 Idrees A. Shah1
Idrees A. Shah1 Hari Prasad1
Hari Prasad1 Sanghita Banerjee1Reuben Thomas Kurien2
Sanghita Banerjee1Reuben Thomas Kurien2 Sudipta Dhar Chowdhury2*
Sudipta Dhar Chowdhury2* Sandhya S. Visweswariah1*
Sandhya S. Visweswariah1*- 1Department of Molecular Reproduction, Development and Genetics, Indian Institute of Science, Bangalore, India
- 2Department of Gastroenterology, Christian Medical College Vellore, Vellore, Tamil Nadu, India
Introduction: Hereditary pancreatitis (HP) is a rare debilitating disease with incompletely understood etio-pathophysiology. The reduced penetrance of genes such as PRSS1 associated with hereditary pancreatitis indicates a role for novel inherited factors.
Methods: We performed whole-exome sequencing of three affected members of an Indian family (Father, Son, and Daughter) with chronic pancreatitis and compared variants with those seen in the unaffected mother.
Results: We identified a novel frameshift mutation in exon 11 of TRPV6 (c.1474_1475delGT; p.V492Tfs*136), a calcium channel, in the patients. Functional characterization of this mutant TRPV6 following heterologous expression revealed that it was defective in calcium uptake. Induction of pancreatitis in mice induced Trpv6 expression, indicating that higher expression levels of the mutant protein and consequent dysregulation of calcium levels in patients with chronic pancreatitis could aggravate the disease.
Discussion: We report a novel frameshift mutation in TRPV6 in an Indian family with HP that renders the mutant protein inactive. Our results emphasize the need to expand the list of genes used currently for evaluating patients with hereditary pancreatitis.
Introduction
Hereditary pancreatitis (HP) is a rare familial genetic disorder that is characterized by recurrent episodes of pancreatitis commonly presented in childhood or early adulthood (Shelton and Whitcomb, 2018). These repeated episodes lead to irreversible morpho-physiological changes, including fibrosis, pancreatic exocrine insufficiency, and diabetes mellitus (Ramsey et al., 2017). Moreover, patients with HP have a ∼50 fold risk of developing pancreatic ductal adenocarcinoma (PDAC) (Rebours et al., 2008). This disease is more common in Caucasians and rare among Asians (Shelton and Whitcomb, 2016).
The involvement of a fine protease–antiprotease balance in the pathogenesis of pancreatitis has been a predominant school of thought so far (Hegyi and Sahin-Tóth, 2017). Inactive precursor forms of digestive proteases secreted by pancreatic acinar cells are flushed from the ductal system in a sodium bicarbonate–rich fluid into the duodenum. However, gain-of-function or mutations that cause misfolding of cationic trypsinogen (PRSS1) lead to its premature intrapancreatic activation, causing autodigestion of the pancreas and pancreatitis (Mayerle et al., 2019). Moreover, an increase in calcium concentrations and compartmental distribution in the acinar cells is a known risk factor for acute pancreatitis (Lee and Papachristou, 2019). Dysregulated Ca2+ levels can cause premature activation of PRSS1 (Krüger et al., 2000), endoplasmic reticulum stress (Sah et al., 2014), or hampered zymogen granule trafficking in the acinar cells (Messenger et al., 2014) resulting in pancreatitis.
Since the pioneering discovery in 1996 of PRSS1 mutation in patients with HP (Whitcomb et al., 1996), other mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), Serine Protease Inhibitor Kazal type 1 (SPINK1), Chymotrypsin C (CTRC), and Carboxypeptidase A1 (CPA1) have been associated with the development of HP (Raphael and Willingham, 2016; Mayerle et al., 2019). Copy number variations (CNV) in PRSS1 have also been reported in hereditary (LaRusch et al., 2012) and chronic pancreatitis (Chen et al., 2008). However, the reduced penetrance of the mutations in PRSS1 and the absence of other known pathogenic mutations in pancreatitis susceptibility genes in some affected individuals (Masamune, 2014) suggest the role of additional yet-undiscovered inherited factors. Moreover, the marked variation in the age of disease onset, associated complications, and known genetic predisposition, suggests the complex nature of the disease as an orchestration of gene-gene and gene-environment interactions (Hasan et al., 2018).
In the current study, we studied an Indian family with autosomal dominant, hereditary pancreatitis in two generations, the Father (F) and his two children Son (S) and Daughter (D) (Figure 1A). Whole-exome sequencing and CNV analysis in the affected trio revealed a novel, functionally relevant mutation in TRPV6 (Transient Receptor Potential Vanilloid subfamily member 6).
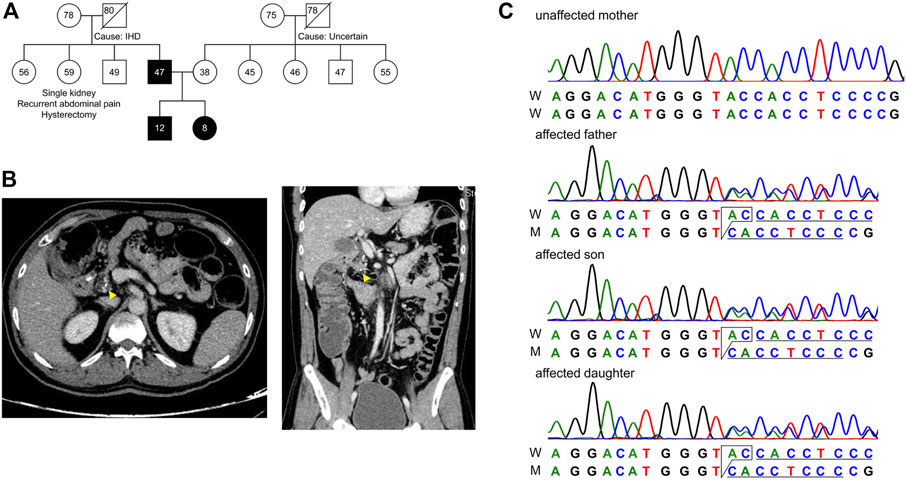
FIGURE 1. Pedigree of the subjects in this study. (A) Extended family pedigree of the subjects. Circle represents females, square represents males and numbers represent age of the individual. Affected trio Father (F), Son (S) and Daughter (D) are marked black, and Mother (M) is unaffected. Except for the Father, none of the first-degree relatives had developed pancreatitis from both paternal and maternal sides of patient S and D. (B) CT scan of the abdomen showing transverse (left) and longitudinal (right) sections, with focal calcium deposition (yellow arrow) in the tail of the pancreas of patient F. (C) Chromatogram representing c.1474_1475delGT in the affected trio as a heterozygous state.
Results
Clinical characterization of the patients
A family with a history of chronic calcific pancreatitis and recurrent pancreatitis in two generations was investigated for an underlying genetic cause. The family pedigree and clinical features of the affected trio are presented in Figure 1A and Table 1. Patient F (Father) had a history of recurrent episodes of abdominal pain since the age of 12. During the episodes the pain was located in the upper abdomen with radiation to the back, lasting for 2–3 days, and occurred at 6–12 months intervals. At the age of 27 he was diagnosed with chronic calcific pancreatitis and underwent a lateral pancreaticojejunostomy. The recent computed tomography (CT) scan of Patient F, now a diabetic, shows evidence of chronic calcific pancreatitis in the head of the pancreas (Figure 1B).
