Abstract
Background: Recently, UNC45 myosin chaperone A (UNC45A) deficiency was identified as a cause of osteo-oto-hepato-enteric syndrome (O2HE) characterized by congenital diarrhea, neonatal cholestasis, deafness, and bone fragility. To date, only a few O2HE cases have been reported in the literature.
Case presentation: Here, we present a child from China diagnosed with O2HE with novel compound heterozygous variants in UNC45A. The patient suffered with neonatal jaundice, cholestasis, and intractable diarrhea after birth. Laboratory tests revealed highly elevated levels of total serum bilirubin (TB), direct bilirubin (DB), and total bile acid (TBA). The patient was managed with ursodeoxycholic acid (UDCA)-based treatments, and the clinical symptoms and abnormal liver functions were significantly relieved. The patient’s hearing was normal, and no sign of bone fragility was observed. Exome sequencing (ES) identified novel compound heterozygote variants c.292C>T (p.Arg98Trp)/c.2534-2545del (p.Leu845-Met848del) in UNC45A, which were inherited from her mother and father, respectively. Both variants are predicted to be deleterious by in silico predictors.
Conclusion: We present an O2HE child from China with novel compound heterozygous variants in UNC45A. Our patient’s clinical manifestations were less severe than those of the previous reported cases, which expands the clinical spectrum of O2HE.
Background
Neonatal cholestasis is defined as the impairment of bile formation or flow that is characterized by intrahepatic cholestasis, jaundice, and conjugated hyperbilirubinemia in newborns and young infants (Feldman and Sokol, 2020). The incidence of neonatal cholestasis is approximately 1:2500 live births (Fawaz et al., 2017). As a multifactorial disease, more than 100 hepatobiliary and/or metabolic disorders have been clarified that are responsible for the occurrence of neonatal cholestasis (Feldman and Sokol, 2020). The leading cause of neonatal cholestasis is biliary atresia (BA), which accounts for 25%–40% of the total cases, and followed by monogenic disorders (∼25%) (Feldman and Sokol, 2020). Defects in different genes related to the regulation of bile acid synthesis, secretion, transport, and metabolism have been linked to neonatal cholestasis in last decades, including pathogenic mutations in bile acid synthesis and conjugating enzyme-coding genes (AKR1D1, CYP7A1, CYP7B1, SLC27A5, etc.), genes coding for canalicular transporters (ATP8B1, ABCB11, ABCB4, etc.), and nuclear hormone receptor gene, NR1H4 (Lam et al., 2010; Paulusma et al., 2010; Srivastava, 2014; Gordo-Gilart et al., 2015; Feldman and Sokol, 2020). In addition, deficiencies in genes of cytoskeleton and tight junctions were identified to be associated with neonatal cholestasis, such as mutations in MYO5B and TJP2 (Sambrotta et al., 2014; Qiu et al., 2017; Ge et al., 2019). However, known gene defects cannot account for all hereditary cholestasis cases, and many patients remain genetically undiagnosed.
Recently, osteo-oto-hepato-enteric syndrome (O2HE) presenting with congenital diarrhea, neonatal cholestasis, deafness, and bone fragility attributed to loss-of-function mutations in UNC45 myosin chaperone A (UNC45A) was described (Esteve et al., 2018; Duclaux-Loras et al., 2022). Here, we report a male infant from China presented with neonatal cholestasis and intractable diarrhea caused by novel compound heterozygous variants of UNC45A.
Case presentation
A three-month-old boy was admitted to our hospital because of a history of jaundice and intractable diarrhea after birth. The boy was born full-term by cesarean section with a birth weight of 2,250 g. He is the second child of a healthy non-consanguineous couple of Chinese Han ethnicity. The family history was unremarkable. His five-year old brother was healthy. Mild jaundice of the skin and the sclera were observed after birth, and diarrhea was characterized by loose, yellow–green, non-bloody stools occurring 3–5 times per day. He was diagnosed with neonatal jaundice and was discharged from the neonatology department after improvement in the serum bilirubin level. However, jaundice and diarrhea persisted, and he was admitted to the local hospital again when he was 3 months old. Laboratory tests showed highly elevated levels of total serum bilirubin (TB 137.8 μmol/L, reference range: 3.4–17.1 μmol/L), direct bilirubin (DB 107.8 μmol/L, reference range: 0–6.8 μmol/L), and total bile acid (TBA 217.3 μmol/L, reference range: 0–10 μmol/L), and then he was transferred to our hospital for further evaluation.
