal production of metabolites in MMA patients has been relatively clear, but proteomic changes in MMA patients are rarely reported.
Isolated MMA patients with mutations in the MUT gene are usually severely ill with poor prognosis, high early mortality, and high lifelong morbidity (Jiang et al., 2020; Liang et al., 2021). The age of death in children with isolated MMA was 2 years, ranging from 5 days to 15 years, and 40% of individuals died and the overall mortality rate was 36%, with all deaths occurring during or after the acute metabolic crisis (Dionisi-Vici et al., 2006). The primary treatment for patients with isolated MMA is dietary restriction of propyl amino acid and carnitine supplementation. Despite treatment, the metabolite profile of the patients with isolated MMA remains abnormal, and the prognosis is still poor. Although several hypotheses have been proposed, the pathologic mechanism of abnormal protein expression in progressive isolated MMA systemic injury remains to be elucidated.
Recently, the characterization of MMA with homocystinuria and cobalamin deficiency type C (cblC) proteome in fibroblasts found that the downregulated proteins were mainly enriched in cellular detoxification (Hannibal et al., 2011). Also, the proteome in circulating lymphocytes in cblC patients has been demonstrated (Caterino et al., 2015). However, the proteomic characteristics of isolated MMA patients are still unclear. The present study investigated the proteome of serum isolated from three isolated MMA patients with MUT treated with a multi-drug combination. These three patients still have abnormal levels of metabolites although receiving treatment. This study of the proteome in isolated MMA patients was performed using the isobaric tag for relative and absolute quantitation (iTRAQ) analysis. Proteome identification helped bridge the gap between the role of MUT gene products and the clinical manifestations of MMA defects in humans.
Materials and methods
Ethics statement
This project was approved by the Ethical Committee of The Sixth Affiliated Hospital of Sun Yat-sen University and adhered to the World Medical Association’s Declaration of Helsinki (WMADH 2008). In this study, MMA analysis was performed with written informed consent from guardians of each pediatric patient, following the approval of The Sixth Affiliated Hospital of Sun Yat-sen University.
Patient selection
The serum for three MMA patients and three healthy subjects from The Sixth Affiliated Hospital of Sun Yat-sen University was collected. Genotypes of MMA donors are listed in Table 1; the virulence gene of three MMA patients is mut gene. Clinical diagnosis of MMA patients relies upon specialized metabolic testing, which is performed by the Genetic Metabolic Disease Detection Laboratory of the Sixth Affiliated Hospital of Sun Yat-sen University. Table 2 listed the detailed clinical features of MMA patients determined by gas–liquid chromatography and mass spectrometry and also listed the detail of laboratory biochemical parameters. Serum from normal control and patients with confirmed MMA were subjected to iTRAQ assay for investigating the differentially expressed proteins (DEPs). Moreover, to verify the clinical characteristics of candidate DEPs, we subsequently collected the clinical biochemical data from 14 MMA patients and 15 non-MMA and uninfected patients. These 29 samples were in addition to those analyzed in the iTRAQ experiment. The information of these 29 samples is shown in Supplementary Table S1. All frozen tissue samples in liquid nitrogen were stored at −80°C.

TABLE 1. List of MMA donors with their genotypes and clinical features used in this study.
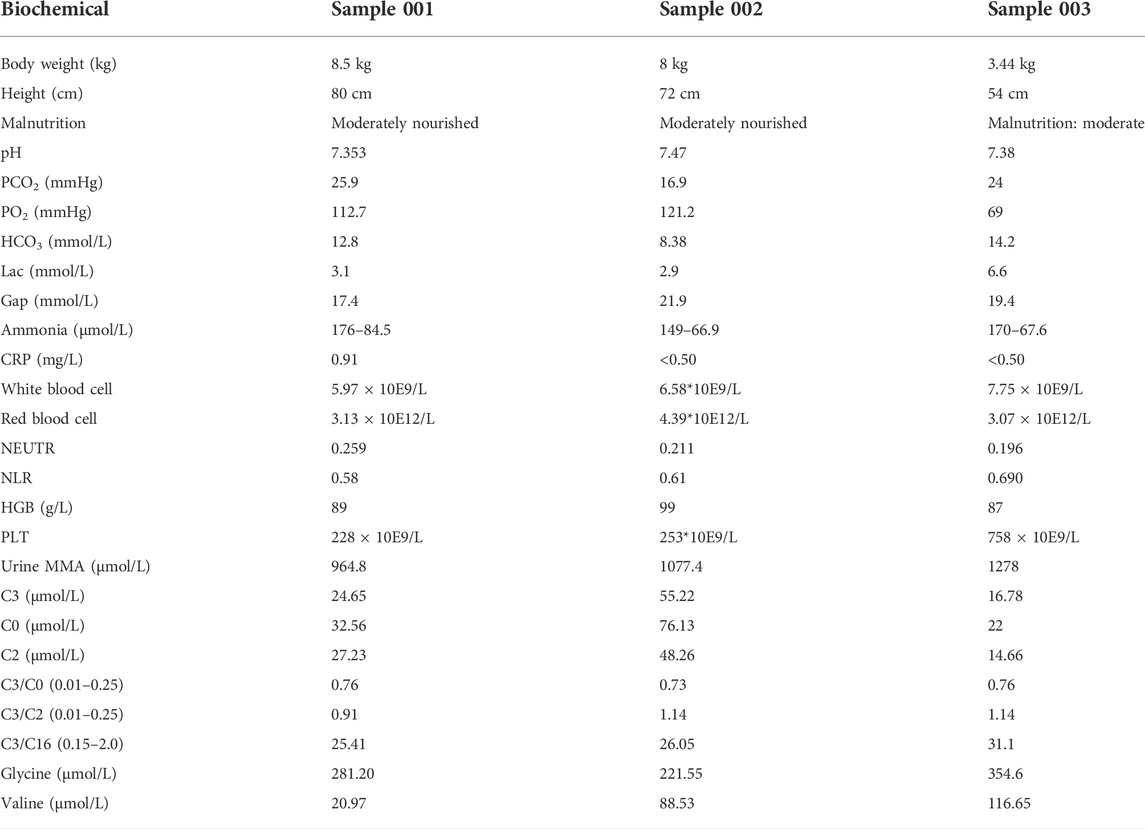
TABLE 2. Clinical information of MMA patients prior to sampling.