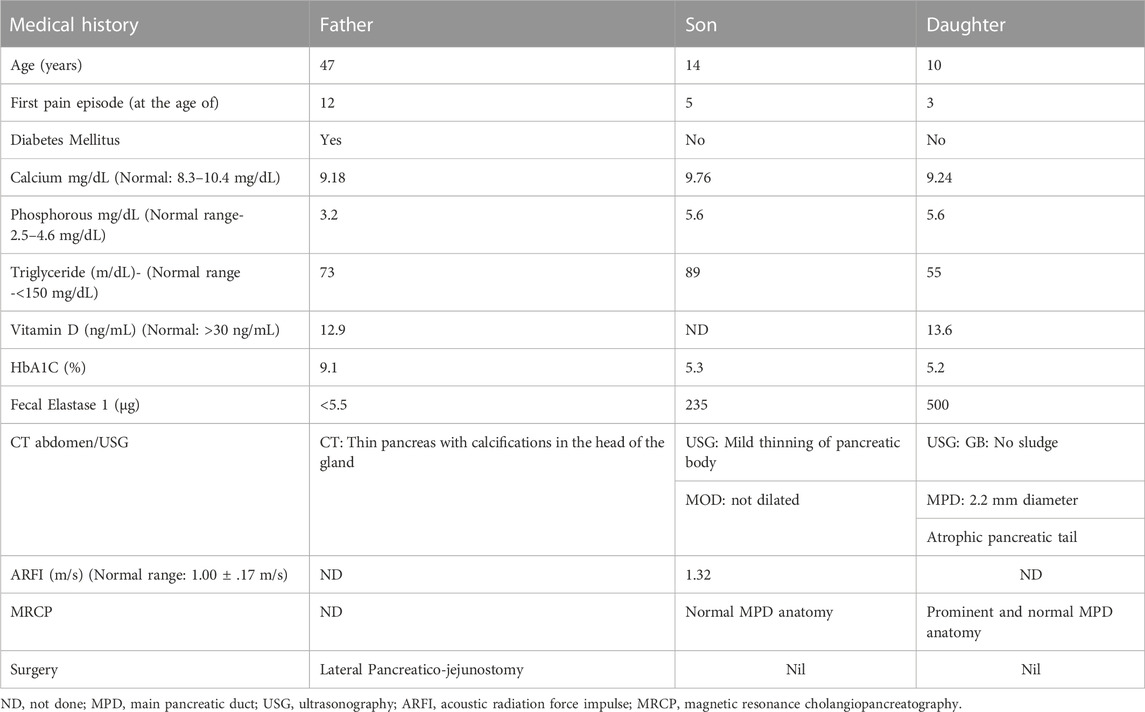
TABLE 1. Clinical history of the affected patients (Father (F), Son (S), and Daughter (D).
The other two affected members of the family, Son (S) and Daughter (D) reported the first episode of pancreatitis at the age of 3 and 5, respectively. Patient S has suffered eight episodes of acute pancreatitis each more than 6 months apart while patient D suffered multiple episodes of acute pancreatitis. CT of abdomen and endoscopic ultrasonography (EUS) of the patient S did not reveal any features of chronic pancreatitis, however, Acoustic Radiation Force Impulse Imaging (ARFI) of the pancreas revealed an increased shear wave velocity in the pancreas, suggesting pancreatic fibrosis (Sanjeevi et al., 2020). An ultrasound abdomen of patient D showed an atrophic tail of the pancreas and a prominent main pancreatic duct (2.2 mm), suggestive of chronic pancreatitis. While patients F had a history of occasional alcohol use from the age of 21, an evaluation for an etiology for recurrent acute pancreatitis in the affected trio did not reveal any specific cause.
The Mother (M) of the patient S and D does not have any history of pancreatic disorder or diabetes mellitus. No other first degree relative of the primary patient F had developed pancreatitis (Figure 1A). Patients had low serum Vitamin D levels and children presented moderately elevated serum phosphorous levels (Table 1).
Whole exome sequencing identifies novel mutation in TRPV6
To identify genes predisposing the subjects to hereditary pancreatitis, we performed WES on the affected trio–father, son, and daughter, and the unaffected mother. Out of the 44,322 variants called by WES, 710 novel and/or rare missense variants with gAD MAF score of ≤.01 (Karczewski et al., 2020), present in the affected trio and not in the mother were processed further for bioinformatic analysis. Interestingly, we found no mutations that passed quality control (see Materials and Methods) or the expected segregation pattern, in genes known to be associated with pancreatitis such as PRSS1, CFTR, SPINK, CPA1, CTRC, CLDN2, and PLINP.
Copy number variant analysis identified a copy gain of the initial segment of the CBS gene (58kb, POS: 44475162–44533170, GRCh37). No CNVs in the trio were found in genes hitherto known to be associated with pancreatitis.
We found a novel mutation (2 bp deletion) in the transient receptor potential cation channel subfamily V member 6, TRPV6 (c.1474_1475delGT; p.V492Tfs*136), in the affected trio (Table 2). The deletion was confirmed by Sanger sequencing (Figure 1C), and found in a heterozygous state in the affected individuals. The frameshift that resulted from the 2 bp deletion that lies in the ion transport domain (Figure 2A) would generate an altered amino acid sequence from p.V492 such that the mutant protein extends to p.626 position before reaching a stop codon. The sequence beyond V492 bears no resemblance to the wild type TRPV6 (Figure 2B).
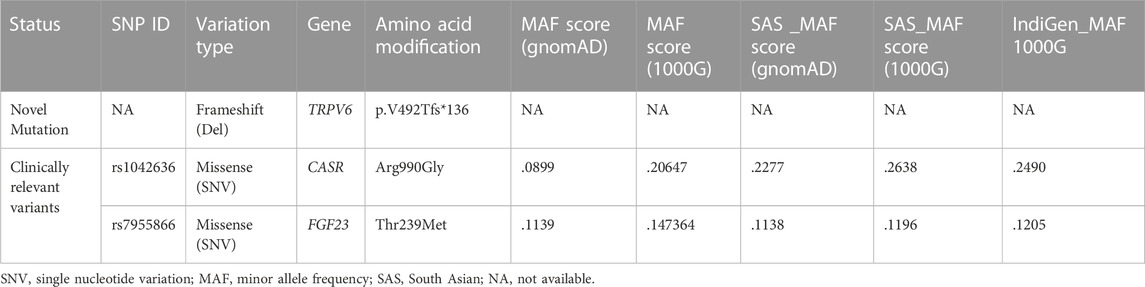
TABLE 2. List of mutations present in the affected patients (FSD), their allele frequencies in the different populations, including data from recent 1000 Indian genomes (Indigenome).