On admission, the patient’s body temperature was 36.8°C, heart rate was 137 per min, respiratory rate was 27 per min, and blood pressure was 88/57 mmHg. He had a normal weight of 4.8 kg and a height of 58 cm. The skin of the face, trunk, and limbs and the sclera were mildly yellow. The heart and lungs were normal, the abdomen was soft, the liver was 4 cm below the ribs, and the spleen was not touched under the ribs. The muscle strength and tension of the limbs and the neurological examination were unremarkable. The binaural auditory brainstem response (ABR) wave V threshold was 25 dBHL. The liver biochemical profile revealed elevated levels of alanine aminotransferase (ALT 391 U/L, reference range: 5–40 U/L), aspartate aminotransferase (AST 565 U/L, reference range: 8–40 U/L), gamma-glutamyltransferase (GGT 81 U/L, reference range: 7–32 U/L), TB 136.7 μmol/L (reference range: 3.4–17.1 μmol/L), DB 104.4 μmol/L (reference range: 0–6.8 μmol/L), and TBA 195 μmol/L (reference range: 0–10 μmol/L) (Table 1). The detection of liver-damaging pathogens was negative, including Epstein–Barr virus (EBV), cytomegalovirus (CMV), herpes simplex virus (HSV), and hepatitis A, B, and C viruses. Abdominal ultrasound indicated hepatomegaly with diffuse changes. Hepatobiliary scintigraphy with single-photon emission computed tomography (SPECT) revealed impaired hepatic uptake and clearance function and delayed biliary excretion. Magnetic resonance imaging (MRI) showed that the gallbladder was filled, and the common bile duct was less visible. BA was excluded by laparoscopic common bile duct exploration. The diagnosis of intrahepatic cholestasis was made, and the patient was treated with symptomatic therapies of oral ursodeoxycholic acid (UDCA, 50 mg/day), fat-soluble vitamins (vitamin AD, 1800 U/day; vitamin E, 25 mg/day), intravenous compound glycyrrhizin (15 mg/day), and ademetionine 1, 4-butanedisulfonate (250 mg/day).
TABLE 1
| Biochemical index | Reference | 3 months | 4 months | 6 months | 9 months | 12 months |
|---|---|---|---|---|---|---|
| TB (μmol/L) | 3.4–17.1 | 136.75 | 78.56 | 12.49 | 3.9 | 5.9 |
| DB (μmol/L) | 0–6.8 | 104.38 | 42.3 | 3.0 | 1.4 | 2.15 |
| ALT (U/L) | 5–40 | 391 | 156 | 68 | 45 | 35 |
| AST (U/L) | 8–40 | 565 | 176 | 74 | 49 | 54 |
| TBA (μmol/L) | 0–10 | 195 | 138 | 11 | 16.22 | 12.62 |
| GGT (U/L) | 7–32 | 81 | 110 | 42 | 23 | 18 |
Liver biochemical profile of the patient.
TB, total serum bilirubin; DB, direct bilirubin; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, gamma-glutamyltransferase; TBA, total serum bile acid.