Protein extraction and isobaric tag for relative and absolute quantitation labeling
Frozen serum samples were extracted by using RIPA lysis buffer (Elabscience, China) and subsequently homogenized by sonication (Scientz) on ice. The homogenate was cleared using centrifugation at 12,000 rpm for 15 min at 4°C. The major proteins of albumin and IgG were removed using ProteoExtract™ Albumin Removal Kit (Cat. No.:122640, MERCK). After that, the protein concentrations of supernatants were determined by Pierce BCA Protein Assay Kit. Subsequently, 20 μg of each sample was taken and tested by SDS-PAGE. The qualified protein sample entered the iTRAQ labeling stage. Total protein (150 μg) from each sample solution was mixed with 200 μl of 8 mol/L urea in Nanosep Centrifugal Devices (PALL, United Kingdom) and centrifuged at 14,000 g at 20°C for 20 min. All following centrifugation steps were performed applying the same conditions allowing maximal concentration. Samples were incubated in 20 μl of 50 mmol/L iodoacetamide for 30 min in the dark to block reduced cysteine residues followed by centrifugation, and the liquid at the bottom was discarded. The Tris–HCl (pH 8.0) was added to every tube, centrifuged at 12,000 rpm for 10 min, and the solution was discarded at the bottom. Then, 200 μl of 100 mmol/L triethylammonium bicarbonate (TEAB) buffer was added to tubes and centrifuged twice. The solution was subjected to proteolytic digestion (1:50) overnight at 37°C. The digests of peptides were collected by centrifugation following iTRAQ labeling.
Each iTRAQ-label reagent (AB Sciex, United States) was dissolved in 70 μl of isopropanol. The digested-peptide samples were dissolved in 30 μl TEAB buffer (200 mM). Next, the iTRAQ-label reagent was added to the respective peptide mixture for 2 h. The labeling reaction was quenched by the addition of 100 μl of MilliQ water, and six labeled samples were then pooled into one sample according to the manufacturer’s instructions. After pooling, the samples were evaporated by vacuum concentration to remove excess water, TEAB, and isopropanol.
One-dimensional high-pH reversed-phase chromatography separation
The freeze-dried samples were dissolved in 100 μl of mobile phase A (10 nM ammonium formate, 5% acetonitrile, pH 10.0). Peptide separation was performed on a Thermo UltiMate 3000 UHPLC, and the chromatographic column was purchased from Agilent (ZORBAX Extended-C18, 2.1). The detection wavelength was UV 215 nm, and the flow rate was 0.3 ml/min. Mobile phase B (10 nM ammonium formate, 90% acetonitrile, and pH 10.0) separation gradient was linear from 5 to 38% in 80 min. One tube was collected every 1 min within the gradient range, and a total of 16 tubes of elution solution were collected, centrifuged, and dried for LC-MS analysis.
Liquid chromatography tandem mass spectrometry analysis and data reorganization
The LC-MS/MS analysis was conducted by Wininnovate Bio (China). Briefly, the lyophilized peptide fractions were re-suspended in 0.1% formic acid and then loaded into a nanoViper C18 (3 μm, 100Å) trap column, and online chromatography separation was performed on the Easy nLC 1200 system (ThermoFisher, United States). The trapping and desalting procedure was carried out with a volume of 20 μl 100% solvent A (0.1% formic acid). Then, an elution gradient of 5–38% solvent B (80% acetonitrile, 0.1% formic acid) at a flow rate of 300 nl/min (0–50 min, 5–38% B; 60–60 min, 38–100% B) in 60 min was used on an analytical column (50 μm × 15 cm C18-3 μm 100 Å). The data-dependent acquisition (DDA) mass spectrum techniques were used to acquire tandem mass spectrometry (MS) data on a ThermoFisher Q Exactive mass spectrometer (ThermoFisher, United States) fitted with a Nano Flex ion source. Data were acquired using an ion spray voltage of 1.9 kV, and an interface heater temperature of 275°C. The MS was operated with FULL-MS scans. For DDA, survey scans were acquired in 250 ms and up to 20 product ion scans (50 ms) were collected. Normalized collision energy (NCE) was set to 30 eV. Only spectra with a charge state of 2–4 were selected for fragmentation by higher-energy collision energy. Dynamic exclusion was set for 25 s. The proteomic raw data have been deposited on PRIDE Archive (https://www.ebi.ac.uk/pride/archive) with the project accession: PXD034075.
Data reorganization
The MS/MS data were analyzed for protein identification and quantification using the Proteome discoverer (v2.1.0.81). The local false discovery rate was 1.0% after searching against the Human protein database with a maximum of two missed cleavages and one missed termini cleavage. The following settings were selected: oxidation (M), acetylation (Protein N-term), deamidation (NQ), Pyro-glu from E, Pyro-glu from Q for variable modifications as well as carbamidomethylation (C), iTRAQ8plex (N-term), and iTRAQ8plex (K) for fixed modifications. Precursor and fragment mass tolerance were set to 10 ppm and 0.05 Da, respectively. A full list of proteins identified and relatively quantified by iTRAQ is shown in Supplementary Table S4. A full list of peptide groups identified and relatively quantified by iTRAQ is shown in Supplementary Table S5.