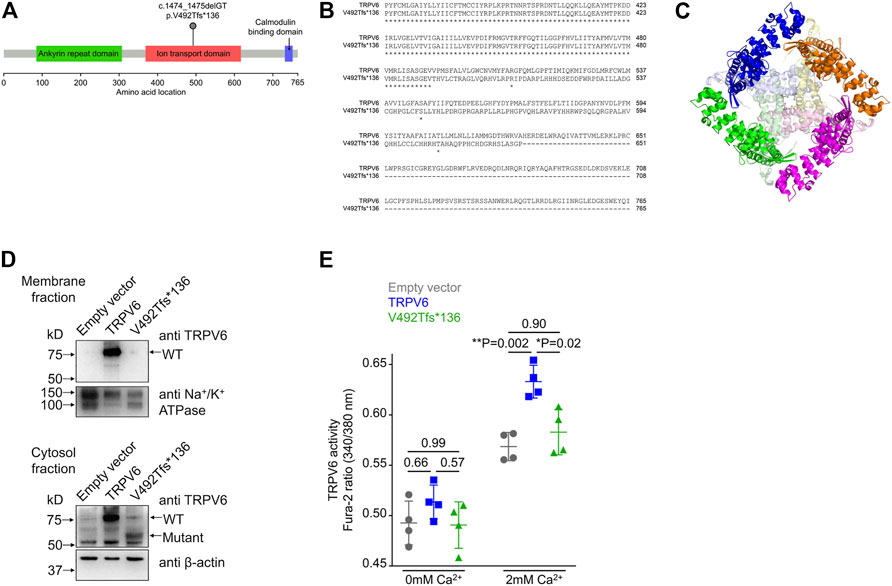
FIGURE 2. Frameshift mutation in TRPV6. (A) Diagrammatic representation of the location of the p.V492Tfs*136 mutation in the ion transport domain of TRPV6. (B) Sequence alignment of wild type and mutant TRPV6 from amino acid 367. Protein sequences of TRPV6 were obtained by translating in silico the mRNA sequences using expasy.org and alignment performed at embnet.vital-it.ch. The alignment shows that the mutant protein is predicted to be 626 amino acids long and there is no sequence similarity between the wild type and the mutant protein beyond amino acid 492. (C) The structure of TRPV6 (PDB ID: 6BO8). Individual chains comprising the native tetramer are shown in magenta, green, orange and blue. The regions beyond amino acid 492 in each chain are shown in lighter shades of pink, light green, pale yellow and light blue, respectively. The mutant protein loses the central part of the oligomer which constructs the pore. (D) Western blot of membrane (top) and cytosolic (bottom) fractions prepared from HEK 293T cells transfected with either the empty vector, or plasmid harboring either the wild type TRPV6 or mutant TRPV6 cDNA. An antibody recognizing the N-terminus of the TRPV6 protein was used. Na+/K+ ATPase and β-actin were used for normalizing the amount of protein taken for western blot analysis. (E) Cytosolic Ca2+ as assessed by a ratiometric Fura-2 assay in cells transiently transfected with the empty vector, or plasmids harboring either the wild type or mutant TRPV6 cDNA. No significant differences in the basal or resting Ca2+ levels were observed in cells expressing either the wild type or p.V492Tfs*136 TRPV6 when compared to the empty vector. Expression of wild type TRPV6 in cells significantly increased Ca2+ uptake on exposure to 2 mM Ca2+, whereas p.V492Tfs*136 failed to increase Ca2+ uptake. Each data point represents the average of 16 measurements taken from different regions of a microplate well. The data shown is the mean ± standard deviation of data from a single experiment, which is representative of experiments performed twice. p values shown were calculated using one-way ANOVA with Welch’s correction.
Functionally defective variant in transient receptor potential vanilloid subfamily member 6 (TRPV6) is associated with hereditary pancreatitis
TRPV6 is a constitutively active Ca2+ channel (Venkatachalam and Montell, 2007) and belongs to the vanilloid subfamily of transient receptor potential channels. It is the most selective TRP channel for Ca2+ (Nilius and Flockerzi, 2014), and is expressed in certain epithelial cells, where it has been proposed to play a role in Ca2+reabsorption. The precise physiological function of TRPV6 in most tissues remains largely unknown [reviewed in Fecher-Trost et al. (2017)]. In humans, the TRPV6 gene is expressed in pancreatic ductal cells (Segerstolpe et al., 2016), where it is thought to be involved in Ca2+ uptake from the duct lumen (Masamune et al., 2020). TRPV6 mutations have very recently been identified in pancreatitis patients of Japanese, Chinese, and European ethnicity, but there have been no reports from the South Asian population (Masamune et al., 2020; Zou et al., 2020; Oracz et al., 2021; Hamada et al., 2022). A preliminary characterization of the few variants so far reported to be associated with pancreatitis demonstrated deficient calcium transport or decreased levels of protein expression (Masamune et al., 2020; Zou et al., 2020; Oracz et al., 2021; Hamada et al., 2022). Examination of the structure of human TRPV6 predicted that as a consequence of the two base pair deletion seen in our patients, the truncation would delete the central Ca2+ channel forming domain (Figure 2C).
We examined the expression and localization of wild type (WT) or p.V492Tfs*136 TRPV6 in HEK293T cells following transient transfection of plasmids containing the corresponding cDNAs. Wild type TRPV6 (Mr ∼80 kDa) was localized to both the membrane and cytosolic fractions (Figure 2D). However, the mutant protein, which migrated at a predicted molecular weight of ∼60 kDa, was not detected in the membrane fraction, but localized to the cytosol (Figure 2D).
Following transfection of wild type or p.V492Tfs*136 TRPV6 cDNAs in HEK293T cells, cytosolic Ca2+ levels were measured by a ratiometric Fura-2 assay. While no differences in the resting Ca2+ levels were evident in cells expressing wild type or p.V492Tfs*136 TRPV6 as compared to the empty vector control, significantly higher Ca2+ uptake was observed in cells expressing wild type TRPV6 in response to 2 mM Ca2+ than that seen in cells transfected with an empty vector. Cells transfected with the p.V492Tfs*136 variant, however, failed to display increased Ca2+ uptake in response to 2 mM Ca2+ (Figure 2E). Together, these studies suggest that the p.V492Tfs*136 TRPV6 variant is a loss-of- function protein which is not localized to the cell surface and defective in Ca2+ transport.
Variants in additional genes in the affected trio that could contribute to clinical presentation
Genetic mutations associated with pancreatitis could be influenced by variants in additional disease-modifying genes. The calcium-sensing receptor (CASR) is a plasma membrane protein, expressed to moderately high levels in the endocrine cells in the pancreas (Baron et al., 2016). CASR, encoded by CASR, regulates intracellular calcium levels based on extracellular Ca2+ concentrations. We noted a gain-of-function variant in CASR (p.R990G) in patients that was absent in the mother (Supplementary Figure S1 and Table 2) and present in a heterozygous state. The CASR R990G variant has been reported to be significantly associated with chronic pancreatitis and in subjects who reported moderate alcohol consumption (Muddana et al., 2008). This variant is found to be common in different populations (Table 2), and increased Ca2+ levels in cells in response to lower concentrations of extracellular calcium in comparison with wild type CASR (Vezzoli et al., 2007). However, both loss and gain-of-function mutations of the CASR gene have been associated with pancreatitis (Hasan et al., 2018), and recent studies even suggest that variants do not modify the risk for chronic pancreatitis (Takats et al., 2021).