To further investigate the etiology of intrahepatic cholestasis, exome sequencing (ES) using genomic DNA extracted from peripheral blood was performed on a HiSeqX10 (Illumina, United States) platform. Novel compound heterozygote variants c.292C>T (p.Arg98Trp)/c.2534-2545del (p.Leu845-Met848del) in UNC45A were identified and were further confirmed by Sanger sequencing (Figure 1). No other potentially pathogenic variants were observed in known genes related to cholestasis. Genotyping of the unaffected family members by Sanger sequencing showed that his mother carries the c.292C>T (p.Arg98Trp) variant, his father carries the c.2534-2545del (p.Leu845-Met848del) variant, and both variants are not detected in his brother (Figure 1). Variant c.292C>T (p.Arg98Trp) is found four times (4/282,568) in heterozygotes, and c.2534-2545del (p.Leu845-Met848del) is reported only once (1/250,666) in the heterozygote state in the gnomAD database. In silico tools predicted that the c.292C>T (p.Arg98Trp) missense variant is deleterious (PROVEAN score, -6.989; PolyPhen-2 score, 1.000). The c.2534-2545del (p.Leu845-Met848del) variant leads to a deletion of four amino acids in the C-terminal UCS domain, and it is predicted to be deleterious with a PROVEAN score of -16.547.
FIGURE 1
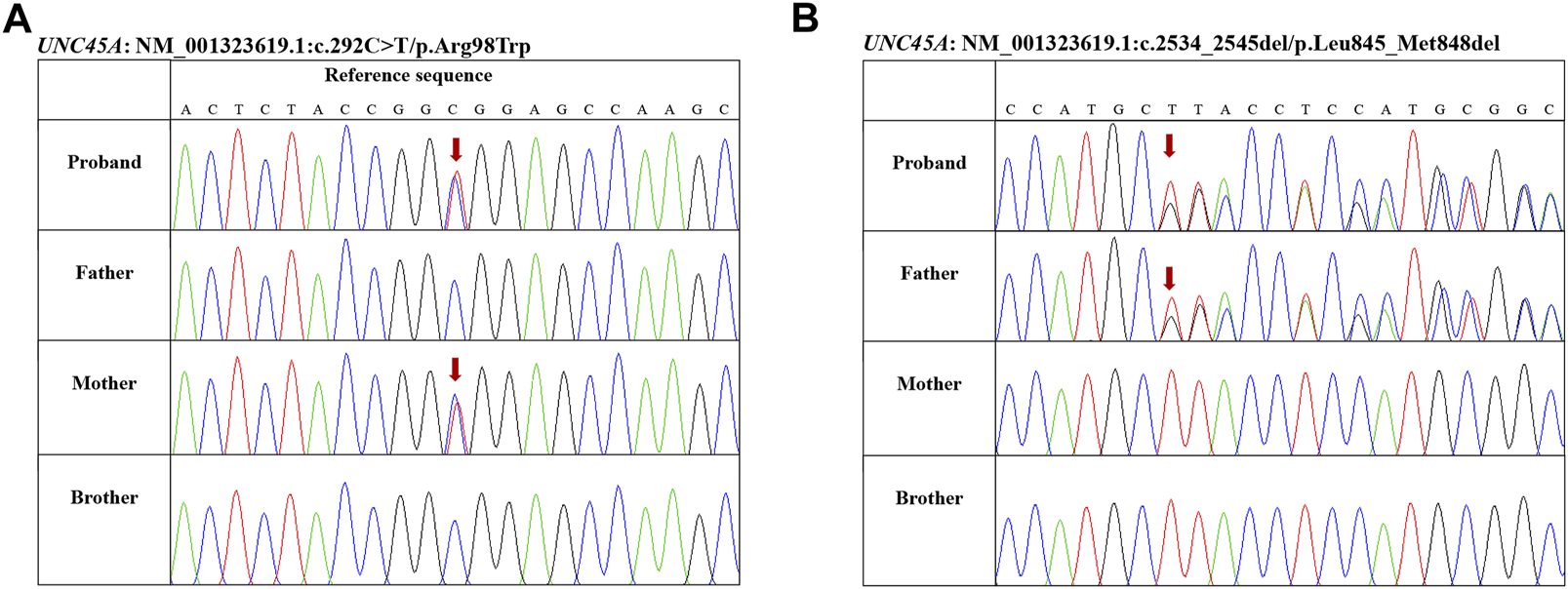
Genotyping of the family members by Sanger sequencing. (A) Proband and his mother carrying variant c.292C>T (p.Arg98Trp). (B) Proband and his father carrying variant c.2534-2545del (p.Leu845-Met848del).