Statistical analysis of isobaric tags for relative and absolute quantitation data
DEPs were screened by t-test of the “limma” R package, and proteins with p-value < 0.05 and fold change (FC) ≥ 1.2 were considered DEPs.
Functional annotation
For functional annotation, the proteomic data were normalized, and then the protein expression of each sample was analyzed for differences to screen the DEPs using a t-test provided by the “limma” R package. The screening criterion of DEPs was that fold change (FC) ≥ 1.2 and the p-value < 0.05, between the two groups. Next, cluster analysis, principal component analysis (PCA), and partial least squares discrimination analysis (PLS-DA) were performed on DEPs, and they were referred to the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) database, among which the “ggplot2” R package was used for volcano plots, heatmap, and PCA. The “ropls” R package was used for PLS-DA. For gene set enrichment analysis (GSEA), the whole genome was ranked from the largest to smallest based on the logFC of DEPs and matched to the KEGG pathway database, to draw a GSEA plot. A protein–protein interaction (PPI) network was constructed by Cytoscape version 3.6.1 using DEPs.
Results
Quantitative proteomic profiling of serum of patients with isolated methylmalonic acidemia by isobaric tags for relative and absolute quantitation labeling
Serum of patients with isolated MMA and healthy donators was subjected to proteomic analysis to elucidate proteomic changes. A total of 10306 peptides were identified, of which 9722 were uniquely mapped to known sequences of our iTRAQ proteomic results. Moreover, we identified 770 proteins whose average molecular weight was between 24 and 80 kDa (Figure 1A). PCA and PLS-DA analysis showed an obvious separation in the control group and mut group with the 95% confidence intervals, implicating that proteins were significantly separated (Figures 1B,C). Therefore, these data were used for further analysis.
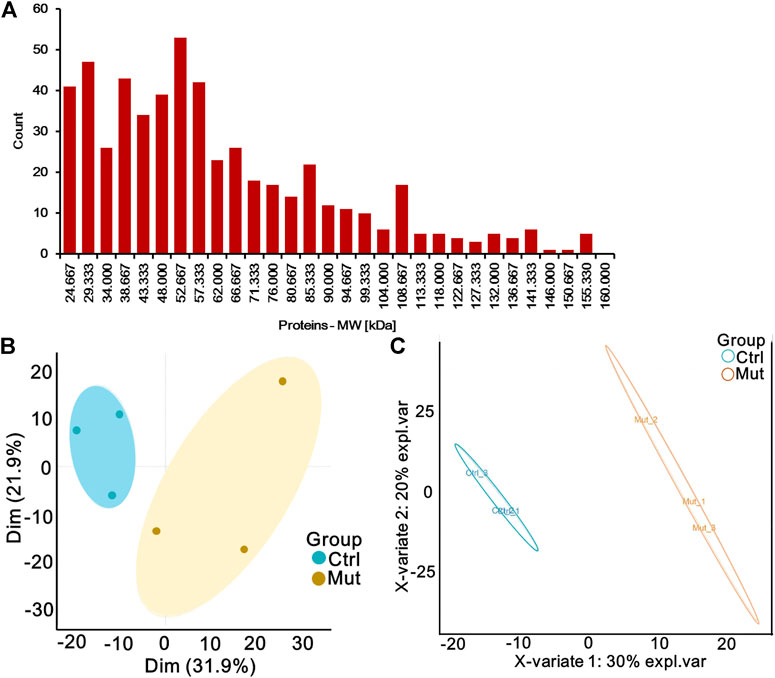
FIGURE 1. Quantitative proteomic profiling of serum of patients with MMA by iTRAQ labeling. (A) Histogram of the distribution of the average molecular weight of protein. (B) Principal component analysis (PCA) diagram of six samples. Oval indicates the 95% confidence intervals. (C) Partial least-squares discriminant analysis (PLS-DA) diagram of six samples based on all proteins. Oval indicates the 95% confidence intervals.
Differential proteomics analysis
To search for MMA-induced specific proteins, proteome differential analysis was performed. As shown in the volcano plot, MMA induction upregulated the expression of 58 proteins and downregulated the expression of 36 proteins (Figure 2A). MMA and control groups were analyzed by the heatmap overview showing a good separation between the two groups, as well as confirmed by the PLSDA (Figures 2B,C). All the details of DEPs are listed in Supplementary Table S2, compared with the control group, the most significantly upregulated proteins were C-reactive protein (CRP), immunoglobulin, collagen alpha-2 (XI) chain (COL11A2), GTP-binding protein SAR1a (SAR1A), and dipeptidyl peptidase 1 (CTSC), and the top five most significantly downregulated proteins were all immunoglobulins, followed by apolipoprotein (APO), tubulin beta chain (TUBB), and cholesteryl ester transfer protein (CETP).