Of clinical relevance in our study is a variant we identified in fibroblast growth factor 23 (FGF23; Supplementary Figure S1). FGF23 is a bone-derived endocrine hormone that orchestrates phosphate and calcium homeostasis, along with parathyroid hormone (PTH) and 1,25-dihydroxy vitamin D. FGF23 binds to its receptor complex FGFR/Klotho in the kidney (Rodríguez, 2020). FGF23 regulates Vitamin D biosynthesis in two ways. First, it inhibits 25-hydroxyvitamin D-1α-hydroxylase (CYP27B1), which converts 25-hydroxyvitamin D into biologically active 1,25-dihydroxyvitamin D and second, stimulates 1,25-dihydroxyvitamin D3 24-hydroxylase (CYP24A1), which accelerates 1,25-dihydroxyvitamin D inactivation (and degradation of 25-hydroxyvitamin D). Together, these FGF23-mediated effects decrease the concentrations of active 1,25-dihydroxyvitamin D, leading to a reduced intestinal absorption of calcium and, to a lesser extent, intestinal phosphate absorption [reviewed in Edmonston and Wolf (2020)]. The mutation in FGF23 (rs7955866; p.T239M) present in our patients has been proposed to stabilize the FGF23 protein and cause enhanced downstream signaling (Rendina et al., 2012). Although FGF23 is not expressed in the human pancreas (Segerstolpe et al., 2016), elevated serum FGF23 levels could lead to low serum Vitamin D levels seen in the affected patients in the family. Indeed, deficiency in Vitamin D levels has been shown to predict the severity of pancreatitis (Huh et al., 2019).
Trpv6 is upregulated in caerulein-induced acute pancreatitis in mice
Expression of a mutant protein may have more deleterious effects if its levels are modulated during pancreatic inflammation. We therefore monitored transcript levels of TRPV6 in the mouse pancreas following onset of acute pancreatitis in a well-established model of cerulein-induced pancreatitis (Figure 3A) (Zhao et al., 2018). Instead of the six to seven hourly injections usually administered to these mice, we sacrificed mice after three injections, to monitor changes that occur during the onset of acute pancreatitis. Caerulein administration significantly increased serum amylase and lipase activities compared with control mice that received saline (Figure 3B). Histological analysis of the sections from control and cerulein-treated mice showed increased pancreatic edema, intracellular vacuoles in acinar cells and increased infiltration of inflammatory cells in caerulein-treated mice (Figure 3C).
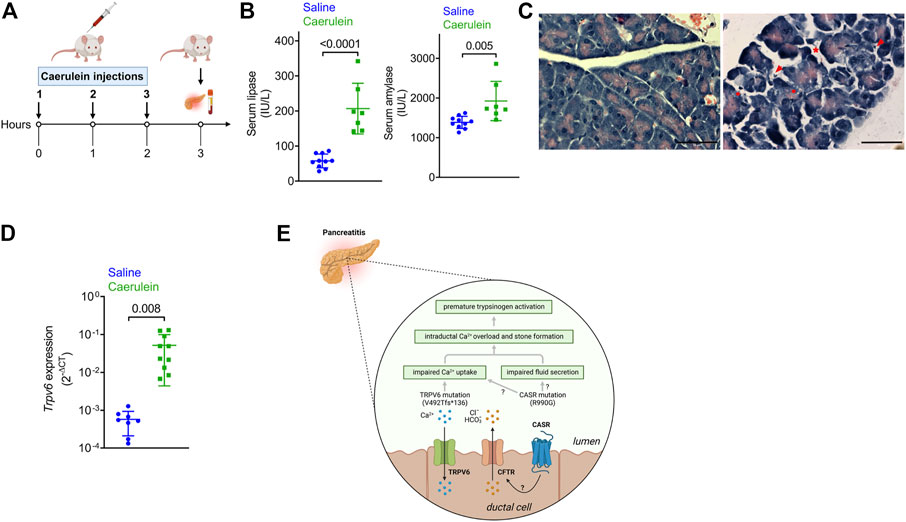
FIGURE 3. Expression of the genes in the pancreas following caerulein-induced early onset acute pancreatitis. (A) Schematic depicting the experimental protocol for caerulein-induced acute pancreatitis. The Figure was created with Biorender. (B) Serum lipase (left) and amylase (right) levels in mice injected with saline or caerulin. Data shown are mean ± standard deviation from experiments performed at least thrice and data points represent individual mice. (C) Representative images of hematoxylin and eosin-stained pancreatic tissue sections from mice injected with saline or caerulein. Arrows indicated infiltrated immune cells. * denotes edema and circles represent vacuolized acinar cells. Scale bar = 50 μm. (D). Real time quantitative PCR (RTqPCR) analysis of Trpv6 expression following caerulean-induced pancreatitis. Each point on the scatter plots represents data from a single mouse. The data are presented as the mean SD, and p values were calculated using the Student’s t-test. (E). Proposed mechanism underlying the role of defective TRPV6 in pancreatitis. Loss of function of TRPV6 in patients harboring the frameshift mutation can lead to impaired Ca2+ uptake from the ductal lumen. This can cause increased ductal Ca2+ concentrations, premature activation of trypsinogen (the product of PRSS1) and subsequent cellular damage and autodigestion. CASR is proposed to respond to high Ca2+ concentrations in the ductal lumen by triggering fluid and ion secretion, possibly through regulation of CFTR activity. The activating CASR p.R990G mutation sensitizes the receptor to Ca2+ thereby increasing fluid and ion secretion into the ducts leading to edema. The CASR p.R990G mutation may also cause impaired Ca2+ uptake in the pancreatic duct, as has been reported in the renal tubules. Based on this model, the cumulative effects of both TRPV6 and CASR mutations may cause Ca2+ overload in the ductal lumen and formation of stones, both of which are known risk factors for pancreatitis. The Figure was created with Biorender.
Trpv6 was significantly upregulated in early stages of caerulein-induced acute pancreatitis (Figure 3D). Notably, while CASR is reported to be expressed in the human pancreas (Baron et al., 2016), Casr and Fgf23, were not detected in RTqPCR (data not shown), potentially indicating species differences. Translating the observation that induction of pancreatitis in mice robustly induced TRPV6 expression, we propose that the aberrant activity of mutant TRPV6 may initiate pancreatitis and its severity due to increased expression during active disease.
Discussion
PRSS1-linked HP is more common in North American, North-East Asian, and North European ethnicities, but is rare or absent in African or Indian populations (Chandak et al., 2004). We did not observe mutations or CNVs in PRSS1, or mutations in other genes that are associated with pancreatitis, in this study. Instead, we report for the first time, a novel heterozygous frameshift mutation caused by a 2bp deletion in TRPV6 (p.V492Tfs*136) in the affected trio, along with a heterozygous CASR variation (p.R990G).