The patient was discharged with alleviated jaundice and significantly improved liver function indexes after 1 month of symptomatic treatments. Maintenance treatments with oral UDCA (50 mg/day), compound glycyrrhizin (12.5 mg/day), and fat-soluble vitamins (vitamin AD, 1800 U/day; vitamin E, 25 mg/day) were recommended to be continued further after discharge from the hospital. During the recent 9-month follow-up period, he had complete clinical and biochemical recovery except for slightly elevated levels of AST and TBA (Table 1). At this writing, he is 1 year old and remains stable with a normal growth (weight, 8,750 g; height, 74 cm) after drug withdrawal for 3 months. He can walk with the help of an instrument, and no sign of bone fragility was observed. His language development is normal, and repeated hearing testing was unremarkable.
Discussion
As a clinical feature common to numbers of hepatobiliary and metabolic diseases, neonatal cholestasis is a condition that requires prompt diagnosis for achieving optimal outcomes. Genetic testing is widely used to define the hereditary etiology of neonatal cholestasis cases without anatomic obstruction of the biliary system. Recently, loss-of-function mutations in UNC45A was reported to be related with a syndrome named O2HE associating cholestasis, diarrhea, impaired hearing, and bone fragility (Esteve et al., 2018; Duclaux-Loras et al., 2022). To the best of our knowledge, we are the first to report an O2HE case from China with novel compound heterozygous variants in UNC45A. The main clinical manifestation of our patient was neonatal cholestasis and persistent diarrhea. ES identified novel compound heterozygote variants c.292C>T (p.Arg98Trp)/c.2534-2545del (p.Leu845-Met848del) in UNC45A, which were inherited from his mother and father, respectively. Both variants were defined as damaging/likely pathogenic according to the guidelines of the American College of Medical Genetics (ACMG) (Kalia et al., 2017).
To date, a total of 10 O2HE patients from eight families with UNC45A loss-of-function mutations were described in the literature (Esteve et al., 2018; Duclaux-Loras et al., 2022). We briefly summarized the characteristics of the reported O2HE patients with UNC45A mutations in Table 2. The most common clinical symptoms of the reported O2HE patients were severe diarrhea (9/10), followed by neonatal cholestasis (8/10), and bone fracture and deafness were presented in six and four patients, respectively (Table 2). Parenteral nutrition and enteral nutrition were required to treat severe diarrhea. In contrast, our patient only suffered with mild diarrhea (3–5 times/day) and recovered along with cholestasis relief at the age of 6 months. In addition to oral UDCA and fat-soluble vitamins, the patient was treated with intravenous compound glycyrrhizin and ademetionine 1, 4-butanedisulfonate. Glycyrrhizin is an active ingredient extracted from licorice, a widely used herbal medicine in traditional Chinese medicine with hepatoprotective and detoxifying properties (Li et al., 2019). The active metabolic product of glycyrrhizin, glycyrrhetinic acid, could relieve cholestasis by regulating bile acid transporters and is anti-inflammatory (Li et al., 2019; Yan et al., 2021). Ademetionine 1, 4-butanedisulfonate injection is a preparation with S-adenosyl-L-methionine (SAMe) as the main active ingredient. SAMe is an important methyl donor that participates in multiple cellular methyltransferase reactions present for an article type that does not allow ones reactions (Lieber and Packer, 2002). It has been shown that SAMe supplementation restores hepatic glutathione (GSH) deposits and attenuates liver injury (Lieber and Packer, 2002; Mato and Lu, 2007). Both compound glycyrrhizin and ademetionine 1, 4-butanedisulfonate injections are recommended for the treatment of intrahepatic cholestasis in China (National Center for Clinical Research of InfectiousD, 2022). Since some O2HE patients’ hearing loss was diagnosed in the late age, further hearing testing is necessary to determine the possible hearing impairment of the patient. At the time of this writing, no sign of bone fragility was observed. Although the patient has remained stable for 3 months with normal growth after discontinuation of drug treatment, he should be monitored closely to be aware of symptom recurrence and late-onset manifestations. For genetic variants in UNC45A, a total of 15 variants were observed, including four homozygous variants and seven compound heterozygote variants, and no genotype and phenotype correlations were identified (Table 2).