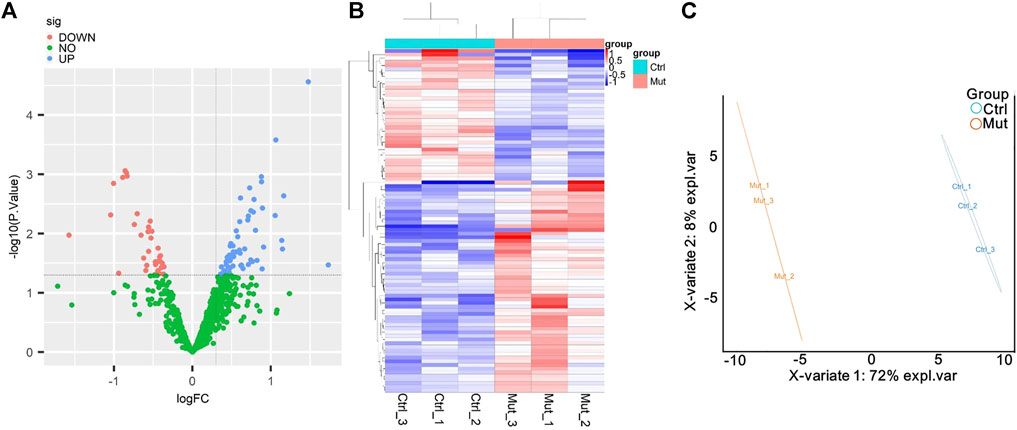
FIGURE 2. Analysis of the differentially expressed proteins (DEPs) between the MMA patients and healthy control. (A) Volcano plot of DEPs. The blue dot represents DEPs upregulated, and the red dot represents DEPs downregulated in the MMA patients compared with the healthy control. (B) Cluster heatmaps of significant DEPs. MUT represents the serum that was obtained from the MMA patients, and Ctrl represents the serum obtained from the healthy control. Each column represents a sample, and the row shaded in red represents upregulated, while shaded in blue represents downregulated when MMA patients vs. healthy control. (C) PL-SDA diagram of six samples based on DEPs. Oval indicates the 95% confidence intervals.
Functional analysis of differentially expressed proteins
To further explore the potential functions of DEPs, GO, and KEGG predictions were performed. GO functional analysis showed that DEPs were mainly related to immune function (such as neutrophil-mediated immunity, neutrophil activation, neutrophil degranulation, neutrophil activation involved in immune response) and membrane protein function (such as vesicle lumen, cytoplasmic vesicle lumen, collagen-containing extracellular matrix) (Figure 3A). The KEGG pathway analysis showed that six DEPs were significantly enriched in the lysosome, including CTSC, tartrate-resistant acid phosphatase type 5 (ACP5), lysosomal protective protein (CTSA), cathepsin B (CTSB), prosaposin (PSAP), and tripeptidyl-peptidase 1 (TPP1), these six DEPs were downregulated in MMA patients (Figure 3B, Supplementary Figure S1). Also, four DEPs were significantly enriched in cholesterol metabolism, including APOE, APOC2, APOB, CETP, and these four DEPs were downregulated in MMA patients (Figure 3B, Supplementary Figure S2). We also found that the classical immune pathway of the NOD-like receptor pathway was also significantly enriched by these DEPs. Moreover, we used GSEA to further explore the potential function of DEPs, consistent with the KEGG results, the lysosome pathway was also observed in the GSEA-plot with the top five significant enrichment pathways, as well as an autoimmune disease pathway of systemic lupus erythematosus (Figure 3C). These results suggested that the disease progression of MMA may be involved not only in immune disorders but also related to abnormal lysosomal function and cholesterol metabolism.
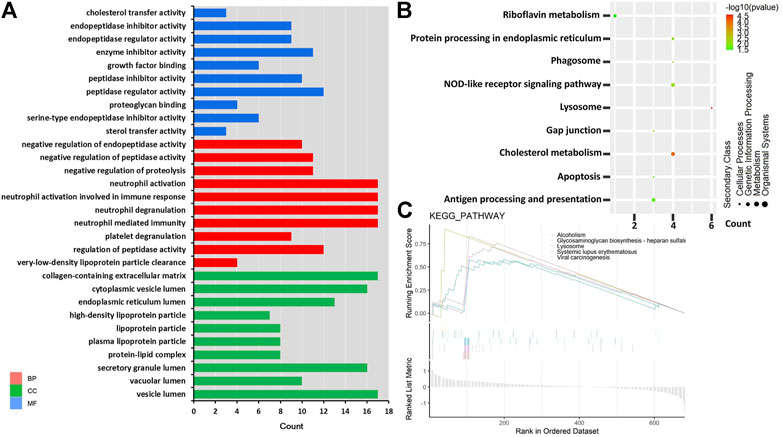
FIGURE 3. GO and KEGG pathway enrichment based on DEPs. (A) Top 30 GO terms in three domains. Red shows DEPs enriched in the biological process (BP); blue shows DEPs enriched in the molecular function (MF); and green shows DEPs enriched in the cellular component (CC). The left is the name of the GO term, while the right column represents the counts of DEPs. (B) KEGG enrichment items. The left is the name of the KEGG pathway, the right represents enrichment, and the size of the solid circle indicates the number of genes. (C) GSEA of identified DEPs. The abscissa is the list of whole genes sorted by logFC; the upper half of the ordinate is divided into enrichment scores, the lower half is divided into log2FC, and the pathway type is represented by the color label.
Differentially expressed proteins associated with lipid metabolic diseases
Furthermore, we predicted the relationship between DEPs and human disease. As shown in Figure 4, the diseases most significantly associated with these DEPs were as follows: congenital disorders of lipid/glycolipid metabolism, lipid metabolism phenotypes, LDL cholesterol, lipoprotein-associated phospholipase A2 activity and mass, and HDL cholesterol. In addition, other diseases related to lipid metabolism were also predicted, such as triglycerides and cholesterol total. These results imply that DEPs are widely involved in lipid-abnormal metabolic diseases.