Calcium signaling plays a pivotal role in the regulated secretion of the digestive enzymes, and fluids by the exocrine pancreas (Gerasimenko et al., 2018). Dysregulated Ca2+ levels in the pancreatic acinar cells are fundamental to the pathogenesis of pancreatitis by causing premature activation of PRSS1 (Krüger et al., 2000), ER stress (Sah et al., 2014) or hampered zymogen granule trafficking in the acinar cells (Messenger et al., 2014). TRPV6, a Ca2+ channel is thought to be involved in the reuptake of calcium from the duct lumen in the pancreas (Masamune et al., 2020). Defective TRPV6 that is seen in our family can result in elevated levels of Ca2+ in the duct lumen and consequent Ca2+ dysregulation in the cytoplasm. Elevated Ca2+ levels in the duct may cause premature activation of PRSS1 in the duct leading to ductal stress and cell damage (Figure 3E). Recent studies that have reported an association of TRPV6 mutations with chronic pancreatitis in multinational, multiethnic cohorts strengthen a causative link between mutant TRPV6 and pancreatitis (Masamune et al., 2020; Zou et al., 2020; Oracz et al., 2021; Hamada et al., 2022).
The gain of function CASR mutation p.R990G (Vezzoli et al., 2007), present in our patients makes the receptor more sensitive to extracellular Ca2+ and could lead to high intracellular Ca2+ levels (Vezzoli et al., 2007). CASR is expressed in the endocrine pancreas and to lower levels in pancreatic ductal cells in humans (Baron et al., 2016), suggesting that mutations in both TRPV6 and CASR may have additive or synergistic effects in the ducts. Although the precise role of CASR in pancreatic ductal cells is unknown, it has been proposed that it responds to high calcium concentrations in the juice by triggering fluid and ion secretion and ductal flushing, possibly through regulation of CFTR activity, thereby preventing stone formation and pancreatitis (Mayerle et al., 2019) (Figure 3E). Furthermore, the p.R990G variant has been shown to inhibit Ca2+ uptake from the lumen in the renal tubule leading to hypercalciuria and formation of kidney stones (Vezzoli et al., 2007). A similar pathomechanism may exist in pancreatic ductal cells, such that individuals harboring the 990G variant allele would show decreased Ca2+ uptake (Figure 3E). This combined effect of mutations in TRPV6 and CASR would lead to intra-ductal Ca2+ overload, an established causal factor for premature activation of pancreatic enzymes and pancreatitis (Figure 3E). Furthermore, there is a strong relationship between increased intraductal Ca2+ concentration, precipitation of calcium salts in the duct lumen, and stone formation, which could independently predispose to pancreatitis (Figure 3E) (Mayerle et al., 2019).
Our data also shows that TRPV6 mRNA is upregulated in mice during caerulein-induced, early-onset acute pancreatitis. Masamune et al. (2020) has reported that caerulein-induced pancreatitis was exacerbated in TRPV6mut/mut mice compared to wild type mice suggesting that functional TRPV6 is important for protection against pancreatitis.
Low Vitamin D levels in the serum have been shown to predict the severity of chronic pancreatitis (Huh et al., 2019). Our patients also show low levels of serum Vitamin D, perhaps contributed by the activating and stabilizing mutation in FGF23 (p.T239M) as discussed earlier. An increase in serum FGF23 level is also associated with elevated blood phosphate in vivo [reviewed in Noonan and White (2019)]. FGF23 knock out mice experiments revealed that the paramount physiological function of FGF23 is its role in the regulation of Vitamin D levels via CYP27B1, and CYP24A1 transcription, and not its phosphaturic function (Erben, 2018). Therefore, the FGF23 p.T239M mutation present in our patients might explain low vitamin D levels and marginally higher levels of phosphate in the serum of patient S and D.
In conclusion, we report a novel frameshift mutation in TRPV6 in an Indian family with hereditary pancreatitis that renders the mutant protein inactive. We also find a relevant mutation in CASR in this family. Unlike earlier reports on HP where mutations in PRSS1 and SPINK1 have been the focus so far, our results suggest that the disease can be caused by mutations, which may act in synergy. Upregulation of the TRPV6 wild-type allele in heterozygous carriers by vitamin D administration has been reported earlier (Nilius and Flockerzi, 2014) and could provide a promising therapeutic approach for pancreatitis research in the future (Sahin-Tóth, 2020).
Materials and methods
Subjects
Three members of a family, Father (46 years), Son (11 years), and Daughter (08 years) presenting episodes of recurrent acute pancreatitis were recruited at Christian Medical College (CMC), Vellore, Tamil Nadu, India. Unlike the father, who had the first episode at 12 years of age and has progressed to the chronic stage of disease, children had an early onset (less than 5 years). The unaffected mother of the children aged 38 years was taken as a control for the study.
Sample collection
The study was reviewed and approved by the Institutional Ethics Committees of both Christian Medical College (CMC) Vellore (IRB No. 11254), and the Indian Institute of Science (IISc), Bangalore (IHEC No: 2-23012019). 2–3 mL of whole blood was collected from all four subjects by trained nurses at CMC Vellore in EDTA-coated vials and stored in −80°C before it was shipped in dry ice to the IISc, Bangalore for further analysis. The medical and family history of all the subjects was also recorded. Written informed consent was taken from all the subjects.
Whole exome analysis
DNA was isolated from the whole blood using QIAamp® DNA Blood Mini Kit (Qiagen). Genomic DNA from each subject was subjected to whole-exome analysis (Clevergene, Bangalore). One microgram of genomic DNA was taken for library preparation using Next DNA II Library preparation Kit for Illumina® (New England Biolabs). The genomic library was enriched by using xGEN Research panel V1.0 (Integrated DNA Technologies) as per the manufacturer’s protocol before sequencing on the Illumina HiSeq platform to generate 2 × 150 bp reads. The quality check of sequence data was done using FastQC and MultiQC (Babraham-Bioinformatics-FastQC; Ewels et al., 2016) software. The adapter sequences, low-quality reads were removed using fast p (Chen et al., 2018). To call variants, Genome Analysis Tool Kit (GTAK) best practices were followed. In brief, the quality trimmed reads were mapped to the human reference genome build hg19 with the use of BWA and MEM algorithm (Li and Durbin, 2009). PCR duplicates were removed from the mapped reads using Picard tools (Broad Institute). The base qualities were recalibrated using GATK v4.0.12.0 (Van der Auwera et al., 2013) and Genome Variant Call Formats (GVCFs) were generated for each sample. The GVCFs were combined before variants were called. The variant quality scores were recalibrated to obtain the probability that an SNP is a true genetic variant and not a sequencing or data processing artifact.
All annotations for the pass filtered variants were extracted from Variant Effect Predictor (VEP) (McLaren et al., 2016) (ensemble.org). All the missense variants present in the affected trio (FSD), not present in the mother, with gAD allele frequency of ≤.01 were considered for further analysis. The variants that had a Combined Annotation Dependent Depletion (CADD) score of ≥20 were retained (Rentzsch et al., 2019). In the genetic analysis, only the variants that presented high genotype confidence and were predicted to be deleterious by five algorithms, namely SIFT (Kumar et al., 2009), PolyPhen-2 (Adzhubei et al., 2010), MetaSVM (Kim et al., 2017), MCAP (Jagadeesh et al., 2016), and MutationTaster (Schwarz et al., 2010) were considered for further analysis. Novel mutations in the affected trio, where allele frequency and prediction of being deleterious were not available on VEP, were retained during the analysis. The allele frequencies of the variants that came from the final analysis were also checked in the Indian 1000 genome (Indigenome project) (Jain et al., 2021). All genetic variants of interest were further validated by Sanger sequencing.