TABLE 2
| Variant and consequence | Origin | Gender | Onset age | Age at writing | Diarrhea | Cholestasis | Bone fragility | Deafness | |
|---|---|---|---|---|---|---|---|---|---|
| P1[10] | c.784C>T:p.Arg262* | European descent | Girl | 4 days | 5 years | Yes, need TPN | No | No | Yes |
| c.1268T>A:p.Val423Asp | |||||||||
| P2[10] | c.2581C>T:p.Gln861* | Tunisia | Girl | 15 days | 23 years | No | Yes, resolved at 2.5 years | 23 times bone fractures | Yes, diagnosed at 5 years |
| c.2633C>T:p.Ser878Leu | |||||||||
| c.2734T>G:p.Cys912GIy | |||||||||
| P3[10] | c.2581C>T:p.Gln861* | Tunisia | Girl | 7 days | 18 years | Yes, need TPN | Yes, resolved at 3 years | Yes | Yes, diagnosed in the teens |
| c.2633C>T:p.Ser878Leu | |||||||||
| c.2734T>G:p.Cys912GIy | |||||||||
| P4[10] | c.247C>T:p.Arg83Trp | Turkish | Girl | 15 days | 5 years | Yes, need TPN | Yes, resolved at 3 years | Yes, two times bone fractures | No |
| c.983G>T:p.Gly328Val | |||||||||
| P5[11] | c.710T>C:p.Leu237Pro (homozygous) | Turkish | Girl | 3 weeks | 6 years | Yes, hypovolemic shock | Yes | Yes, two times bone fractures | No |
| P6[11] | c.721C>T:p.Arg241* | United Kingdom | Girl | 1 week | 2 years | Yes, need TPN | No | No | No |
| c.2182G>A:p.Glu728Lys | |||||||||
| P7[11] | c.1452delinsGCA:p.Asp484Glufs*17 | French | Boy | 1 week | 22 years | Yes, need EEN, intermittent until 10 years | Yes | Yes, two times bone fracture | Yes |
| c.2512G>C:p.Ala838Pro | |||||||||
| P8[11] | c.689C>G:p.Thr230Arg (homozygous) | French (West Indies) | Girl | 4 days | 10.5 years | Yes, need TPN and small bowl transplantation | No | No | No |
| P9[11] | c.710T>C:p.Leu237Pro (homozygous) | Turkish | Girl | 1 day | Died at day 93 | Yes, need TPN | Yes | ND | No |
| P10[11] | c.710T>C:P.Leu237Pro (homozygous) | Turkish | Girl | 5 days | 3 months | Yes, need TPN | Yes | Yes, one time bone fracture | ND |
| P11a | c.292C>T:p.Arg98Trp | China | Boy | 1 day | 1 year | Yes, resolved at 6 months | Yes, resolved at 6 months | No | Yes |
| c.2534-2545del:p.Leu845-Met848del |
Summary of clinical and genetic features of mutations in the UNC45A leading to O2HE.
Case reported in this study. UNC45A, UNC45 myosin chaperone A; EEN, exclusive enteral nutrition; ND, not determined; TPN, total parenteral nutrition; O2HE, osteo-oto-hepato-enteric syndrome.
UNC45 is a member of the evolutionary conserved UNC45/Cro1/She4p (UCS) protein family. As an important myosin cochaperone, UNC45 participates in the regulation of a variety of myosin-driven processes, such as cytokinesis, endocytosis, and muscle organization (Lee et al., 2014). There are two related UNC45 proteins (UNC45A and UNC45B) in vertebrates with a sequence identity of 55%, and UNC45A is a ubiquitously expressed protein in different organs, while UNC45B is only expressed in muscle cells (Lee et al., 2014). UNC45 contains three recognizable domains: a C-terminal UCS domain that binds to myosin II motor, a central domain with unknown function, and an N-terminal tetratricopeptide repeat (TPR) domain that interacts with heat shock protein 90 (Hsp90) (Venolia et al., 1999).