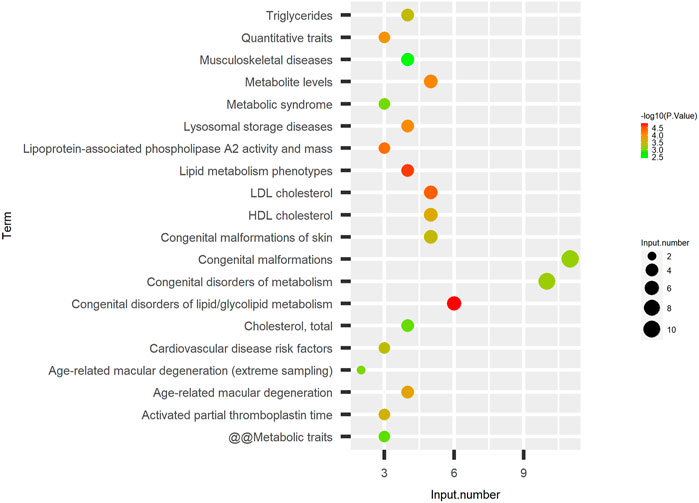
FIGURE 4. DEPs associated with lipid metabolic diseases. Disease enrichment analysis of identified DEPs. The left is the name of the disease, the right represents enrichment, and the size of the solid circle indicates the number of genes; the color of the solid circle indicates the–log10 (p-value) of enrichment.
Protein–protein interaction network analysis
Subsequently, we interrogated the interaction relationship of these DEPs. The UNIPROT_ACCESSION of DEPs was input in Cytoscape, the data were retrieved from the HPIDB database to plot the protein interaction network, and the results as shown in Figure 5. APOE is not only a target of itself but also a target protein of other APO family members. TPP1, which is significantly downregulated in MMA patients, is mainly a target protein of BCAR1, while the downregulated protein of TUBB interacts with multiple DEPs at the same time. Interestingly, both APO and TPP1 have been reported to be involved in lipid metabolism (Ghosh et al., 2017). It follows that there is a tight interaction relationship between the DEPs, which may co-act together on MMA progression, and this contribution may be related to lipid metabolism.
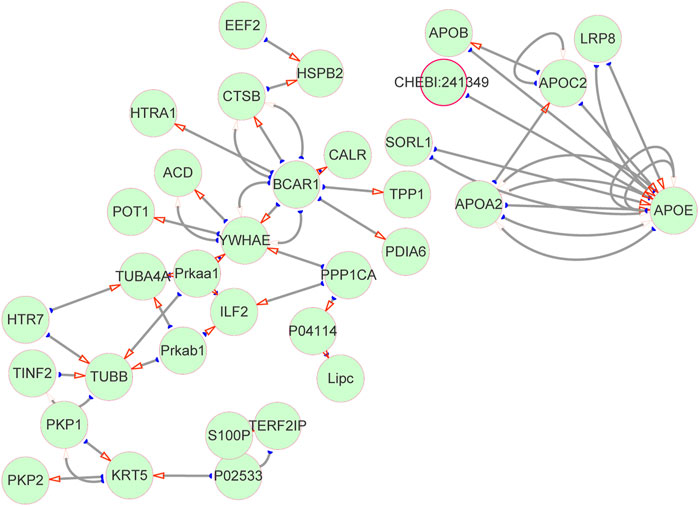
FIGURE 5. Protein–protein interaction network. DEPs interaction network diagram. The green circles represent proteins, and the red arrows point to target proteins (target proteins of DEPs), and the corresponding ones are source proteins (DEPs).
C-reactive protein was positively correlated with methylmalonic acidemia
To explore the reliability of the proteomic data, we expanded the sample size. We compared the levels of CRP in the serum of 14 MMA patients and 15 non-MMA (uninfected) patients, these values were in a range (MMA: 0.5–23.09; non-MMA: 0.5–2.41) consistent with the finding in serum levels of CRP (MMA: < 0.5, 0.91; no-MMA: < 0.5, 0.52) in serum of Table 2 and Supplementary Table S3. The results showed that CRP levels were significantly up-regulated in MMA patients compared with those of non-MMA patients (Figure 6A). Further analysis showed that serum CRP levels were significantly positively correlated with MMA levels and methylcitric acid levels in MMA patients (Figures 6B,C). In fact, we have also analyzed the correlation between CRP content and patient age, gender, C3, C0, and other indicators, but no correlation was found. Together, these results support our proteomic data and suggest a positive correlation between CRP and MMA. The CRP validation is consistent with clinical metadata shown for the patients and thus gives confidence to the iTRAQ dataset as a whole.
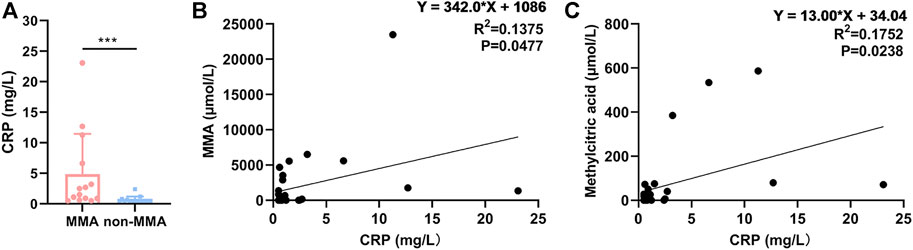
FIGURE 6. CRP was positively correlated with MMA. (A) Levels of CRP in serum of 14 MMA patients and 15 non-MMA (uninfected) patients. (B) Correlation analysis of serum CRP and MMA. (C) Correlation analysis of serum CRP and methylcitric acid.