Copy number variant analysis
One microgram of gDNA was subjected to an array-based CNV analysis (InfiniumOmni5Exome-4v1-3, Illumina) performed by Macrogen Inc., South Korea.
Bioinformatics
Wild type and mutant protein sequence of TRPV6 was obtained in silico by translating the wild type and mutant (having desired 2 bp deletion) mRNA (NM_018646.6) sequence using the translation tool of expasy.org. Sequence alignment of both the protein sequences was done using embnet.vital-it.ch. The published cryo-EM structure of human TRPV6 (McGoldrick et al., 2018) (PDB ID: 6BO8) was used to identify regions deleted in the mutant TRPV6. RNA sequencing data sets for the study included (a) E-MTAB-5061 with single cell RNA-seq analysis of human pancreas from healthy individuals and (b) GSE84133 with single cell RNA-seq analysis of mouse and human pancreas.
Cell culture, plasmids and antibodies
HEK293T cells were obtained from ATCC and grown in DMEM/high glucose/sodium pyruvate containing 10% fetal bovine serum in a 5% CO2 incubator at 37°C. The TRPV6 cDNA in pLenti6.3/V5-Dest backbone was obtained from the DNASU plasmid repository (HsCD00860923). TRPV6 cDNA was then subcloned into pBKS vector using SpeI and XhoI sites for mutagenesis. Mutant TRPV6 was generated by site-directed mutagenesis using two primers 5′ CAGCGGGGAGGTGACCCATGTCCTTTGC 3′ and 5 GCAAAGGACATGGGTCACCTCCCCGCTG 3′. The TRPV6 cDNA with 2bp desired deletion was cloned back into the pLenti6.3/V5-Dest backbone. The loss of the KpnI restriction site resulting from the mutation was used for screening, and the plasmid was confirmed by sequencing. Membrane and cytosolic proteins from HEK293T cells transiently transfected with empty vector, WT and mutant TRPV6 cDNA constructs were extracted and Western blotting was performed as previously described (Mishra et al., 2021). Antibodies used were anti-TRPV6 from Abclonal (A16128) that detects amino acids 1–100 of human TRPV6, anti-Na+/K+ ATPase from Abcam (ab76020), anti-β-actin from Cell Signalling Technology (4970S), and HRP conjugated anti-rabbit IgG from Sigma-Aldrich (A0545).
Measurement of cytosolic Ca2+ changes
Cytosolic Ca2+ was assessed as previously described (Prasad et al., 2019), with modifications. Briefly, cells were washed with PBS and loaded with Fura-2 AM (Invitrogen F1221) at 1 μg/mL in calcium-containing buffer (2 mM CaCl2, 126 mM NaCl, 4.5 mM KCl, 2 mM MgCl2, 10 mM glucose, 20 mM HEPES, pH 7.4). After 30 min at room temperature, cells were washed three times for 5 minutes in calcium-containing buffer without Fura-2 AM and incubated for another 30 min to allow for Fura-2 AM de-esterification. Cells were replaced with calcium-free buffer (126 mM NaCl, 4.5 mM KCl, 2 mM MgCl2, 10 mM glucose, 20 mM HEPES, pH 7.4), and resting calcium levels and calcium uptake after the addition of 2 mM Ca2+ were measured by exciting cells at 340 nm and 380 nm and emission captured at 510 nm using a microplate reader (Tecan Infinite M200 PRO). Ratios of 340/380 nm emissions were was calculated and plotted.
Caerulein induced early-onset, acute pancreatitis
All procedures were carried out in agreement with the Control and Supervision Rules, 1998 of Ministry of Environment and Forest Act (Government of India), and the Institutional Animal Ethics Committee of the Indian Institute of Science (Approval CAF/Ethics/547/2017). All animals were bred and housed in the same vivarium. Mice were housed in a clean air facility in multiple cages and separated on the basis of sex and genotype. The temperature was maintained at 22°C ± 2°C, humidity was maintained at 55% ± 10%, and the mice were maintained on a 12-h light/dark cycle. Mice had access to laboratory chow and water ad libitum. Chow was procured from Altromin International (Germany) and contained ∼24% protein, 6% oil, and 3% dietary fibers.
Pancreatitis was induced in overnight fasted 8–10 weeks old female C57BL6/N mice by the administration of three repeated intraperitoneal hourly injections of caerulein (Merck; 50 µg/kg body weight) (Reed and Gorelick, 2014). Control mice were administered phosphate buffered saline. Mice were sacrificed 1 hour after the last injection. The pancreas was quickly isolated and stored in pre-cooled RNA-later (Merck) for 6–7 h at 4°C before storing at −80°C. RNA was prepared from ∼20 mg of the tissue using the RNAeasy RNA extraction Kit (Qiagen) following manufacturer’s instructions. The expression profile of mutant genes of the affected trio was assessed by quantitative real-time PCR using SYBR Premix Ex Taq (Tli RNase H Plus; Takara Bio) on a CFX96 Touch real-time PCR detection system (Bio-Rad). The housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (Gapdh) was used for normalization. Primers used for real-time PCR are Trpv6 forward primer: 5′- TCTGCAGATGGTTCCACAGAC-3′; Trpv6 reverse primer: 5′-AGGGCTGCTATGTGAAGTGC-3′, Gapdh forward primer: 5′-CAACTCCCTCAAGATTGTCAGCAA-3′; and Gapdh reverse primer: 5′-GGCATGGACTGTGGTCATGA-3′).
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by the Institutional Ethics Committees of both Christian Medical College (CMC) Vellore (IRB No. 11254), and the Indian Institute of Science (IISc), Bangalore (IHEC No: 2-23012019). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. The animal study was reviewed and approved by Institutional Animal Ethics Committee of the Indian Institute of Science (Approval CAF/Ethics/547/2017).
Author contributions
SD, RT, and SV conceived of the study; IS, HP, and SB collected samples, performed experiments and analyzed data; IS, HP, SD, and SV wrote the manuscript and all authors approved of the final version of the manuscript.
Funding
This work has been supported by a Margdarshi Fellowship awarded by the DBT-Wellcome Trust India Alliance to SV (IA/M/16/1/502). SV is a JC Bose National Fellow (SB/S2/JCB-18/2013) and is a recipient of a Collaborative Grant for Research Professors from the Royal Society, United Kingdom. IS was a recipient of the DS Kothari, Postdoctoral Fellowship by UGC, New Delhi. HP is an Early Career Fellow supported by the Wellcome Trust DBT India Alliance (IA/E/17/1/503665).
Acknowledgments
The authors acknowledge the support of the paramedical staff at CMC Vellore, during sample collection.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1058057/full#supplementary-material
References
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7 (4), 248–249. doi:10.1038/nmeth0410-248
Babraham-Bioinformatics-FastQC (2019). A quality control tool for high throughput sequence data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (Accessed June, 2019).