Functional studies revealed that loss-of-function mutations in UNC45A attenuate or abolish UNC45A activity, leading to gut development and functional defects (Esteve et al., 2018). Lechuga et al. (2022) showed that defects in UNC45A disrupted epithelial barrier integrity, impaired the assembly of epithelial adherence and tight junctions, and attenuated cell migration by disorganizing actomyosin bundles at epithelial junctions and the migrating cell edge. It has been shown that the loss expression of UNC45A or replacement of the endogenous UNC45A gene with the UNC45A gene carrying the loss-of-function mutation associated with O2HE led to a significantly reduced expression of its cochaperone, myosin VB, in intestinal and liver epithelial cells (Li et al., 2022). As mentioned previously, O2HE patients displayed heterogeneous clinical features, and no obvious genotype and phonotype correlations were observed. Similarly, the heterogeneity presentation has been described in MYO5B-associated microvillus inclusion disease (MVID) patients (Perry et al., 2014). The loss of myosin VB expression is most likely responsible for severe diarrhea in MVID. In accordance with other reported cytoskeleton and tight junction gene defects associated with intrahepatic cholestasis (MYO5B and TJP2) (Sambrotta et al., 2014; Qiu et al., 2017; Ge et al., 2019), cholestasis presented in O2HE patients may be caused by UNC45A mutation-mediated epithelial barrier integrity disruption. Furthermore, mutated UNC45A may affect other myosin proteins involved in precise arrays of mechanosensitive microvilli-like stereocilia crowning the auditory hair cells (e.g., myosin IIIa and MYO15A) that lead to hearing loss (Lelli et al., 2016; Li et al., 2022). UNC45A is required for proper folding of MYO15A. MYO15A is necessary for elongation and maintenance of inner ear hair cell stereocilia (Bird et al., 2014). Stereocilia are required for mechanotransduction of sound. Variants of MYO15A are associated with human deafness DFNB3 (Wang et al., 1998). Thus, UNC45A deficiency-related O2HE is considered a new variant of MVID (Duclaux-Loras et al., 2022).
Conclusion
In summary, we are the first to report a Chinese male O2HE child with novel compound heterozygous variants in UNC45A. The clinical manifestations of our patient were less severe than those of the previous reported cases, which expands the clinical spectrum of O2HE. Future studies are needed to further investigate the molecular mechanisms of UNC45A deficiency.
Statements
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by the Regional Ethical Review Board of Shanghai Children’s Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
YW and YX conceived the study. RW and YW drafted the manuscript. RY, TZ, and YX acquired, analyzed, and interpreted the clinical data. YW and TZ edited the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the grant from the Natural Science Foundation of Shanghai (Grant Number: 22ZR1451800).
Acknowledgments
The authors thank the patient’s family for participating and supporting this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ABCB, adenosine triphosphate-binding cassette subfamily B; AKR1D1, steroid A-ring 5 beta-reductase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; ATP8B1, ATPase class I type 8B member 1; BA, biliary atresia; CYP7, cholesterol 7alpha-hydroxylase; DB, direct bilirubin; EBV, Epstein–Barr virus; ES, exome sequencing; GGT, gamma-glutamyltransferase; MVID, microvillus inclusion disease; MYO5B, motor myosin VB; NR1H4, nuclear receptor subfamily 1, group H, member 4; O2HE, osteo-oto-hepato-enteric syndrome; SLC27A5, solute carrier family 27 member 5; TB, total serum bilirubin; TBA, total bile acid; TJP2, tight junction protein 2; TPR, tetratricopeptide repeat; UDCA, ursodeoxycholic acid; UNC45A, UNC45 myosin chaperone A.