Discussion
Isolated MMA is an inborn error of metabolism caused by the impaired isomerization of L-MCM to succinyl-CoA mutase. Normally, methylmalonic acid is the product of odd-chain fatty acids, some branched-chain amino acids, and cholesterol via the catabolic pathway of the propionyl coenzyme A to Krebs cycle (Richard et al., 2006). MUT gene mutations prevent the proper breakdown of these molecules, leading to the accumulation of a large number of compounds in the tissue that eventually form the pathologic characteristics of MMA. Considering Since the fact that the clinical diagnosis of MMA relies on metabolite detection, at present, the alteration of the metabolome caused by mutations in the MUT gene has been greatly recognized. In this study, we characterized the protein expression profiles of three isolated MMA patients and three healthy controls by iTRAQ technology in an attempt to connect the relationship between MUT mutations and abnormal protein expression at the global level in MMA patients.
In our study, a total of 94 DEPs were identified between MMA patients and healthy control, including 58 up-regulated and 36 down-regulated DEPs in MMA patients. Among them, compared with the control group, the most significantly up-regulated proteins were CRP and immunoglobulins, and the top five most significantly down-regulated proteins were all different types of immunoglobulins. Immunoglobulins appear repeatedly in the differential protein expression profiles of MMA patients and healthy control, which emphasizes that the immune environment may be strongly altered in MMA patients. This is consistent with the clinical outcome of MMA patients. Wong et al. (1992) reported that the population of B-lymphocytes was aggressively diminished and the ratio of CD4/CD8 was reversed in MMA infants which were regarded as having severe immunodeficiency. Altun et al. (2021) also proved that immunoglobulins and various immune cell types (including memory B cells, recent thymic emigrants, and Naïve helper T cells) exhibit marked defects. Importantly, intravenous administration of immunoglobulin has a dramatic rescue effect on the adverse clinical manifestations of MMA (Aikoh et al., 1997). Abnormal metabolism of MMA is located in mitochondria and leads to the failure of the citric acid cycle and affects the mitochondria function (Forny et al., 2021). It is reported that mitochondrial function determines individual B-cell fates, as well as their immunoglobulins (Jang et al., 2015). Therefore, immunoglobulins may be a potential idea to reverse-interrogate the consequences of MUT mutations in MMA patients.
In addition, CRP is massively elevated in the plasma of patients with MMA, and we speculate that there may be a point of association. It is generally agreed that CRP is highly conserved and is part of innate immune function, and occurs locally in inflamed or damaged tissues. As part of inflammation, elevated CRP is associated with cardiovascular disease, which is well-established by the Centers for Disease Control and the American Heart (Black et al., 2004). Wang et al. (2020) demonstrated that cardiovascular disease was strongly associated with mitochondria-derived MMA. Thus, the inflammatory response may be responsible for the accumulation of CRP protein in MMA serum. However, Del Giudice and Gangestad, (2018) proposed a new theory that CRP may exist in the absence of inflammation and have a net anti-inflammatory effect. In this case, elevated CRP indicates that the body is investing in protecting, preserving, and/or repairing somatic tissue. Depending on the state of the organism, maintenance may translate into responses including inflammation or tolerance. We conjecture that the currently recognized function of CRP may be only the tip of the iceberg and that the increase of CRP in MMA patients may be either a mechanism of clearance of damaged mitochondria or an unknown one. Collectively, CRP is one of the ways to understand the pathological mechanism of MMA.
Moreover, we found significant enrichment of DEPs in cholesterol metabolism pathways and lipid-related diseases. Similar results were found in zebrafish, where cobalamin C (cblC) deficiency caused by mutations in MMA resulted in deregulated expression of genes involved in cholesterol metabolism as demonstrated by Sloan et al. (2020). This can be explained by the physiological function of MCM enzymes. The MCM enzyme is encoded by the MUT gene and is also required for the degradation of a variety of amino acids, odd-chain fatty acids, and cholesterol (Keyfi et al., 2016). Thus, MMA patients with MUT mutations have abnormal cholesterol metabolic pathways. In addition, Manoli and Venditti (2005) reviewed that the major secondary complications of MMA include mental retardation, metabolic stroke, progressive impairment of renal function, pancreatitis, acrodermatitis-enteropathica-like lesions, and functional immune impairment. Interestingly, the occurrence of these diseases is associated with cholesterol or lipid abnormalities. For example, studies have shown that abnormal mutations in cholesterol biosynthetic genes are a possible cause of intellectual disability (Besnard et al., 2019). Mental retardation is also accompanied by a significant accumulation of lipids (Elmahgoub et al., 2009). The total cholesterol levels and incidence of severe acute pancreatitis present a U-shaped relationship (Hong et al., 2020). Therefore, the disease predicted by DEPs may be similar to the complications of MMA, with the common denominator of abnormal cholesterol or lipid metabolism. Summarily, cholesterol metabolism may be one of the important bridges between communicating MUT gene mutations and MMA disease.
The CRP validation is consistent with clinical metadata shown for the patients and thus gives confidence to the iTRAQ dataset as a whole. However, the clinical validation of only one DEP also limits the generalizability of the results of this study to a certain extent. Therefore, in the further continuation, we will continue to explore the expression patterns and functions of four DEPs, including two upregulated DEPs (CTSC and SAR1A) and two downregulated DEPs (lactotransferrin and CETP) in MMA patients compared with healthy control. As mentioned in the results, DEPs associated with lipid metabolic diseases, which prompted us to examine the relationship between lipid-related DEPs and MMA progression. Therefore, CTSC (Rufinatscha et al., 2017), SAR1A (Sane et al., 2019), lactotransferrin (Kovacic et al., 2014), and CETP (Trinder et al., 2021) were selected for future validation due to their significant p-value and their correlation with lipid metabolism. We expect to provide more theoretical bases and evidence for clinical determination.