Baron, M., Veres, A., Wolock, S. L., Faust, A. L., Gaujoux, R., Vetere, A., et al. (2016). A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst. 3 (4), 346–360. doi:10.1016/j.cels.2016.08.011
Broad Institute (XXXX). Picard-tools. Broad Institute. Available at: https://broadinstitute.github.io/picard/.
Chandak, G., Idris, M., Reddy, D., Mani, K., Bhaskar, S., Rao, G., et al. (2004). Absence of PRSS1 mutations and association of SPINK1 trypsin inhibitor mutations in hereditary and non-hereditary chronic pancreatitis. Gut 53 (5), 723–728. doi:10.1136/gut.2003.026526
Chen, J., Masson, E., Le Maréchal, C., and Férec, C. (2008). Copy number variations in chronic pancreatitis. Cytogenet Genome Res. 123 (1-4), 102–107. doi:10.1159/000184697
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34 (17), i884–i890. doi:10.1093/bioinformatics/bty560
Edmonston, D., and Wolf, M. (2020). FGF23 at the crossroads of phosphate, iron economy and erythropoiesis. Nat. Rev. Nephrol. 16 (1), 7–19. doi:10.1038/s41581-019-0189-5
Erben, R. G. (2018). Physiological actions of fibroblast growth factor-23. Front. Endocrinol. 9, 267. doi:10.3389/fendo.2018.00267
Ewels, P., Magnusson, M., Lundin, S., and Käller, M. (2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32 (19), 3047–3048. doi:10.1093/bioinformatics/btw354
Fecher-Trost, C., Wissenbach, U., and Weissgerber, P. (2017). TRPV6: From identification to function. Cell Calcium 67, 116–122. doi:10.1016/j.ceca.2017.04.006
Gerasimenko, J. V., Peng, S., Tsugorka, T., and Gerasimenko, O. V. (2018). Ca2+ signalling underlying pancreatitis. Cell Calcium 70, 95–101. doi:10.1016/j.ceca.2017.05.010
Hamada, S., Masson, E., Chen, J. M., Sakaguchi, R., Rebours, V., Buscail, L., et al. (2022). Functionally deficient TRPV6 variants contribute to hereditary and familial chronic pancreatitis. Hum. Mutat. 43 (2), 228–239. doi:10.1002/humu.24315
Hasan, A., Moscoso, D. I., and Kastrinos, F. (2018). The role of genetics in pancreatitis. Gastrointest. Endosc. Clin. N. Am. 28 (4), 587–603. doi:10.1016/j.giec.2018.06.001
Hegyi, E., and Sahin-Tóth, M. (2017). Genetic risk in chronic pancreatitis: the trypsin-dependent pathway. Dig. Dis. Sci. 62 (7), 1692–1701. doi:10.1007/s10620-017-4601-3
Huh, J. H., Kim, J. W., and Lee, K. J. (2019). Vitamin D deficiency predicts severe acute pancreatitis. United Eur. Gastroenterol. J. 7 (1), 90–95. doi:10.1177/2050640618811489
Jagadeesh, K. A., Wenger, A. M., Berger, M. J., Guturu, H., Stenson, P. D., Cooper, D. N., et al. (2016). M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 48 (12), 1581–1586. doi:10.1038/ng.3703
Jain, A., Bhoyar, R. C., Pandhare, K., Mishra, A., Sharma, D., Imran, M., et al. (2021). IndiGenomes: a comprehensive resource of genetic variants from over 1000 Indian genomes. Nucleic Acids Res. 49 (D1), D1225–D1232. doi:10.1093/nar/gkaa923
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141, 456 humans. Nature 581 (7809), 434–443. doi:10.1038/s41586-020-2308-7
Kim, S., Jhong, J-H., Lee, J., and Koo, J-Y. (2017). Meta-analytic support vector machine for integrating multiple omics data. BioData Min. 10 (1), 2–14. doi:10.1186/s13040-017-0126-8
Krüger, B., Albrecht, E., and Lerch, M. M. (2000). The role of intracellular calcium signaling in premature protease activation and the onset of pancreatitis. Am. J. Pathol. 157 (1), 43–50. doi:10.1016/S0002-9440(10)64515-4
Kumar, P., Henikoff, S., and Ng, P. C. (2009). Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4 (7), 1073–1081. doi:10.1038/nprot.2009.86
LaRusch, J., Barmada, M. M., Solomon, S., and Whitcomb, D. C. (2012). Whole exome sequencing identifies multiple, complex etiologies in an idiopathic hereditary pancreatitis kindred. JOP 13 (3), 258–262.
Lee, P. J., and Papachristou, G. I. (2019). New insights into acute pancreatitis. Nat. Rev. Gastroenterol. Hepatol. 16 (8), 479–496. doi:10.1038/s41575-019-0158-2
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Masamune, A. (2014). Genetics of pancreatitis: the 2014 update. Tohoku J. Exp. Med. 232 (2), 69–77. doi:10.1620/tjem.232.69
Masamune, A., Kotani, H., Sörgel, F. L., Chen, J-M., Hamada, S., Sakaguchi, R., et al. (2020). Variants that affect function of calcium channel TRPV6 are associated with early-onset chronic pancreatitis. Gastroenterol 158 (6), 1626–1641.e8. doi:10.1053/j.gastro.2020.01.005
Mayerle, J., Sendler, M., Hegyi, E., Beyer, G., Lerch, M. M., and Sahin-Tóth, M. (2019). Genetics, cell biology, and pathophysiology of pancreatitis. Gastroenterology 156 (7), 1951–1968. doi:10.1053/j.gastro.2018.11.081
McGoldrick, L. L., Singh, A. K., Saotome, K., Yelshanskaya, M. V., Twomey, E. C., Grassucci, R. A., et al. (2018). Opening of the human epithelial calcium channel TRPV6. Nature 553 (7687), 233–237. doi:10.1038/nature25182
McLaren, W., Gil, L., Hunt, S. E., Riat, H. S., Ritchie, G. R., Thormann, A., et al. (2016). The ensembl variant effect predictor. Genome Biol. 17 (1), 122–214. doi:10.1186/s13059-016-0974-4
Messenger, S. W., Falkowski, M. A., and Groblewski, G. E. (2014). Ca²⁺-regulated secretory granule exocytosis in pancreatic and parotid acinar cells. Cell Calcium 55 (6), 369–375. doi:10.1016/j.ceca.2014.03.003
Mishra, V., Bose, A., Kiran, S., Banerjee, S., Shah, I. A., Chaukimath, P., et al. (2021). Gut-associated cGMP mediates colitis and dysbiosis in a mouse model of an activating mutation in GUCY2C. J. Exp. Med. 218 (11), e20210479. doi:10.1084/jem.20210479
Muddana, V., Lamb, J., Greer, J. B., Elinoff, B., Hawes, R. H., Cotton, P. B., et al. (2008). Association between calcium sensing receptor gene polymorphisms and chronic pancreatitis in a US population: role of serine protease inhibitor kazal 1type and alcohol. World J. Gastroenterol. 14 (28), 4486–4491. doi:10.3748/wjg.14.4486
Nilius, B., and Flockerzi, V. (2014). Mammalian transient receptor potential (TRP) cation channels. Berlin, Germany: Springer.