References
1
BirdJ. E.TakagiY.BillingtonN.StrubM. P.SellersJ. R.FriedmanT. B. (2014). Chaperone-enhanced purification of unconventional myosin 15, a molecular motor specialized for stereocilia protein trafficking. Proc. Natl. Acad. Sci. U. S. A.111 (34), 12390–12395. 10.1073/pnas.1409459111
2
Duclaux-LorasR.LebretonC.BertheletJ.Charbit-HenrionF.NicolleO.Revenu De CourtilsC.et al (2022). UNC45A deficiency causes microvillus inclusion disease-like phenotype by impairing myosin VB-dependent apical trafficking. J. Clin. Invest.132 (10), e154997. 10.1172/JCI154997
3
EsteveC.FrancescattoL.TanP. L.BourchanyA.De LeusseC.MarinierE.et al (2018). Loss-of-Function mutations in UNC45A cause a syndrome associating cholestasis, diarrhea, impaired hearing, and bone fragility. Am. J. Hum. Genet.102 (3), 364–374. 10.1016/j.ajhg.2018.01.009
4
FawazR.BaumannU.EkongU.FischlerB.HadzicN.MackC. L.et al (2017). Guideline for the evaluation of cholestatic jaundice in infants: Joint recommendations of the north American society for pediatric gastroenterology, hepatology, and nutrition and the European society for pediatric gastroenterology, hepatology, and nutrition. J. Pediatr. Gastroenterol. Nutr.64 (1), 154–168. 10.1097/MPG.0000000000001334
5
FeldmanA. G.SokolR. J. (2020). Recent developments in diagnostics and treatment of neonatal cholestasis. Semin. Pediatr. Surg.29 (4), 150945. 10.1016/j.sempedsurg.2020.150945
6
GeT.ZhangX.XiaoY.WangY.ZhangT. (2019). Novel compound heterozygote mutations of TJP2 in a Chinese child with progressive cholestatic liver disease. BMC Med. Genet.20 (1), 18. 10.1186/s12881-019-0753-7
7
Gordo-GilartR.AnduezaS.HierroL.Martinez-FernandezP.D'agostinoD.JaraP.et al (2015). Functional analysis of ABCB4 mutations relates clinical outcomes of progressive familial intrahepatic cholestasis type 3 to the degree of MDR3 floppase activity. Gut64 (1), 147–155. 10.1136/gutjnl-2014-306896
8
KaliaS. S.AdelmanK.BaleS. J.ChungW. K.EngC.EvansJ. P.et al (2017). Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of medical genetics and genomics. Genet. Med.19 (2), 249–255. 10.1038/gim.2016.190
9
LamP.SorokaC. J.BoyerJ. L. (2010). The bile salt export pump: Clinical and experimental aspects of genetic and acquired cholestatic liver disease. Semin. Liver Dis.30 (2), 125–133. 10.1055/s-0030-1253222
10
LechugaS.Cartagena-RiveraA. X.KhanA.CrawfordB. I.NarayananV.ConwayD. E.et al (2022). A myosin chaperone, UNC-45A, is a novel regulator of intestinal epithelial barrier integrity and repair. FASEB J.36 (5), e22290. 10.1096/fj.202200154R
11
LeeC. F.MelkaniG. C.BernsteinS. I. (2014). The UNC-45 myosin chaperone: From worms to flies to vertebrates. Int. Rev. Cell Mol. Biol.313, 103–144. 10.1016/B978-0-12-800177-6.00004-9
12
LelliA.MichelV.Boutet De MonvelJ.CorteseM.Bosch-GrauM.AghaieA.et al (2016). Class III myosins shape the auditory hair bundles by limiting microvilli and stereocilia growth. J. Cell Biol.212 (2), 231–244. 10.1083/jcb.201509017
13
LiQ.ZhouZ.SunY.SunC.KlappeK.VanI. S. C. D. (2022). A functional relationship between UNC45A and MYO5B connects two Rare diseases with shared enteropathy. Cell Mol. Gastroenterol. Hepatol.14 (2), 295–310. 10.1016/j.jcmgh.2022.04.006
14
LiX.SunR.LiuR. (2019). Natural products in licorice for the therapy of liver diseases: Progress and future opportunities. Pharmacol. Res.144, 210–226. 10.1016/j.phrs.2019.04.025