In conclusion, the present study characterized the protein expression profile of isolated MMA patients with MUT mutation and identified DEPs compared with healthy controls. These DEPs were mainly enriched in GO entries associated with an immune function such as neutrophil-mediated immunity, neutrophil activation, neutrophil degranulation, neutrophil activation involved in immune response, and membrane protein function. KEGG revealed that these DEPs were mainly enriched in lysosome and cholesterol metabolism pathways. Moreover, these DEPs contributed to lipid metabolic diseases. The DEPs and their pathways may be useful targets for new therapies to alleviate disease symptoms, importantly, candidate DEPs, such as CRP and immunoglobulins, and are associated with specific pathways were can be measured by routine tests (already available) and thus implemented in a clinical setting.
Data availability statement
The data presented in this study are deposited in the repository, accession number: PXD034075 (PRIDE Archive, https://www.ebi.ac.uk/pride/archive). The other data found in the article/Supplementary Material.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Sixth Affilia ted Hospital of Sun Yat sen University (Ethics Committee Batch no. 2019ZSLYEC-105). Written informed consent to participate in this study was provided by the p articipant’s legal guardian/next of kin.
Author contributions
Conceptualization and Funding acquisition, HH, and XL; Data curation, SL, XG, and CS; bioinformatics analyses, SL, CS, HX, and YZ; prepared the samples, YC, XL, XG, and HX; extracted serum protein, CS, YZ, and FM; Writing-original draft, SL, YC, and CS, Writing-review & editing, HH, XX, and FM. All authors read and approved the final manuscript, contributed to the article, and approved the submitted version.
Funding
This study was supported by the Guangzhou Science and Technology Project (202102080683).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.765637/full#supplementary-material
References
Aikoh, H., Sasaki, M., Sugai, K., Yoshida, H., and Sakuragawa, N. (1997). Effective immunoglobulin therapy for brief tonic seizures in methylmalonic acidemia. Brain Dev. 19 (7), 502–505. doi:10.1016/S0387-7604(97)00059-4
Altun, I., Kiykim, A., Zubarioglu, T., Burtecene, N., Hopurcuoglu, D., Topcu, B., et al. (2021). Altered immune response in organic acidemia. Pediatr. Int. 64, e15082. doi:10.1111/ped.15082
Besnard, T., Sloboda, N., Goldenberg, A., Kury, S., Cogne, B., Breheret, F., et al. (2019). Biallelic pathogenic variants in the lanosterol synthase gene LSS involved in the cholesterol biosynthesis cause alopecia with intellectual disability, a rare recessive neuroectodermal syndrome. Genet. Med. 21 (9), 2025–2035. doi:10.1038/s41436-019-0445-x
Black, S., Kushner, I., and Samols, D. (2004). C-Reactive protein. J. Biol. Chem. 279 (47), 48487–48490. doi:10.1074/jbc.R400025200
Caterino, M., Pastore, A., Strozziero, M. G., Di Giovamberardino, G., Imperlini, E., Scolamiero, E., et al. (2015). The proteome of cblC defect: in vivo elucidation of altered cellular pathways in humans. J. Inherit. Metab. Dis. 38 (5), 969–979. doi:10.1007/s10545-014-9806-4
Del Giudice, M., and Gangestad, S. W. (2018). Rethinking IL-6 and CRP: why they are more than inflammatory biomarkers, and why it matters. Brain Behav. Immun. 70, 61–75. doi:10.1016/j.bbi.2018.02.013
Dionisi-Vici, C., Deodato, F., Roschinger, W., Rhead, W., and Wilcken, B. (2006). Classical' organic acidurias, propionic aciduria, methylmalonic aciduria and isovaleric aciduria: long-term outcome and effects of expanded newborn screening using tandem mass spectrometry. J. Inherit. Metab. Dis. 29 (2-3), 383–389. doi:10.1007/s10545-006-0278-z
Elmahgoub, S. M., Lambers, S., Stegen, S., Van Laethem, C., Cambier, D., Calders, P., et al. (2009). The influence of combined exercise training on indices of obesity, physical fitness and lipid profile in overweight and obese adolescents with mental retardation. Eur. J. Pediatr. 168 (11), 1327–1333. doi:10.1007/s00431-009-0930-3
Forny, P., Horster, F., Ballhausen, D., Chakrapani, A., Chapman, K. A., Dionisi-Vici, C., et al. (2021). Guidelines for the diagnosis and management of methylmalonic acidaemia and propionic acidaemia: first revision. J. Inherit. Metab. Dis. 44 (3), 566–592. doi:10.1002/jimd.12370
Ghosh, A., Rangasamy, S. B., Modi, K. K., and Pahan, K. (2017). Gemfibrozil, food and drug administration-approved lipid-lowering drug, increases longevity in mouse model of late infantile neuronal ceroid lipofuscinosis. J. Neurochem. 141 (3), 423–435. doi:10.1111/jnc.13987
Han, L. S., Huang, Z., Han, F., Wang, Y., Gong, Z. W., Gu, X. F., et al. (2017). Eight novel MUT loss-of-function missense mutations in Chinese patients with isolated methylmalonic academia. World J. Pediatr. 13 (4), 381–386. doi:10.1007/s12519-016-0085-z