Noonan, M. L., and White, K. E. (2019). FGF23 synthesis and activity. Curr. Mol. Biol. Rep. 5 (1), 18–25. doi:10.1007/s40610-019-0111-8
Oracz, G., Zarod, M., Ewers, M., Laumen, H., Gambin, T., Kaminski, P., et al. (2021). Loss of function TRPV6 variants are associated with chronic pancreatitis in nonalcoholic early-onset Polish and German patients. Pancreatology 21 (8), 1434–1442. doi:10.1016/j.pan.2021.09.005
Prasad, H., Dang, D. K., Kondapalli, K. C., Natarajan, N., Cebotaru, V., and Rao, R. (2019). NHA2 promotes cyst development in an in vitro model of polycystic kidney disease. J. Physiol. 597 (2), 499–519. doi:10.1113/JP276796
Ramsey, M. L., Conwell, D. L., and Hart, P. A. (2017). Complications of chronic pancreatitis. Dig. Dis. Sci. 62 (7), 1745–1750. doi:10.1007/s10620-017-4518-x
Raphael, K. L., and Willingham, F. F. (2016). Hereditary pancreatitis: current perspectives. Clin. Exp. Gastroenterol. 9, 197–207. doi:10.2147/CEG.S84358
Rebours, V., Boutron-Ruault, M-C., Schnee, M., Férec, C., Maire, F., Hammel, P., et al. (2008). Risk of pancreatic adenocarcinoma in patients with hereditary pancreatitis: a national exhaustive series. Am. J. Gastroenterol. 103 (1), 111–119. doi:10.1111/j.1572-0241.2007.01597.x
Reed, A., and Gorelick, F. (2014). “Animal models of chronic pancreatitis,” in Pancreapedia: The exocrine pancreas knowledge base. doi:10.3998/panc.2014.1
Rendina, D., Esposito, T., Mossetti, G., De Filippo, G., Gianfrancesco, F., Perfetti, A., et al. (2012). A functional allelic variant of the FGF23 gene is associated with renal phosphate leak in calcium nephrolithiasis. J. Clin. Endocrinol. Metab. 97 (5), E840–E844. doi:10.1210/jc.2011-1528
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J., and Kircher, M. (2019). CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47 (D1), D886–D94. doi:10.1093/nar/gky1016
Rodríguez, M. (2020). FGF23: Is it another biomarker for phosphate–calcium metabolism? Adv. Ther. 37 (2), 73–79. doi:10.1007/s12325-019-01181-4
Sah, R. P., Garg, S. K., Dixit, A. K., Dudeja, V., Dawra, R. K., and Saluja, A. K. (2014). Endoplasmic reticulum stress is chronically activated in chronic pancreatitis. J. Biol. Chem. 289 (40), 27551–27561. doi:10.1074/jbc.M113.528174
Sahin-Tóth, M. (2020). Channelopathy of the pancreas causes chronic pancreatitis. Gastroenterol 158 (6), 1538–1540. doi:10.1053/j.gastro.2020.03.027
Sanjeevi, R., John, R. A., Kurien, R. T., Dutta, A. K., Simon, E. G., David, D., et al. (2020). Acoustic radiation force impulse imaging of pancreas in patients with early onset idiopathic recurrent acute pancreatitis. Eur. J. Gastroenterol. Hepatol. 32 (8), 950–954. doi:10.1097/MEG.0000000000001732
Schwarz, J. M., Rödelsperger, C., Schuelke, M., and Seelow, D. (2010). MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7 (8), 575–576. doi:10.1038/nmeth0810-575
Segerstolpe, Å., Palasantza, A., Eliasson, P., Andersson, E-M., Andréasson, A-C., Sun, X., et al. (2016). Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab. 24 (4), 593–607. doi:10.1016/j.cmet.2016.08.020
Shelton, C., and Whitcomb, D. C. (2018). Hereditary chronic pancreatitis: Molecular pattern, clinical consequences, and management principles. Pancreas Integr. Textb. Basic Sci. Med. Surg. 40 (50), 374.
Shelton, C. A., and Whitcomb, D. C. (2016). “Hereditary pancreatitis,” in Pancreapedia: The exocrine pancreas knowledge base.
Takats, A., Berke, G., Szentesi, A., Farkas, G., Izbeki, F., Eross, B., et al. (2021). Common calcium-sensing receptor (CASR) gene variants do not modify risk for chronic pancreatitis in a Hungarian cohort. Pancreatology 21 (7), 1305–1310. doi:10.1016/j.pan.2021.08.012
Van der Auwera, G. A., Carneiro, M. O., Hartl, C., Poplin, R., Del Angel, G., Levy-Moonshine, A., et al. (2013). From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinforma. 43 (1), 11. doi:10.1002/0471250953.bi1110s43
Venkatachalam, K., and Montell, C. (2007). TRP channels. Annu. Rev. Biochem. 76, 387–417. doi:10.1146/annurev.biochem.75.103004.142819
Vezzoli, G., Terranegra, A., Arcidiacono, T., Biasion, R., Coviello, D., Syren, M., et al. (2007). R990G polymorphism of calcium-sensing receptor does produce a gain-of-function and predispose to primary hypercalciuria. Kidney Int. 71 (11), 1155–1162. doi:10.1038/sj.ki.5002156
Whitcomb, D. C., Gorry, M. C., Preston, R. A., Furey, W., Sossenheimer, M. J., Ulrich, C. D., et al. (1996). Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat. Genet. 14 (2), 141–145. doi:10.1038/ng1096-141
Zhao, Q., Wei, Y., Pandol, S. J., Li, L., and Habtezion, A. (2018). STING signaling promotes inflammation in experimental acute pancreatitis. Gastroenterology 154 (6), 1822–1835. doi:10.1053/j.gastro.2018.01.065
Keywords: pancreatitis, CaSR, FGF23, WES - whole-exome sequencing, TRPV6 (transient receptor potential cation channel subfamily V member 6)
Citation: Shah IA, Prasad H, Banerjee S, Kurien RT, Chowdhury SD and Visweswariah SS (2023) A novel frameshift mutation in TRPV6 is associated with hereditary pancreatitis. Front. Genet. 13:1058057. doi: 10.3389/fgene.2022.1058057
Received: 30 September 2022; Accepted: 23 December 2022;
Published: 09 January 2023.
Edited by:
Obul Reddy Bandapalli, Hopp Children’s Cancer Center Heidelberg (KiTZ), GermanyReviewed by:
Alex M. Mawla, University of California, Davis, United StatesUlrich Wissenbach, Saarland University, Germany
Copyright © 2023 Shah, Prasad, Banerjee, Kurien, Chowdhury and Visweswariah. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sudipta Dhar Chowdhury, c3VkaXB0YWRoYXJjaG93ZGh1cnlAZ21haWwuY29t; Sandhya S. Visweswariah, c2FuZGh5YUBpaXNjLmFjLmlu