15
LieberC. S.PackerL. (2002). S-adenosylmethionine: Molecular, biological, and clinical aspects-an introduction. Am. J. Clin. Nutr.76 (5), 1148S–1150S. 10.1093/ajcn/76/5.1148S
16
MatoJ. M.LuS. C. (2007). Role of S-adenosyl-L-methionine in liver health and injury. Hepatology45 (5), 1306–1312. 10.1002/hep.21650
17
National Center for Clinical Research of Infectious, D (2022). Expert consensus on the diagnosis and treatment of intrahepatic cholestasis (2021 edition). Zhonghua Gan Zang Bing Za Zhi30 (2), 137–146. 10.3760/cma.j.cn501113-20220119-00033
18
PaulusmaC. C.ElferinkR. P.JansenP. L. (2010). Progressive familial intrahepatic cholestasis type 1. Semin. Liver Dis.30 (2), 117–124. 10.1055/s-0030-1253221
19
PerryA.BensallahH.Martinez-VinsonC.BerrebiD.ArbeilleB.SalomonJ.et al (2014). Microvillous atrophy: Atypical presentations. J. Pediatr. Gastroenterol. Nutr.59 (6), 779–785. 10.1097/MPG.0000000000000526
20
QiuY. L.GongJ. Y.FengJ. Y.WangR. X.HanJ.LiuT.et al (2017). Defects in myosin VB are associated with a spectrum of previously undiagnosed low gamma-glutamyltransferase cholestasis. Hepatology65 (5), 1655–1669. 10.1002/hep.29020
21
SambrottaM.StrautnieksS.PapouliE.RushtonP.ClarkB. E.ParryD. A.et al (2014). Mutations in TJP2 cause progressive cholestatic liver disease. Nat. Genet.46 (4), 326–328. 10.1038/ng.2918
22
SrivastavaA. (2014). Progressive familial intrahepatic cholestasis. J. Clin. Exp. Hepatol.4 (1), 25–36. 10.1016/j.jceh.2013.10.005
23
VenoliaL.AoW.KimS.KimC.PilgrimD. (1999). unc-45 gene of Caenorhabditis elegans encodes a muscle-specific tetratricopeptide repeat-containing protein. Cell Motil. Cytoskelet.42 (3), 163–177. 10.1002/(SICI)1097-0169(1999)42:3<163:AID-CM1>3.0.CO;2-E
24
WangA.LiangY.FridellR. A.ProbstF. J.WilcoxE. R.TouchmanJ. W.et al (1998). Association of unconventional myosin MYO15 mutations with human nonsyndromic deafness DFNB3. Science280 (5368), 1447–1451. 10.1126/science.280.5368.1447
25
YanM.GuoL.YangY.ZhangB.HouZ.GaoY.et al (2021). Glycyrrhetinic acid protects alpha-naphthylisothiocyanate- induced cholestasis through regulating transporters, inflammation and apoptosis. Front. Pharmacol.12, 701240. 10.3389/fphar.2021.701240
Summary
Keywords
O2HE, cholestasis, compound heterozygote variants, UNC45A, case report
Citation
Wang R, Wang Y, Yu R, Xu W, Zhang T and Xiao Y (2023) Case report: Osteo-oto-hepato-enteric syndrome caused by UNC45A deficiency. Front. Genet. 13:1079481. doi: 10.3389/fgene.2022.1079481
Received
25 October 2022
Accepted
19 December 2022
Published
09 January 2023
Volume
13 - 2022
Edited by
Babak Behnam, National Sanitation Foundation International, United States
Reviewed by
Loreto Hierro, University Hospital La Paz Research Institute (IdiPAZ), Spain
Thomas Friedman, National Institute on Deafness and Other Communication Disorders (NIH), United States
Updates
Copyright
© 2023 Wang, Wang, Yu, Xu, Zhang and Xiao.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongmei Xiao, xiaoym@shchildren.com.cn
†These authors have contributed equally to this work
This article was submitted to Genetics of Common and Rare Diseases, a section of the journal Frontiers in Genetics
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.