Hannibal, L., DiBello, P. M., Yu, M., Miller, A., Wang, S., Willard, B., et al. (2011). The MMACHC proteome: hallmarks of functional cobalamin deficiency in humans. Mol. Genet. Metab. 103 (3), 226–239. doi:10.1016/j.ymgme.2011.03.008
He, R., Zhang, H., Kang, L., Li, H., Shen, M., Zhang, Y., et al. (2020). Analysis of 70 patients with hydrocephalus due to cobalamin C deficiency. Neurology 95 (23), e3129–e3137. doi:10.1212/WNL.0000000000010912
Hong, W., Zimmer, V., Basharat, Z., Zippi, M., Stock, S., Geng, W., et al. (2020). Association of total cholesterol with severe acute pancreatitis: a U-shaped relationship. Clin. Nutr. 39 (1), 250–257. doi:10.1016/j.clnu.2019.01.022
Jang, K. J., Mano, H., Aoki, K., Hayashi, T., Muto, A., Nambu, Y., et al. (2015). Mitochondrial function provides instructive signals for activation-induced B-cell fates. Nat. Commun. 6, 6750. doi:10.1038/ncomms7750
Jiang, Y. Z., Shi, Y., Shi, Y., Gan, L. X., Kong, Y. Y., Sun, L. Y., et al. (2020). Factors influencing in-hospital death for pediatric patients with isolated methylmalonic acidemia: a nationwide inpatient database analysis. Orphanet J. Rare Dis. 15 (1), 154. doi:10.1186/s13023-020-01446-0
Keyfi, F., Sankian, M., Moghaddassian, M., Rolfs, A., and Varasteh, A. R. (2016). Molecular, biochemical, and structural analysis of a novel mutation in patients with methylmalonyl-CoA mutase deficiency. Gene 576 (1 Pt 2), 208–213. doi:10.1016/j.gene.2015.10.002
Kovacic, B., Hoelbl-Kovacic, A., Fischhuber, K. M., Leitner, N. R., Gotthardt, D., Casanova, E., et al. (2014). Lactotransferrin-Cre reporter mice trace neutrophils, monocytes/macrophages and distinct subtypes of dendritic cells. Haematologica 99 (6), 1006–1015. doi:10.3324/haematol.2013.097154
Liang, L., Shuai, R., Yu, Y., Qiu, W., Shen, L., Wu, S., et al. (2021). A rare mutation c.1663G > A (p.A555T) in the MMUT gene associated with mild clinical and biochemical phenotypes of methylmalonic acidemia in 30 Chinese patients. Orphanet J. Rare Dis. 16 (1), 22. doi:10.1186/s13023-020-01632-0
Manoli, I., Sloan, J. L., and Venditti, C. P. (1993). Isolated methylmalonic acidemia. Seattle, Seattle (WA): University of Washington.
Manoli, I., and Venditti, C. P. (2005). Methylmalonic acidemia. GeneReviews™. Seattle: University of Washington. Available at: https://www.research gate.net/publication/221964133.
Richard, E., Monteoliva, L., Juarez, S., Perez, B., Desviat, L. R., Ugarte, M., et al. (2006). Quantitative analysis of mitochondrial protein expression in methylmalonic acidemia by two-dimensional difference gel electrophoresis. J. Proteome Res. 5 (7), 1602–1610. doi:10.1021/pr050481r
Rufinatscha, K., Radlinger, B., Dobner, J., Folie, S., Bon, C., Profanter, E., et al. (2017). Dipeptidyl peptidase-4 impairs insulin signaling and promotes lipid accumulation in hepatocytes. Biochem. Biophys. Res. Commun. 485 (2), 366–371. doi:10.1016/j.bbrc.2017.02.071
Sane, A., Ahmarani, L., Delvin, E., Auclair, N., Spahis, S., Levy, E., et al. (2019). SAR1B GTPase is necessary to protect intestinal cells from disorders of lipid homeostasis, oxidative stress, and inflammation. J. Lipid Res. 60 (10), 1755–1764. doi:10.1194/jlr.RA119000119
Sloan, J. L., Achilly, N. P., Arnold, M. L., Catlett, J. L., Blake, T., Bishop, K., et al. (2020). The vitamin B12 processing enzyme, mmachc, is essential for zebrafish survival, growth and retinal morphology. Hum. Mol. Genet. 29 (13), 2109–2123. doi:10.1093/hmg/ddaa044
Trinder, M., Wang, Y., Madsen, C. M., Ponomarev, T., Bohunek, L., Daisely, B. A., et al. (2021). Inhibition of Cholesteryl ester transfer protein preserves high-density Lipoprotein cholesterol and improves survival in sepsis. Circulation 143 (9), 921–934. doi:10.1161/CIRCULATIONAHA.120.048568
Wang, S., Liu, Y., Liu, J., Tian, W., Zhang, X., Cai, H., et al. (2020). Mitochondria-derived methylmalonic acid, a surrogate biomarker of mitochondrial dysfunction and oxidative stress, predicts all-cause and cardiovascular mortality in the general population. Redox Biol. 37, 101741. doi:10.1016/j.redox.2020.101741
Keywords: methylmalonic acidemia, iTRAQ, differential proteome, cholesterol metabolism, immunoglobulins, methylmalonyl coenzyme A mutase
Citation: Li S, Shi C, Cai Y, Gu X, Xiong H, Liu X, Zhang Y, Xiao X, Ma F and Hao H (2022) Serum differential proteomic profiling of patients with isolated methylmalonic acidemia by iTRAQ. Front. Genet. 13:765637. doi: 10.3389/fgene.2022.765637
Received: 27 August 2021; Accepted: 01 July 2022;
Published: 29 August 2022.
Edited by:
Hui Xiong, First Hospital, Peking University, ChinaReviewed by:
Hao Hu, Guangzhou Medical University, ChinaCaroline Evans, The University of Sheffield, United Kingdom
Copyright © 2022 Li, Shi, Cai, Gu, Xiong, Liu, Zhang, Xiao, Ma and Hao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hu Hao, aGFvaHVAbWFpbC5zeXN1LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship