
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 04 January 2023
Sec. Stem Cell Research
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.1056114
This article is part of the Research Topic Molecular and Genetic Control of Muscle Stem Cell Fate Decision View all 4 articles
 Giulio Cossu1,2,3*
Giulio Cossu1,2,3* Rossana Tonlorenzi2*
Rossana Tonlorenzi2* Silvia Brunelli4*
Silvia Brunelli4* Maurilio Sampaolesi5,6*
Maurilio Sampaolesi5,6* Graziella Messina7*
Graziella Messina7* Emanuele Azzoni4
Emanuele Azzoni4 Sara Benedetti8
Sara Benedetti8 Stefano Biressi9Chiara Bonfanti7Laricia Bragg1Jordi Camps10
Stefano Biressi9Chiara Bonfanti7Laricia Bragg1Jordi Camps10 Ornella Cappellari11
Ornella Cappellari11 Marco Cassano12Fabio Ciceri2Marcello Coletta6Diego Covarello13Stefania Crippa14
Marco Cassano12Fabio Ciceri2Marcello Coletta6Diego Covarello13Stefania Crippa14 M. Gabriella Cusella-De Angelis15Luciana De Angelis6Arianna Dellavalle16
M. Gabriella Cusella-De Angelis15Luciana De Angelis6Arianna Dellavalle16 Jordi Diaz-Manera17
Jordi Diaz-Manera17 Daniela Galli18Francesco Galli1Cesare Gargioli19
Daniela Galli18Francesco Galli1Cesare Gargioli19 Mattia F. M. Gerli20Giorgia Giacomazzi21
Mattia F. M. Gerli20Giorgia Giacomazzi21 Beatriz G. Galvez22Hidetoshi Hoshiya23Maria Guttinger24Anna Innocenzi2M. Giulia Minasi25Laura Perani2Stefano C Previtali2Mattia Quattrocelli26Martina Ragazzi27
Beatriz G. Galvez22Hidetoshi Hoshiya23Maria Guttinger24Anna Innocenzi2M. Giulia Minasi25Laura Perani2Stefano C Previtali2Mattia Quattrocelli26Martina Ragazzi27 Urmas Roostalu28Giuliana Rossi29Raffaella Scardigli30Dario Sirabella31Francesco Saverio Tedesco32Yvan Torrente8Gonzalo Ugarte32
Urmas Roostalu28Giuliana Rossi29Raffaella Scardigli30Dario Sirabella31Francesco Saverio Tedesco32Yvan Torrente8Gonzalo Ugarte32In 2002 we published an article describing a population of vessel-associated progenitors that we termed mesoangioblasts (MABs). During the past decade evidence had accumulated that during muscle development and regeneration things may be more complex than a simple sequence of binary choices (e.g., dorsal vs. ventral somite). LacZ expressing fibroblasts could fuse with unlabelled myoblasts but not among themselves or with other cell types. Bone marrow derived, circulating progenitors were able to participate in muscle regeneration, though in very small percentage. Searching for the embryonic origin of these progenitors, we identified them as originating at least in part from the embryonic aorta and, at later stages, from the microvasculature of skeletal muscle. While continuing to investigate origin and fate of MABs, the fact that they could be expanded in vitro (also from human muscle) and cross the vessel wall, suggested a protocol for the cell therapy of muscular dystrophies. We tested this protocol in mice and dogs before proceeding to the first clinical trial on Duchenne Muscular Dystrophy patients that showed safety but minimal efficacy. In the last years, we have worked to overcome the problem of low engraftment and tried to understand their role as auxiliary myogenic progenitors during development and regeneration.
In vertebrates, skeletal myoblasts originate from dorsal somites in the trunk and from presomitic paraxial mesoderm in the head. Bones rather originate from ventral somite in the trunk, lateral mesoderm in the limbs and neural crest in the head. This notion came from pioneering chick-quail transplantation studies (Couly et al., 1993; Monsoro-Burq et al., 1994; Chevallier et al., 1977; Christ et al., 1977).
Skeletal muscle development starts right after the onset of somitogenesis and is completed at the end of post-natal growth (Buckingham 2017), though adult muscle maintains the ability to regenerate after injury. Therefore, it was safe to postulate the existence of different progenitor cells that could undergo different fates (differentiation versus proliferation) in the same micro-environment to accommodate the need of providing early motility and structure to the embryo while increasing the mass of the tissue (Cossu and Molinaro, 1987). In fact, muscle and bone cells stop dividing when differentiated, therefore new cells must be added by fusion to the growing fibres or apposition to the growing bone. Canonical progenitors, active throughout growth, are osteoblasts underneath the endosteum layer (Bianco 2014) and satellite cells underneath the endomysium (Engquist and Zammit, 2021). However, it has been shown that bone pericytes in trabecular bone have the ability to give rise to new osteocytes (Bianco 2014) and we showed that pericytes of skeletal muscle contribute to post-natal development of multinucleated muscle fibres (Dellavalle et al., 2011).
In the mid 90’ the first transgenic mice expressing the LacZ reporter were generated, thanks to the work of Jean Francois Nicholas (Sanes et al., 1986). For the first time a long-lasting dream of mammalian embryologists came true: a cell autonomous, eventually tissue specific, inheritable marker that would allow following the fate of specific cell types (e.g. myogenic) in vivo. The senior author of this review had imported to his lab, then in Rome, MLC1/3F-nLacZ mice (Kelly et al., 1995) from Margaret Buckingham lab in Pasteur. All and only striated muscle cells in these mice express the LacZ gene with a nuclear localization signal. When we co-cultured unlabelled C2C12 myoblasts with LacZ + fibroblasts, we found LacZ + myotubes, easily identified by a nuclear blue staining with X-Gal that reveals the activity of LacZ-encoded β-galactosidase. LacZ + fibroblasts, cultured alone or with other cells, did not show any staining. We concluded that fibroblasts, as then we would define any non-myogenic cell in skeletal muscle (for an overview of muscle interstistial cells, see Table 1) did not possess myogenic potential on their own, but may be recruited to myogenesis by adjacent differentiating myogenic cells, either by fusion or by short-range paracrine signals (conditioned medium did not work) that may render fibroblasts competent to fuse. We published this paper in 1995 (Salvatori et al., 1995), and almost simultaneously two similar papers by the groups of Diane Watt and Louis Garcia (Breton et al., 1995; Gibson et al., 1995) reported very similar data, in agreement with our observation. As it always happens with reports that disagree with current dogmas, the papers were simply ignored or dismissed as “tissue culture artefacts”. Thus, we decided to test whether the phenomenon might also occur in vivo without any intermediate culture. So, we transplanted bone marrow from MLC1/3F-nLacZ mice into wt adult animals. We observed that, following an acute injury to muscle but not under basal conditions, nuclei expressing LacZ were detected in regenerated muscle fibres (Ferrari et al., 1998). The frequency was low but reproducible in all mice and reproduced in other laboratories who tried to use bone marrow derived cells to repair muscle in dystrophic mice (Gussoni et al., 1999), though the frequency was too low to justify any realistic hope. Our paper had an impact (cited more than 2.400 times so far) and opened the controversial field of cell plasticity. Within months many publications appeared in high profile journals, often supported only by few low-resolution immune-fluorescence analysis, showing that virtually any stem/progenitor cell of our body may give rise to any differentiated progeny. This led to premature clinical translation, especially for heart diseases, eventually to be repaired with bone marrow derived cells (Chien et al., 2019). Overall, the field was mudded and results dismissed as technical tissue culture artefacts (Bonfanti et al., 2012).
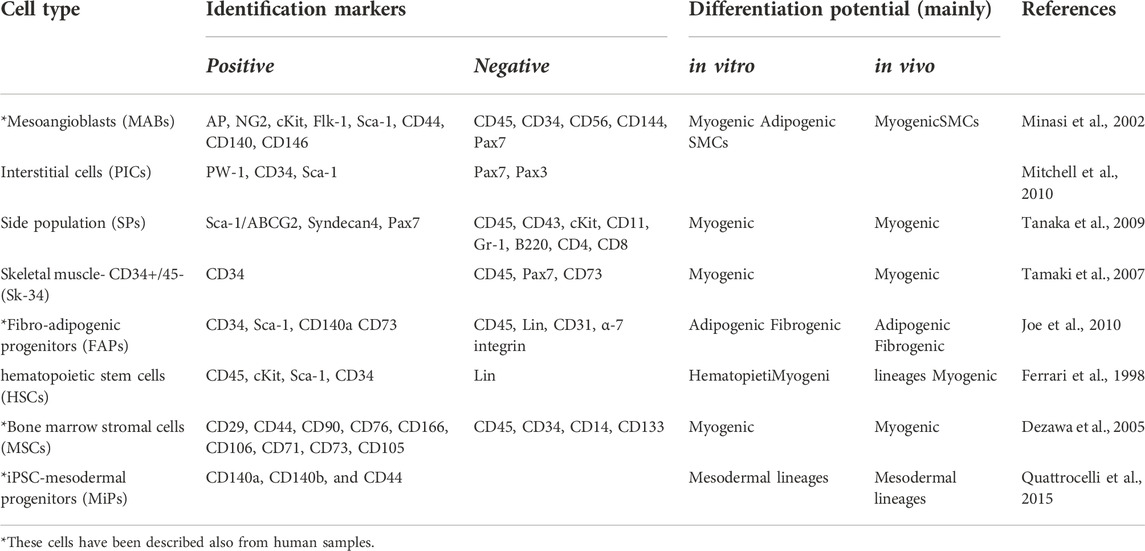
TABLE 1. Adult stem/progenitor cells involved in skeletal muscle regeneration, beside satellite cells and other muscle derived myogenic cells; MABs, mesoangioblasts; PICs, PW1(+)/Pax7 (-) interstitial cells; SP, side population; FAPs, Fibro-adipogenic progenitors; HSCs, hematopoietic stemcells; MSCs, Bone marrow stromal cells; MiPs mesodermal iPSC-derived progenitors; SMCs, smooth muscle cells.
In the meanwhile, we decided to investigate where these progenitors originate from during mouse development. In the absence of any specific marker, we isolated and cultured as tissue explants somites, lateral plates, neural tube, heart, and aorta. After a week, we cloned embryonic cells outgrown from the explants and observed that clones from dorsal aorta had the clear morphology of satellite cells and co-expressed early endothelial and myogenic markers such as MyoD. Of notice the culture of the same undissociated explant did not differentiate into skeletal muscle cells. Years later, we showed that explants of embryonic aorta from MLC1/3F-nLacZ mice, when cultured on C2C12 or primary wt myoblasts, would give rise to LacZ + nuclei in hybrid myotubes. This process was stimulated by Noggin and inhibited by BMP (Ugarte et al., 2012). Moreover, when lateral plate from these embryos at early somite stage (E 8.5) was isolated and cultured onto C2C12 myogenic cells, several myotubes containing LacZ + blue nuclei could be observed, similarly, though at a lesser extent, than when paraxial mesoderm was co-cultured with the same C2C12 cells. In contrast, when lateral plates or paraxial mesoderm were cultured with wt neural tubes, only paraxial mesoderm gave rise to very many LacZ + mononucleated myocytes, while lateral plate did not (Figure 1). This result showed that the lateral plate, from which the vasculature originates, contains cells that cannot be induced to skeletal myogenesis by the neural tube as the newly formed somites (Vivarelli and Cossu, 1986) but can be “recruited” to myogenesis by differentiating muscle cells.
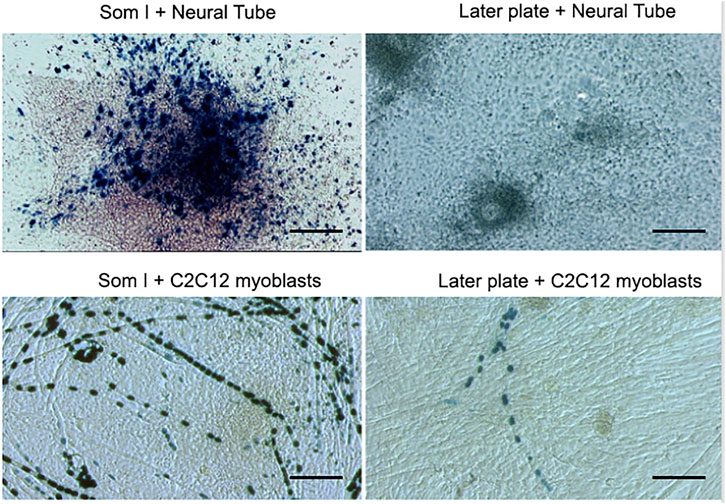
FIGURE 1. A co-culture of paraxial (Som I) mesoderm or Lateral Plate from MLC1/3F-nLacZ embryos with either neural tube from wt embryos or with unlabelled C2C12 myoblasts. After 5 days, cultures were fixed and stained with X-Gal.
This led us to believe that an early endothelial progenitor may be endowed with skeletal myogenic potency, to be revealed once the cells were exposed to conditions promoting skeletal myogenesis (De Angelis et al., 1999). We confirmed these data with quail-chick transplantation experiments where quail, but also mouse dorsal aorta was transplanted into an early chick limb bud: cells from the transplanted aorta gave rise to most mesodermal tissues, including skeletal muscle (Minasi et al., 2002). When cells were isolated from the mouse aorta and cloned on feeder layers, they gave rise to clonal lines expressing both endothelial and pericyte markers that can proliferate indefinitely.
Transcriptomic analysis of these clones revealed a specific molecular signature, slightly overlapping with that of other stem cells but clearly different from embryonic fibroblasts (Tagliafico et al., 2004). The differentiation potential of these clonal lines was explored in vitro and in vivo assays. Dorsal aorta clones were able to promptly differentiate into smooth muscle cells in response to TGFβ1 and into osteoblast-like cells in response to BMP2, in accordance with their high level expression of many members of the TGFβ/BMP pathway. These cells also differentiated into adipocytes in response to insulin but were unable to undertake a chondrogenic pathway, under standard cartilage inducing conditions. Most importantly these cell lines would not differentiate spontaneously into skeletal muscle, unless co-cultured with myogenic cells or transplanted into regenerating skeletal muscle.
Because of their vascular markers and their ability to differentiate into several mesoderm tissues we named these cells “meso-angioblasts”, subsequently simplified into “mesoangioblasts” (MABs). Whether these MABs were a sub-population of endothelial progenitor cells, such as the EPC, described in (Gulati et al., 2003; Hur et al., 2004), or a subtype of pericytes (Cossu and Bianco, 2003), expressing also early endothelial markers remained unclear at the time.
Endothelium had already been shown at the time to be a crossway of several cell lineages during embryogenesis. Fate-mapping and imaging studies of the developing dorsal aorta and work on embryonic stem cells, led to the discovery and characterization of the hemogenic endothelium, a specialized embryonic endothelial population that gives rise to the precursors of hematopoietic stem cells (HSCs) and therefore essentially to all the adult blood (Lancrin et al., 2009; Boisset, et al., 2010; Zovein, et al., 2010; Swiers et al., 2013).
A first strategy to investigate the endothelial embryonic origin of MABs in the dorsal aorta, was to adopt a genetic lineage tracing approach. To genetically label endothelial-derived cell populations, we used the Cdh5-CreERT2 transgenic mouse developed in the lab of Ralph Adams, that expresses a Tamoxifen (TAM)-inducible Cre recombinase (CRE-ERT2) under the control of VE-Cadherin (VE-Cad) regulatory sequences (Wang, et al., 2010). By inducing the expression of the Cre recombinase at E8.5 in a double transgenic Cdh5-CreERT2; R26R-EYFP, we were able to permanently label a population of progenitors with mesodermal potency, that originates from VE-Cad + endothelial cells within the yolk sac and in the placenta and are found later in the embryo proper (Azzoni et al., 2014). These VE-cad + derived cells, expressing hematopoietic (CD45+) but not myeloid/macrophage markers (CD11b-), displayed, in the developing embryos, a tissue distribution overlapping with the one observed with transplantation of MABs cell lines (smooth and skeletal muscle, dermis).
Most interestingly, isolated VE-Cad + -derived CD45+ CD11b-cells displayed MABs features in vitro and in vivo assays. They could indeed differentiate in osteoblasts and smooth muscle when treated with BMP or TGFβ respectively and could give rise to MyHC + myotubes in co-culture with myoblasts. In addition, they were able to contribute to post-natal muscle regeneration in vivo when transplanted in the muscle of wt or dystrophic mice. However, like the dorsal aorta cell lines, VE-Cad + -derived CD45+ CD11b-cells were unable to spontaneously differentiate in skeletal muscle in vitro, and no VE-cad-derived cells could be detected in skeletal muscle after birth (Azzoni et al., 2014). All this evidence suggested that embryonic MABs are a transient sub-population of hemogenic endothelium-derived cells endowed with a mesoderm multi-lineage differentiation potential that is restricted to the embryonic and foetal normal development but is not maintained in postnatal life (Figure 2).
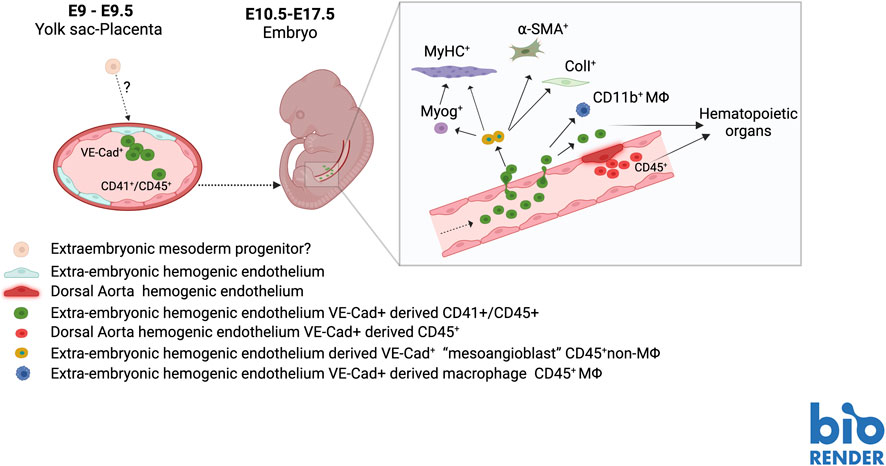
FIGURE 2. Hypothetical scheme of the origin of MABs in during mouse embryo development.
Indeed, when we isolated what we thought were the same progenitors from post-natal murine and human muscle, we noticed that they only expressed pericyte and not endothelial markers. Moreover, they would tend to differentiate spontaneously into smooth muscle (the default pathway of pericytes) and skeletal muscle but no other mesoderm tissues, suggesting a restriction of potency concomitant with a tissue-specific commitment. Indeed, many years later, Paolo Bianco and others showed that there are no identical mesenchymal stem cells: pericytes isolated from human bone would differentiate, using rigorous assays, only into osteocytes, chondrocytes and adipocytes, the components of bone, beside smooth muscle. Conversely, pericytes isolated from human skeletal muscle would differentiate into skeletal and smooth muscles, as stated above (Sacchetti et al., 2016).
A possible interpretation of these data would postulate that vessel associated progenitors may contribute to the post-embryonic growth of tissues. In mammals all organs are formed during embryogenesis at the end of which, a whale, a man and a mouse have very similar size (Roston et al., 2013). Thus, most of the body is built during foetal and post-natal growth by cell division, mainly for epithelia, or by cell addition mainly for solid mesoderm such as bone and skeletal muscles, where differentiated cells no longer divide (Bianco and Cossu, 1999).
To carry on a lineage tracing analysis of vessel-associated postnatal progenitors and assess their in vivo contribution to the growth of the skeletal muscle, we needed a specific marker. Microarray analysis of in vitro expanded MAB cell populations from post-natal and juvenile muscle showed the expression of pericyte markers (e.g., NG2, MCAM and PDGF-Rb) which almost invariably are also expressed, though at low level, by skeletal myogenic progenitors (Tagliafico et al., 2004; Dellavalle et al., 2007). In other words, there is not a unique marker, such as “MyoD” for skeletal myogenic cells, that uniquely identifying these progenitors. Tissue Non-specific Alkaline Phosphatase (TNAP) was the only marker we found not shared with myoblasts. However, as all pericyte markers, also TNAP is expressed on many other cell types, including endothelium, and is not expressed in all pericytes. This prevented a prospective isolation of MABs and confined lineage tracing studies to the use of a TNAP-CreERT mouse that we generated in collaboration with Sharaghim Tajbakhsh (Dellavalle et al., 2011). In addition, we could not lineage trace TNAP + pericytes before birth because TNAP is expressed by many other cell types, whereas postnatally its expression is restricted to pericytes in humans and to pericytes and endothelium in rodents (Safadi et al., 1991). By administering Tamoxifen to P6 pups, we observed labelling of small vessels only, as predicted. However, after 1 month many muscle fibres were LacZ+, indicating that some cell previously expressing TNAP had fused with growing muscle fibres. We also noticed that contribution varied from almost 10% of positive fibres (i.e., containing nuclei derived from TNAP + pericytes) in the diaphragm to less than 1% in the Tibialis anterior. This is interesting in light of subsequent observations showing a higher contribution of satellite cells to these muscles in comparison with hind limb muscle (Keefe et al., 2015; Pawlikowski et al., 2015). Indeed we detected also LacZ + satellite cells, indicating a Mabs contribution to the satellite cell pool, but this was too low to account for the reported difference in satellite cell abundance among these different muscles. Following acute injury or in a dystrophic background, the percentage of positive fibres increased in proportion to the labelling observed in healthy muscle. Most importantly the contribution of pericytes to skeletal muscle could be observed only during post-natal growth. When adult animals (P30 or older) were treated with Tamoxifen, the contribution of TNAP + cells to muscle fibres was negligible. And this also explains the apparent discrepancy with a subsequent paper, claiming that TBX18 + pericytes do not contribute to any non-vascular tissue (Guimarães-Camboa et al., 2017). This work was based on Tamoxifen administration at P30, so that contribution to skeletal muscle could not be observed because it occurs, at least for TNAP + pericytes, only during post-natal growth (Figure 3).
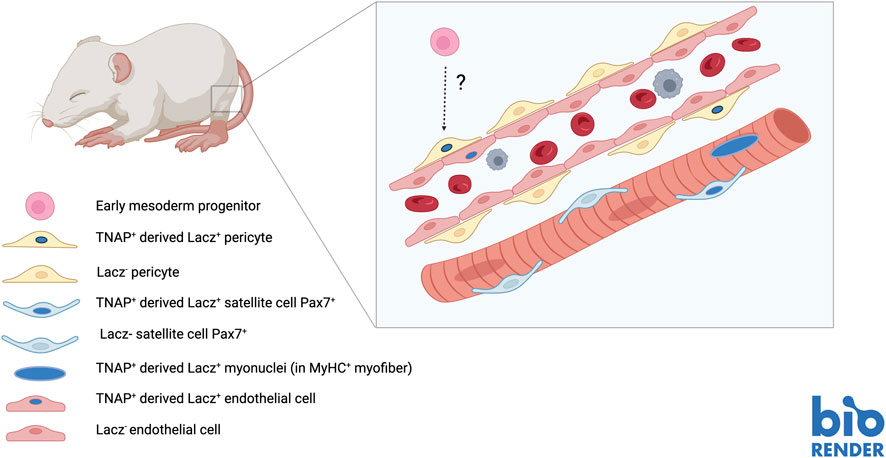
FIGURE 3. Contribution of TNAP + pericyte (MABs) to developing foetal muscle and satellite cells.
These results are in agreement with our hypothesis that a vessel-associated progenitor, most likely an adventitial pericyte (TNAP+), may generate cells fated to differentiate into one or another type of solid mesoderm, depending upon the tissue where the vessel would ingress during foetal angiogenesis, thus being recruited by local differentiating cells (Cossu and Bianco, 2003). Whether these postnatal TNAP + vessel progenitors have a common mesodermal ancestor with the embryonic hemogenic endothelium derived MABs is still unknown, due to the lack of specific early markers for lineage tracing.
Regardless, we have been using the term “mesoangioblast”, MABs, to indicate not only the cultured cells derived from either embryonic endothelial progenitors and pericytes in the post-natal muscle, but also their in vivo counterparts, even though this is not strictly correct as, for example, embryonic stem cells do not exist as such in the inner cell mass of the blastocysts but represent their in vitro-derived counterpart.
In the general context of the pericyte/myogenic cell relationship, the above data supported the idea that pericytes may differentiate into myogenic cells during post-natal muscle growth and this contribution may be enhanced by acute or chronic injury. However, pericytes alone were not sufficient to sustain muscle regeneration when all satellite cells were ablated, also considering that a possible by-stander effect (Bi et al., 1993): pericytes and satellite cells are often juxtaposed across the muscle basal lamina.
On the other hand, we observed that committed myogenic cells could be recruited to a pericyte fate by signals emanated by the endothelium such as Dll4 and PDGF-BB. Of notice both embryonic and foetal myoblasts (Cappellari et al., 2013) but also post-natal satellite cells (Gerli et al., 2019) could be converted to pericytes but maintained expression of Myf5, indicating that their myogenic identity had not been erased.
Thus, one may ask why there is this bi-directional differentiation potency and what role it may fulfil during development or in pathological situation (Stallcup, 2013). During development the equilibrium should usually be skewed towards skeletal muscle, overwhelming pericytes quantitatively, so that non myogenic cells may be recruited to increase the number of myogenic cells. However, when vessels grow into these developing or regenerating muscles, they may, in turn, recruit surrounding mesoderm cells, including myoblasts, to a pericyte fate, needed to stabilise the vessel (Figure 4)
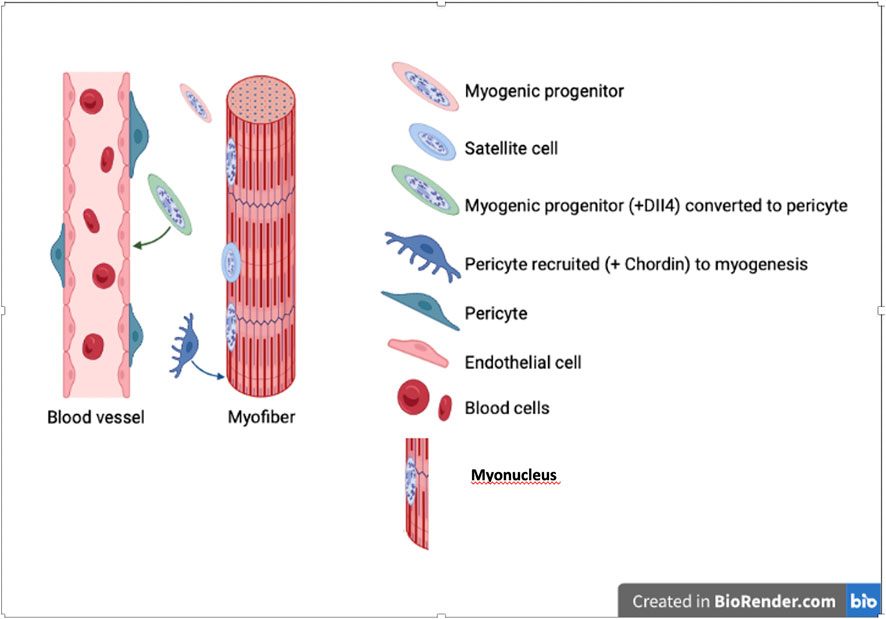
FIGURE 4. Bi-directional differentiation of myogenic cells into pericytes and vice versa. Myogenic cell nuclei are represented in salt and peper gray, while nuclei of non myogenic cells in uniform solid colour.
The potential therapeutic applications of MABs for cell therapy of muscle disease made it particularly important to understand how their acquisition of a skeletal muscle fate is regulated.
Indeed, although the myogenic potential of MABs was progressively demonstrated in vitro and in vivo, the molecules regulating this process were still unknown. Genome-wide RNA expression analysis, performed on embryonic MABs, revealed a characteristic pattern of expression that correlates with the biological features and the developmental potency of these cells (Tagliafico et al., 2004). Interestingly, MABs were found to express Pax3 at a high level. The paired box/homeodomain transcription factors of the Pax family play important roles in the formation of tissues and organs during embryonic development, being implicated in neurogenesis in dorsal regions of the central nervous system, also by regulating neural crest and its derivatives, and playing a key role in skeletal myogenesis. In the latter context, Pax3 is expressed in paraxial mesoderm and marks multipotent cells of the dorsal somite, the dermomyotome, from which skeletal muscle is derived. Endothelial and smooth muscle cells are also derived from paraxial mesoderm, and the same Pax progenitor in the dermomyotome can give rise to both the smooth muscle cells of blood vessels and skeletal muscle (Bajard et al., 2006; Buckingham, 2017). We therefore generated embryonic MABs from Pax3 null embryos (Messina et al., 2009). The absence of Pax3 in multipotent MABs did not affect their morphology, as also their typical gene expression profile required for their proliferation or survival as also their differentiation into the most mesodermal derivatives was not compromised. On the contrary, lack of Pax3 strongly impaired skeletal muscle differentiation, thus suggesting a crucial role of Pax3 in the commitment of embryonic mouse MABs to an inducible myogenic fate. Notably, Pax7 was dispensable, at least in the embryonic period. Consequently, MABs isolated from PAX3PAX3−FKHR-IRESnLacZ/+ mouse embryos, where the oncogenic fusion protein Pax3-FKHR rescues the early Pax3 mutant phenotype by over-activating Pax3 targets (Relaix et al., 1996), when cocultured with human satellite cells, differentiated into multinucleated myotubes with higher efficiency (20%–30% versus 5%–10%) than wt MABs. The fundamental role of Pax3 in MAB biology was also confirmed in vivo in Sgca null dystrophic animal with IA transplanted MABs (Messina et al., 2009). Therefore, our first study aimed at the identification of the molecular pathway(s) regulating MAB myogenic potential showed that Pax3, but not Pax7, regulates the differentiation of MABs into skeletal muscle, consistent with a possible paraxial mesoderm origin of these cells in the embryonic dorsal aorta (Esner et al., 2006; Messina et al., 2009).
However, signalling factors originating from differentiating muscle cells are necessary to induce embryonic MABs into skeletal myogenic programme, and their regulatory targets remain to be defined. Additionally, Pax3 seems not to be necessary for adult mouse MABs, as also for dog and human ones, supporting the hypothesis that embryonic and post-natal MABs are indeed similar but not identical cells.
In 2015, a cross-analysis of independent microarrays performed on different human and murine MAB populations revealed that PW1 is expressed in MABs regardless of species origin and age of isolation (Tagliafico et al., 2004; Bonfanti et al., 2015). PW1, also identified as Peg3 (Paternally expressed gene3), initially isolated following a screening to identify factors involved in skeletal muscle commitment as well as in a screen for parentally imprinted genes (Relaix et al., 2003), is expressed at high levels upon gastrulation and is downregulated during foetal and perinatal development (Kuroiwa et al., 1996). In postnatal skeletal muscle, PW1 expression is restricted to muscle satellite cells and to a subpopulation of interstitial cells (PICs, PW1-interstitial cells), which display myogenic potential (Mitchell et al., 2010; Pannerec et al., 2013). Moreover, PW1 expression identifies multiple adult stem and progenitor cell populations from a wide array of adult tissues including skin, gut, blood and central nervous system (Besson., 2011). We demonstrated that both mouse and human MABs displaying high levels of PW1 show a higher myogenic competence and ability to cross the vessel wall upon systemic delivery in dystrophic animal models. Indeed, lack of PW1 in MABs causes a block in myogenic differentiation, through a mechanism that drives MyoD to degradation via the proteasome machinery and a strong JAM-A dependent adhesion to the endothelium thus preventing MAB extravasation (Bonfanti et al., 2015). These studies from our team represented an important first step in the comprehension of MAB behaviour and competence. Indeed, among the multiple obstacles in using allogeneic cell transplantation for therapeutic ends, a key challenge is to identify the most appropriate donor cell, in addition to requirements related to the immune system such as HLA compatibility. At present, screening the competence of each preparation of MABs is time consuming and costly requiring both in vitro and ultimately in vivo assays. The striking correlation between the level of PW1 expression in MABs and their ability to differentiate into muscle and to cross the endothelium in numerous cell preparations across species and age suggests that assessing PW1 levels may represent a rapid and single step assay for screening donor cell preparations of therapeutic value.
Beside the satellite cells, canonical stem/progenitor cells of skeletal muscle, isolated more than 60 years ago (Mauro, 1961) and recently historically reviewed (Engquist and Zammit 2021), many stem/progenitor cell populations have been isolated and characterized from skeletal muscles in the last 20 years. Table 1, summarises and updates current knowledge on the currently characterized interstitial cells of skeletal muscle, previously known as “fibroblasts”. However, following the unbiased clustering by Smart-seq2 of interstitial cells from healthy and dystrophic mouse muscles, we were able to find distinct cell clusters only for muscle stem cells/satellite cells (MuSCs) and interstitial stem cells (ISCs). This was also observed by other groups who performed scRNA-seq on healthy skeletal muscle (Dell'Orso et al., 2019; Giordani et al., 2019). Within the ISC cluster, markers were found representing different ISC populations, including MABs, fibro-adipogenic progenitors (FAP) and PW1+/Pax7– cells (PICs). However, the different adult stem cells (MABs, FAPs, PICs and so on) are present in the same cluster due to the similarity of single cell transcriptomic profiles, raising the issue of the reciprocal lineage relationship and possible inter-conversion among these stem/progenitors, as previously postulated (Bianco and Cossu, 1999). Indeed, ISCs have been defined by various isolation techniques and functions and have mostly only been compared with MuSCs to show their different origins. Nevertheless, despite these different isolation methods, ISCs share many overlapping markers. For example, FAP, PIC and TWIST2 + mouse progenitors are all positive for SCA-1 that was originally described in MABs (Dellavalle et al., 2007; Mitchell et al., 2010; Liu et al., 2017), which suggests that they may acquire this marker during culture conditions. MABs, PICs and TWIST2+ progenitors have been characterized for the ability to directly participate to adult myogenesis (Mitchell et al., 2010; Dellavalle et al., 2011; Liu et al., 2017) while FAPs only support indirectly adult myogenesis and mainly differentiate into adipocytes and myofibroblasts (Uezumi et al., 2010). Even so, it has been shown that MABs, PICs and TWIST2+ progenitors differentiate also into adipocytes, under different conditions. In addition, HDAC inhibitors can drastically impact FAP differentiation potential, driving them towards a myogenic fate (Saccone et al., 2014). These findings may have been due to different protocols adopted by different laboratories, also because cells were isolated from different muscles and/or different stages of different animal models. Despite that, when we isolated MABs and FAPs from wt and β-sarcoglycan KO mice and compared their cell potency, we observed a myogenic and an adipogenic capacity for both cell types (Camps et al., 2020). All these results highlight the importance of further studies using an unbiased comparison of all these cell populations with simultaneous cell isolation, large scRNA-seq screens, and functional assays in vitro and in vivo to shade light on adult muscle stem/progenitor cell heterogeneity.
Many studies have been directed to improve MAB proliferative capacity, myogenic potency and to overcome the problem of low engraftment; these investigated NOTCH (Quattrocelli et al., 2014) BMP (Costamagna et al., 2016) and PDGF (Camps et al., 2020) signaling, and we also tried to better understand their role during muscle development and regeneration (Quattrocelli et al., 2012; Giacomazzi et al., 2021; Ronzoni et al., 2021). It is clear that aging (Rotini et al., 2018) and the stem cell niche of cachectic muscles (Costamagna et al., 2020) negatively impact on the regenerative capacity of MABs and this should be carefully considered for their future potential therapeutic applications. A promising cell model for targeting muscle tissues is constituted by induced pluripotent stem (iPS) cells (Takahashi et al., 2007). At present, iPS cells can be obtained from a wide variety of somatic cells, including murine (Quattrocelli et al., 2015), equine (Quattrocelli et al., 2016) and human (Giacomazzi et al., 2017) MABs. Originally, iPS cells were reprogrammed from somatic cells by transiently overexpressing pluripotency regulators, such as OCT4, SOX2, KLF4 and cMYC, through retroviral vectors (Takahashi et al., 2007). iPS cells display many pluripotency features, including virtually unlimited proliferation potential and differentiation potency toward derivatives of all embryonic lineages including MAB-like cells (Tedesco et al., 2012). During reprogramming of somatic cells to a pluripotent state, tissue-specific genes and differentiation regulators must be silenced, while pluripotency genes and self-renewal genes must be activated. Hence the reprogramming of MABs to the pluripotent state includes genome-wide modulation of histones and DNA methylation that modify the epigenetic landscape offering, however, a differentiation bias towards myogenic lineages due to a persistent cell memory (Kim et al., 2010) that can be captured by small noncoding RNA signatures (Giacomazzi et al., 2017).
A bio-fabrication approach to restore skeletal muscle loss has been achieved using MABs (Costantini et al., 2021) and the latter are a valuable stem cell source to generate neuromuscular junction models in microfluidic devices (Stoklund Dittlau et al., 2021). These bidimensional cell systems are of great help to study cell-cell interactions and cell signalling and can be genetically manipulated (Santoni de Sio et al., 2008); however, more complex three-dimensional structures are necessary to better mimic the original muscle tissues and to contribute to the maturation of iPS cell derivatives. In fact, organoids as self-organized 3D structures, are composed of different types of cells derived from stem or progenitor cells. In this view, iPS cell technology offers a potential breakthrough, as patient-derived iPS cells can be differentiated toward different cell lineages and yet retain the genetic background of the patients in 2D or 3D cell systems. 3D organoids were developed for a wide range of human tissues or organs for disease modelling and promises to bridge the gap between in vitro monolayer cell cultures and in vivo studies in animal models (Jiang et al., 2022; Jalal t al. 2021). Furthermore, human organoids are employed to study muscle genetic disorders, including Duchenne muscular dystrophy (DMD). After gene editing corrections, it is possible to obtain isogenic controls to better understand the pathological phenotype and to evaluate the effects of new therapeutic strategies more rigorously, during drug screening or genetic interventions (Marini et al., 2022 now published).
During embryonic development, skeletal muscle precursors arise from paraxial mesoderm (Biressi et al., 2007) while splanchnic, cardiogenic mesoderm gives rise to the heart (Abu-Issa and Kirby et al., 2007) with the participation of neural crest cells. During embryonic development, cardiac or skeletal muscle lineages rely on very specific gene expression networks, often recapitulated during adult myogenesis; the regeneration capacity is very limited in the cardiac tissue compared to skeletal muscles and the presence of cardiac progenitor stem cells is still highly debated (Eschenhagen et al., 2017; Chien et al., 2019; Salerno et al., 2022). Preclinical and clinical studies provided arguments for and against the use of various adult stem cell types for therapeutic approaches, including skeletal myoblasts, bone marrow-derived mononuclear cells mesenchymal stem cells, hematopoietic stem cells, endothelial progenitor cells, and cardiac stem/progenitor cells (Duelen and Sampaolesi. 2017; Eschenhagen et al., 2017; Salerno et al., 2022). In the case of MABs isolated from embryonic dorsal aorta, we observed that intra-ventricular injection was effective in reducing postischemic injury via paracrine mechanisms as transplanted cells were unable to differentiate into cardiomyocytes (CMs) (Galli D et al., 2005). Cells with similar features to MABs were isolated from murine (Galvez et al., 2008), human (Galvez et al., 2008), and canine (Cassano et al., 2012) cardiac explants and named cardiac MABs (cMABs). cMABs display pericyte markers together with cardiac transcription factors, differentiate into beating CMs and, after intraventricular delivery in ischemic murine hearts, they contribute directly to the cardiac tissue (Galvez et al., 2009). However, canine, and human MABs showed less proliferative capacity and their cardiac differentiation was modest. Although cMABs may represent a reservoir of cardiac cell progenitors, given the relative abundance of pericytes (from which MABs are derived) around the microvasculature of heart, the fact that they were prone to senescence reduced the enthusiasm for further investigation that was rather directed towards iPS derived cardiac progenitors. Indeed MAB-derived iPS cells show an intrinsic skew towards myogenic commitment, when compared to fibroblast-derived iPS cells and in addition, we could derive from them mesodermal progenitors (MiPs, as iPS cell derived-mesodermal progenitors) able to differentiate in both skeletal and cardiac myocytes, which was unexpected given the different embryonic origin of the 2 cell types (Giacomazzi et al., 2017). MiPs were amenable to gene correction and when systemically injected in dystrophic mice could improve cardiac and skeletal muscle functions. The translational applications of human MiPs for striated muscle repair have not yet been fully addressed but these initial studies are quite promising for a potential therapy to target both cardiac and skeletal dystrophic muscles.
It is largely accepted that murine animal models do not fully resemble human features of DMD degeneration and its disease progression and DMD canine models that provide insight regarding disease pathogenesis and treatment efficacy are expensive and raise many ethical issues. For these reasons, the research of new and more specific treatments aimed to prevent and counteract fibrosis and the resulting cardiomyopathies in DMD patients is hindered. Current advances in organoid technology offered an unprecedented avenue to generate cardiac organoids for studying human heart development in vitro that circumvents the limitations of 2D cell culture systems in terms of CM differentiation and maturation (Guilbert et al., 2021). Following this idea, we have generated DMD cardiac organoids that show fibrosis and adipogenesis as typical degenerative features of dystrophic cardiac phenotype upon long-term dynamic culture (up to 93 days), (Marini et al., 2022). These cardiac organoids were generated from both DMD patient-iPS cells, and its isogenic controls corrected via CRISPR/Cas9 gene editing technology (Duelen et al., 2022), revealing mitochondrial dysfunction and intracellular ROS concentrations and premature death in DMD iPS cell-CMs. The oxidative stress observed in DMD iPS cell-CMs was mainly due to the overexpression of NOX4 an enzyme family member that contributes to a wide range of pathological processes including, excessive ROS production (Duelen et al., 2022).
Despite exhibiting some clinically relevant pathological signs including, calcium overload, oxidative stress, mitochondrial dysfunction, fibrosis, and aberrant fat replacements, DMD cardiac organoid formation is a spontaneous process of CM differentiation from human iPS cells and exhibits 3D cellular structures controlled mainly by Wnt/BMP signaling factors present in culture medium. Thus, current cardiac organoid technologies have limitations due to cell heterogeneity, low reproducibility in terms of size, and in case of DMD organoids premature cell death due to likely culture conditions that enhance dystrophic CM dysfunctions. These shortcomings hamper the robustness of organoids as accurate 3D human disease cell models for toxicology studies and drug screening. It is therefore mandatory to develop advanced 3D human DMD cellular models that adequately exhibit the human disease phenotypes with high controllability, reproducibility and predictivity at an anatomical spatial resolution. Hereof 3D bioprinting technology is able to develop multi-layered models composed of different cell populations and biomaterials that are printed with a highly controlled cytoarchitecture and very short production cycle times. Thus, cardiac multilayer models containing myocardium and endocardium cell populations including endothelial cells could facilitate neovascularization for better post-transplant engraftments (Kawai et al., 2002).
The MAB ability to cross the vessels in the presence of inflammation, made them obvious candidates for systemic cell therapy of muscular dystrophies, and possibly other monogenic disorders of the mesoderm.
We consequently focused on large-scale propagation of cells. The MAB isolation from murine, canine, and human tissue had always been based on a preliminary short-time primary culture of muscle tissue fragments, essential to increase MABs proportion among different cell population outgrowth and allow more efficient selection procedures (Tonlorenzi et al., 2007; Tonlorenzi et al., 2017).
This step was particularly important for human samples, since we could derive MAB cell lines starting from very small skeletal muscles biopsies (sometimes of only 100 mg).
For murine tissues we introduced a mild enzymatic dissociation followed by a cloning step by limited dilution, in presence of a murine feeder layer. Different clones were subsequently screened respect to their proliferation and differentiation efficiency.
Murine MABs could be easily derived and propagated using basic culture media with high FBS content (DMEM supplemented with 20%FBS) in standard culture conditions (5% CO2).
Isolation and propagation of human MABs, in view of clinical use, posed a number of problems, especially regarding propagation and selection.
In most used media and in standard culture conditions human MABs undergo senescence after 10 or at most 20 population doublings (PD). After cloning, life span of derived cell lines was even shorter (less than 10 PD), on top of the fact that the use of a murine feeder layer was not recommendable.
Thanks to the use of “new generation” incubators, we could test low oxygen atmosphere, and found that 5% CO2 and 3–5% O2 tension could mimic more physiologic conditions for MAB cultures.
We than tried to identify a more efficient culture medium and found that Megacell DMEM, supplemented with 5% FBS and 5 ng/ml bFGF supported extensive proliferation, especially under physiological O2 tension, allowing a relevant extension of cells life span.
Under these conditions we could obtain more than a billion cells from a single biopsy. At that stage, cells remained able to differentiate and with a normal karyotype.
More recently we identified Myocult SF as a new serum-free medium that promotes virtually unlimited proliferation of MABs, provided that cells are spared any stress as much as possible (such as introducing the use of a hypoxic station for cell expansion and manipulation).
For what concerns selection, unfortunately MABs, as pericytes derived cells, do not express any unique antigen, except for TNAP whose expression on the surface is not constant so that prospective isolation of MABs is very inefficient.
Our actual selection procedures consist mainly of CD56+ myogenic cell fraction depletion by magnetic microbeads separation and verification of standard mesoderm surface markers.
While efforts to expand human MABs for future clinical use were ongoing, we carried out of pre-clinical work in three mouse and one dog model (Sampaolesi et al., 2003; Sampaolesi et al., 2006; Diaz-Manera et al., 2010; Tedesco et al., 2011), that showed safety and efficacy of this protocol, schematised in Figure 5.
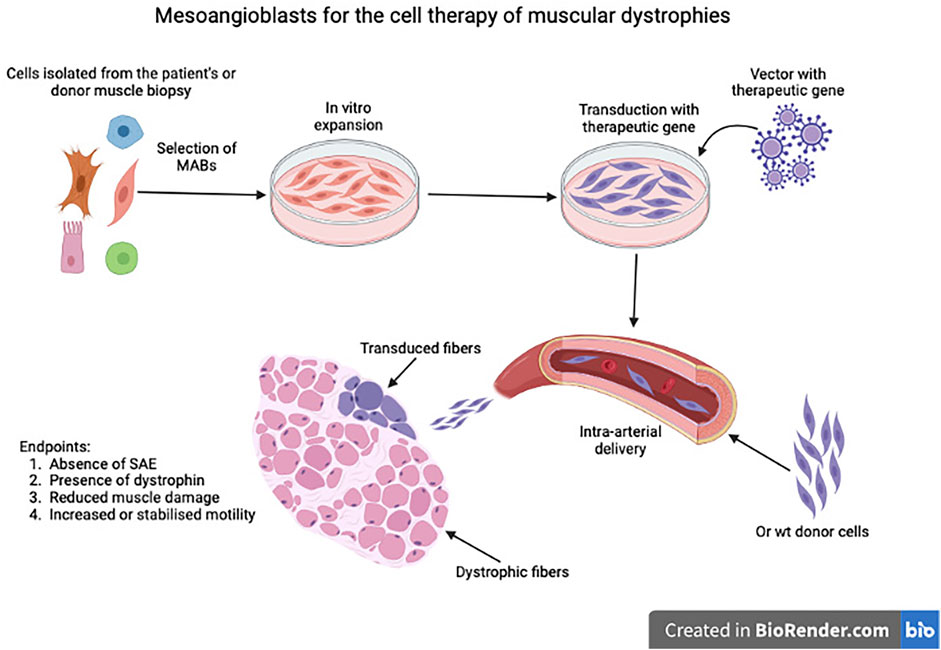
FIGURE 5. Schematic representation of the process leading to isolation, expansion, eventual transduction or mesoangioblasts as Advanced Therapy Medicinal Product (ATMP) and subsequent intra-arterial administration. SAE: Severe Adverse Event.
Results in pre-clinical models were very encouraging with all endpoints of the protocol reached. Both in mice and dogs we observed extensive expression of dystrophin or sarcoglycans or dysferlin, amelioration of the pathology and increased force of contraction, though in no case the levels of heathy animals were reached. Thus, we completed a first in man trial (Cossu et al., 2015) based upon four consecutive intra-femoral arterial administrations of increasing doses of HLA-matched donor MABs (from a sibling) in five DMD patients. We had chosen donor MAB transplantation, based on the superior outcome we observed in dogs, in comparison with autologous cells transduced with a lentivector expressing human micro-dystrophin. The trial showed safety but minimal efficacy, even though we detected, in the youngest patient donor derived dystrophin in the range detected by trials with oligonucleotides for exon skipping (Goemans et al., 2011). Differences with pre-clinical models were mainly due to the advanced age of patients (chosen for safety reasons), the ongoing treatment with steroids (that inhibits MABs adhesion to the endothelium), the lower cell dose and the different posture between human and other mammals, so that targeting leg muscles only is not sufficient to maintain posture and ambulation. The publication of the article reporting the results of the trial was generally interpreted as final evidence that cell therapy would not work for muscular dystrophy, especially with a systemic delivery that would not lead to an engraftment higher than a few percentages of the total population in the best scenario. In contrast, intra-muscular injection of satellite cell-derived progenitors showed some efficacy in localised forms such as OPMD (Oculo-Pharyngeal-Muscular-Dystrophy: Periè et al., 2014) where only pharyngeal muscles were injected.
In the meanwhile, we worked out a way to overcome the unavoidably low engraftment of transplanted cells in tissues where resident, diseased cells cannot be ablated, as in the bone marrow (Cossu et al., 2018).
Various strategies were undertaken to improve MABs engraftment in vivo in models of muscle injury and DMD. Some of these strategies exploited signalling molecules that mediate the crosstalk between the immune system and the muscle regenerative niche upon muscle injury.
Proper skeletal muscle regeneration depends on a highly coordinated sequential events as inflammation phases, tissue formation and maturation, that in turn involve the complex orchestration of tissue-resident and recruited cells. Macrophages (MPs) are the major infiltrating population in injured muscle. A simplified view suggests that activated MPs generate M1 (“classically activated”) mainly involved in the response against pathogens and M2 (“alternatively activated”) cells involved in later phases of inflammation, and tissue regeneration, playing also angiogenic and vascular protective roles in inflamed tissues (Arnold et al., 2007; Brunelli and Rovere-Querini, 2008). Factors produced by macrophages can influence skeletal muscle regeneration by stimulating satellite cell survival, proliferation, migration and differentiation (Chazaud et al., 2003; Hara et al., 2011; Chazaud, 2020). It was postulated that the same signals could promote the recruitment and engraftment of transplanted mesangioblast at the site of muscle injury.
One of such factors is nitric oxide (NO). In the skeletal muscle NO is synthesized from l-arginine by NO synthase (NOS) enzymes (Rigamonti et al., 2013). All NOS deficient mice display impaired muscle regeneration in accordance with NO non redundant role in promoting myogenic precursor cell activation and differentiation (De Palma, et al., 2010; Buono, et al., 2012; Rigamonti et al., 2013).
NO donors where therefore administered to mouse models of acute and chronic muscle injury, transplanted with MABs. In agreement with the original hypothesis, it was shown that a brief pre-treatment of with NO donors was sufficient to enhances MAB ability to migrate to the dystrophic muscle, yielding a recovery of its function (Sciorati et al., 2006). In addition, the combination of NO release and nonsteroidal anti-inflammatory drugs significantly improved the engraftment of MABs in the dystrophic muscles by increasing their homing (Brunelli et al., 2007).
Another chemokine generated at the site of tissue injury is High mobility group box 1 (HMGB1). HMGB1 is a highly conserved DNA binding protein, but in addition to its nuclear activities, HMGB1 also plays a role as a signal of tissue damage or a damage-associated molecular pattern when released passively or actively in the extracellular medium (Scaffidi, et al., 2002). HMGB1 was initially explored as a hypothetical chemoattractant for mesangioblasts in vitro and in vivo. In vitro assays revealed that recruitment of MABs required the secretion of HMGB1 and TNF-α by M1 macrophage (Lolmede et al., 2009). In vivo, it was shown that exogenous HMGB1 is sufficient to attract MABs when injected in healthy muscle, and that it improved their migration in the dystrophic muscle in a RAGE independent way (Palumbo et al., 2004). This is indeed in accordance with recent work that demonstrated that in the regenerating skeletal muscle the activity of HMGB1 is dependent on its redox state: HMGB1 exerts its chemoattractive action only when in a reduced state, via the CXCR4 receptor, while the oxidized disulfide HMGB1 acts as a proinflammatory cytokine signalling through receptors TLR4/MD-2 and RAGE (Venereau et al., 2012; Tirone et al., 2018).
We also studied adhesion of MABs to the endothelium, aiming at implementing trans-endothelial migration and thus enhance cell engraftment. We observed that steroids reduce expression of endothelium adhesion molecules and it is sufficient to remove steroids for a day to increase up to eightfold MAB adhesion in vitro. Interestingly steroids do not cause downregulation of endothelial adhesion molecules such as VCAM but their sialylation that prevents cell binding (Meggiolaro et al. in preparation). In addition, a recent report (Ku et al., 2017) showed that Pioglitazone and Rosiglitazone inhibit endothelial expression of JAMA (Junctional Adhesion Molecule A). Since we had previously shown that silencing JAMA would reduce MAB extravasation (Giannotta et al., 2014 EMBO Mol. Med. 2014) and considering that Pioglitazone is used in paediatric clinical trials, that would be an ideal candidate for a combinatorial therapy.
Even if all the above strategies enhance MAB engraftment in murine models of muscle dystrophy, the extent of engraftment in human muscle would remain so low that improving engraftment alone would not lead to clinical efficacy.
Thus, we completed a study using a new strategy of genetic correction that compensates for the poor engraftment of donor cells in skeletal muscle. This is a cell-mediated exon skipping. DMD MABs are transduced with a lentivector expressing a snRNA engineered to skip exon 51 (De Angelis et al., 2002). Since the snRNA is produced by a donor nucleus, assembles in the cytoplasm and then enters all the neighbouring nuclei, this mechanism amplifies several folds the production of dystrophin both in vitro and in immune deficient DMD mice with a skippable mutation of exon 51 (Galli et al. in preparation) resulting in a much higher production of dystrophin than when healthy cells, that can only produce their own dystrophin, are injected. This is schematically shown in Figure 6.
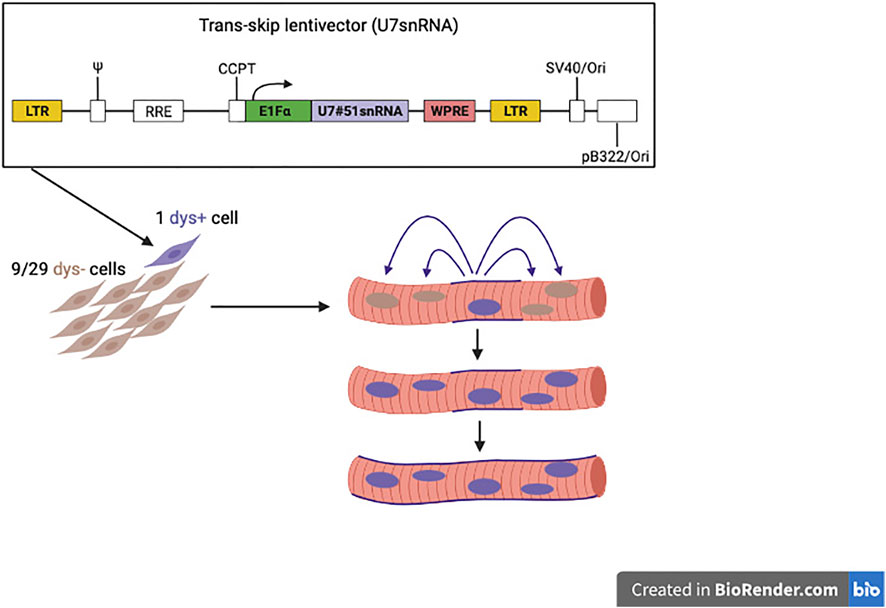
FIGURE 6. Novel strategy to reach quasi-normal expression of dystrophin, with donor cells working as “trojan horses” since the few nuclei penetrated inside the host fibre correct also the resident nuclei, like the few Trojan warriors penetrated inside the horse donated by the Greeks, surprised the Troy residents during the sleep and managed to conquered the city.
We are testing safety of genetically corrected (with the strategy described above) autologous MABs by intramuscular injection in a foot muscle of five non-ambulant DMD patients. In case of dystrophin production ≥10% of a healthy muscle, cells will also be injected in the thumb muscle, whose increased force of contraction would ameliorate the quality of patients’ life. The trial started in Manchester in the fall 2018 but was halted by COVID and the closure of the GMP facility in Manchester and has now restarted utilising a new GMP facility in Switzerland and the San Raffaele Hospital as clinical centre.
Duringthese twenty years, all the authors contributed to the work LDA, MGM, MC, RT and LB developed the methods to isolate and culture mesoangioblasts FC, SCP and YT were responsible for the clinical translation, all other authors studied the biology of these cells both in vitro and after transplantation in animal modelsGC, RT, SB, MS and GM wrote the ms SB and GM drew the figures
Work discussed in this review was supported by a grant from ERC AIG 844952. The views expressed in this publication are those of the authors and not necessarily those of the funding bodies.
This review is dedicated to the late Paolo Bianco, a pioneer in the field, who influenced this as many other areas of research on the mesoderm We also thank Marco Bianchi (San Raffaele Hospital), Irene Bozzoni (University of Rome Sapienza), Margaret Buckingham (Istitut Pasteur), Gill Butler Browne (Sorbonne Universite Inserm Institut de Myologie Paris), Stephan Blot (Ecole Veterinarie de Macon Alfort Roberto Bottinelli (University of Pavia), Emilio Clementi (University of Milan), Elisabetta Dejana (Institute FIRC of Molecular Oncology), Dario Di Francesco (University of Milan), (Thierry Jaffredo Universite Paris VI), Roberto Latini (Istituto MarioNegri Milan), Vincent Mouly (Sorbonne Universite Inserm Institut de Myologie Paris), Mitsuo Oshimura (Tottori University), Giuseppe Peretti Istituto Ortopedico Galeazzi, Milano), Frederique Relaix (Université de Paris Est), Mara Riminucci (University of Rome Sapienza), Patrizia Rovere Querini (San Raffaele Hospital), Verena Schoewel (Max Delbruck, Charité Berlin), Hubert Smeets (Maastricht University), Simone Spüler (Max Delbruck, Charité Berlin), Sharaghim Tajbakhsh (Istitut Pasteur) and Florence van Tienen (Maastricht University) for collaboration and advise through the years
MC is employed by Lunapore. GG and HH by Cell Fiber.Co. GR by Roche and UR by Gupra.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer (KV) declared a shared parent affiliation with the author(s) (MQ) to the handling editor at the time of review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abu-Issa, R., and Kirby, M. L. (2007). Heart field: From mesoderm to heart tube. Annu. Rev. Cell Dev. Biol. 23, 45–68. doi:10.1146/annurev.cellbio.23.090506.123331
Arnold, L., Henry, A., Poron, F., Baba-Amer, Y., Rooijen, N. v., Plonquet, A., et al. (2007). Inflammatory monocytes recruited after skeletal muscle injury switch into anti-inflammatory macrophages to support myogenesis. J. Exp. Med. 204, 1057–1069. doi:10.1084/jem.20070075
Azzoni, E., Conti, V., Campana, L., Dellavalle, A., Adams, R. H., Cossu, G., et al. (2014). Hemogenic endothelium generates mesoangioblasts that contribute to several mesodermal lineages in vivo. Development 141, 1821–1834. doi:10.1242/dev.103242
Bajard, L., Relaix, F., Lagha, M., Rocancourt, D., Daubas, P., and Buckingham, M. E. (2006). A novel genetic hierarchy functions during hypaxial myogenesis: Pax3 directly activates Myf5 in muscle progenitor cells in the limb. Genes Dev. 20, 2450–2464. doi:10.1101/gad.382806
Besson, V., Smeriglio, P., Wegener, A., Relaix, F., Nait Oumesmar, B., Sassoon, D. A., et al. (2011). Smeriglio, P., Wegener, A., Relaix, F., Nait Oumesmar, B., Sassoon, D.A. and, Marazzi, GPW1 gene/paternally expressed gene 3 (PW1/Peg3) identifies multiple adult stem and progenitor cell populations. Proc. Natl. Acad. Sci. U. S. A. 108, 11470–11475. doi:10.1073/pnas.1103873108
Bi, W. L., Parysek, L. M., Warnick, R., and Stambrook, P. J. (1993). In vitro evidence that metabolic cooperation is responsible for the bystander effect observed with HSV tk retroviral gene therapy. Hum Gene Therapy, 4, 725–731. doi:10.1089/hum.1993.4.6-725
Bianco, P. (2014). “Mesenchymal” stem cells. Annu. Rev. Cell Dev. Biol. 30, 677–704. doi:10.1146/annurev-cellbio-100913-013132
Bianco, P., and Cossu, G. (1999). Uno, nessuno e centomila: Searching for the identity of mesodermal progenitors. Exp. Cell Res. 251, 257–263. doi:10.1006/excr.1999.4592
Biressi, S., Molinaro, M., and Cossu, G. (2007). Cellular heterogeneity during vertebrate skeletal muscle development. Dev. Biol. 308, 281–293. doi:10.1016/j.ydbio.2007.06.006
Boisset, J.-C., van Cappellen, W., Andrieu-Soler, C., Galjart, N., Dzierzak, E., and Robin, C. (2010). In vivo imaging of haematopoietic cells emerging from the mouse aortic endothelium. Nature 464, 116–120. doi:10.1038/nature08764
Bonfanti, C., Rossi, G., Tedesco, F. S., Giannotta, M., Benedetti, S., Tonlorenzi, R., et al. (2015). PW1/Peg3 expression regulates key properties that determine mesoangioblast stem cell competence. Nat. Commun. 6, 6364. doi:10.1038/ncomms7364
Bonfanti, P., Barrandon, Y., and Cossu, G. (2012). 'Hearts and bones': The ups and downs of 'plasticity' in stem cell biology. EMBO Mol. Med. 4, 353–361. doi:10.1002/emmm.201200220
Breton, M., Li, Z. L., Paulin, D., Harris, J. A., Rieger, F., Pinçon-Raymond, M., et al. (1995). Myotube driven myogenic recruitment of cells during in vitro myogenesis. Dev. Dyn. 202, 126–136. doi:10.1002/aja.1002020204
Brunelli, S., and Rovere-Querini, P. (2008). The immune system and the repair of skeletal muscle. Pharmacol. Res. 58, 117–121. doi:10.1016/j.phrs.2008.06.008
Brunelli, S., Sciorati, C., D'Antona, G., Innocenzi, A., Covarello, D., Galvez, B. G., et al. (2007). Nitric oxide release combined with nonsteroidal anti-inflammatory activity prevents muscular dystrophy pathology and enhances stem cell therapy. Proc. Natl. Acad. Sci. U. S. A. 104, 264–269. doi:10.1073/pnas.0608277104
Buckingham, M. (2017). Gene regulatory networks and cell lineages that underlie the formation of skeletal muscle. Proc. Natl. Acad. Sci. U. S. A. 114, 5830–5837. doi:10.1073/pnas.1610605114
Buono, R., Vantaggiato, C., Pisa, V., Azzoni, E., Bassi, M. T., Brunelli, S., et al. (2012). Nitric oxide sustains long-term skeletal muscle regeneration by regulating fate of satellite cells via signaling pathways requiring Vangl2 and cyclic GMP. Stem Cells 30, 197–209. doi:10.1002/stem.783
Camps, J., Breuls, N., Sifrim, A., Giarratana, N., Corvelyn, M., Danti, L., et al. (2020). Interstitial cell remodeling promotes aberrant adipogenesis in dystrophic muscles. Cell Rep. 31, 107597. doi:10.1016/j.celrep.2020.107597
Cappellari, O., Benedetti, S., Innocenzi, A., Tedesco, F. S., Moreno-Fortuny, A., Ugarte, G., et al. (2013). Dll4 and PDGF-BB convert committed skeletal myoblasts to pericytes without erasing their myogenic memory. Dev. Cell 24, 586–599. doi:10.1016/j.devcel.2013.01.022
Cassano, M., Berardi, E., Crippa, S., Toelen, J., Barthelemy, I., Micheletti, R., et al. (2012). Alteration of cardiac progenitor cell potency in GRMD dogs. Cell Transpl. 21, 1945–1967. doi:10.3727/096368912X638919
Chazaud, B. (2020). Inflammation and skeletal muscle regeneration: Leave it to the macrophages!. Trends Immunol. 41, 481–492. doi:10.1016/j.it.2020.04.006
Chazaud, B., Sonnet, C., Lafuste, P., Bassez, G., Rimaniol, A.-C., Poron, F., et al. (2003). Satellite cells attract monocytes and use macrophages as a support to escape apoptosis and enhance muscle growth. J. Cell Biol. 163, 1133–1143. doi:10.1083/jcb.200212046
Chevallier, A., Kieny, M., Mauger, A., and Mauger, A. (1977). Limb-somite relationship: Origin of the limb musculature. Development 41, 245–258. doi:10.1242/dev.41.1.245
Chien, K. R., Frisén, J., Fritsche-Danielson, R., Melton, D. A., Murry, C. E., and Weissman, I. L. (2019). Regenerating the field of cardiovascular cell therapy. Nat. Biotechnol. 37, 232–237. doi:10.1038/s41587-019-0042-1
Christ, B., Jacob, H. J., and Jacob, M. (1977). Experimental analysis of the origin of the wing musculature in avian embryos. Anat. Embryol. 150, 171–186. doi:10.1007/BF00316649
Cossu, G., and Bianco, P. (2003). Mesoangioblasts: Vascular progenitors for extravascular mesodermal tissues. Curr. Opin. Genet. Dev. 13, 537–542. doi:10.1016/j.gde.2003.08.001
Cossu, G., Birchall, M., Brown, T., De Coppi, P., Culme-Seymour, E., Gibbon, S., et al. (2018). Lancet commission: Stem cells and regenerative medicine. Lancet 391, 883–910. doi:10.1016/S0140-6736(17)31366-1
Cossu, G., Previtali, S. C., Napolitano, S., Cicalese, M. P., Tedesco, F. S., Nicastro, F., et al. (2015). Intra-arterial transplantation of HLA-matched donor mesoangioblasts in Duchenne muscular dystrophy. EMBO Mol. Med. 7, 1513–1528. doi:10.15252/emmm.201505636
Cossu, G., and Molinaro, M. (1987). Cell heterogeneity in the myogenic lineage. Curr. Top. Dev. Biol. 23, 185–208. doi:10.1016/s0070-2153(08)60625-0
Costamagna, D., Quattrocelli, M., van Tienen, F., Umans, L., de Coo, I. F., Zwijsen, A., et al. (2016). Smad1/5/8 are myogenic regulators of murine and human mesoangioblasts. J. Mol. Cell Biol. 8, 73–87. doi:10.1093/jmcb/mjv059
Costamagna, D., Duelen, R., Penna, F., Neumann, D., Costelli, P., and Sampaolesi, M. (2020). Interleukin-4 administration improves muscle function, adult myogenesis, and lifespan of colon carcinoma-bearing mice. J. Cachexia Sarcopenia Muscle 11, 783–801. doi:10.1002/jcsm.12539
Costantini, M., Testa, S., Fornetti, E., Fuoco, C., Sanchez Riera, C., Nie, M., et al. (2021). Biofabricating murine and human myo-substitutes for rapid volumetric muscle loss restoration. EMBO Mol. Med. 13, e12778. doi:10.15252/emmm.202012778:
Couly, G. F., Coltey, P. M., and Le Douarin, N. (1993). MThe triple origin of skull in higher vertebrates: A study in quail-chick chimeras. Development 117, 409–429. doi:10.1242/dev.117.2.409
De Angelis, F. G., Sthandier, O., Berarducci, B., Toso, S., Galluzzi, G., Ricci, E., et al. (2002). Chimeric snRNA molecules carrying antisense sequences against the splice junctions of exon 51 of the dystrophin pre-mRNA induce exon skipping and restoration of a dystrophin synthesis in Delta 48-50 DMD cells. Proc. Natl. Acad. Sci. U. S. A. 99, 9456–9461. doi:10.1073/pnas.142302299
De Angelis, L., Berghella, L., Coletta, M., Lattanzi, L., Zanchi, M., Cusella-De Angelis, M. G., et al. (1999). C., and Cossu, GSkeletal myogenic progenitors originating from embryonic dorsal aorta co-express endothelial and myogenic markers and contribute to postnatal muscle growth and regeneration. J. Cell Biol. 147, 869–878. doi:10.1083/jcb.147.4.869
De Palma, C., Falcone, S., Pisoni, S., Cipolat, S., Panzeri, C., Pambianco, S., et al. (2010). Nitric oxide inhibition of Drp1-mediated mitochondrial fission is critical for myogenic differentiation. Cell Death Differ. 17, 1684–1696. doi:10.1038/cdd.2010.48
Dell'Orso, S., Juan, A. H., Ko, K.-D., Naz, F., Perovanovic, J., Gutierrez-Cruz, G., et al. (2019). Single cell analysis of adult mouse skeletal muscle stem cells in homeostatic and regenerative conditions. Development 146, dev174177.
Dellavalle, A., Maroli, G., Covarello, D., Azzoni, E., Innocenzi, A., Perani, L., et al. (2011). Pericytes resident in postnatal skeletal muscle differentiate into muscle fibres and generate satellite cells. Nat. Commun. 2, 499. doi:10.1038/ncomms1508
Dellavalle, A., Sampaolesi, M., Tonlorenzi, R., Tagliafico, E., Sacchetti, B., Perani, L., et al. (2007). Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat. Cell Biol. 9, 255–267. doi:10.1038/ncb1542
Dezawa, M., Ishikawa, H., Itokazu, Y., Yoshihara, T., Hoshino, M., Takeda, S., et al. (2005). Bone marrow stromal cells generate muscle cells and repair muscle degeneration. Science 309, 314–317. doi:10.1126/science.1110364
Díaz-Manera, J., Touvier, T., Dellavalle, A., Tonlorenzi, R., Tedesco, F. S., Messina, G., et al. (2010). Partial dysferlin reconstitution by adult murine mesoangioblasts is sufficient for full functional recovery in a murine model of dysferlinopathy. Cell Death Dis. 1, e61. doi:10.1038/cddis.2010.35
Duelen, R., Costamagna, D., Gilbert, G., De Waele, L., Goemans, N., Desloovere, K., et al. (2022). Human iPSC model reveals a central role for NOX4 and oxidative stress in Duchenne cardiomyopathy. Stem Cell Rep. 17, 352–368. doi:10.1016/j.stemcr.2021.12.019
Duelen, R., and Sampaolesi, M. (2017). Stem cell technology in cardiac regeneration: A pluripotent stem cell promise. EBioMedicine 16, 30–40. doi:10.1016/j.ebiom.2017.01.029
Engquist, E. N., and Zammit, P. S. (2021). The satellite cell at 60: The foundation years. J. Neuromuscul. Dis. 8, S183–S203. doi:10.3233/JND-210705
Eschenhagen, T., Bolli, R., Braun, T., Field, L. J., Fleischmann, B. K., Frisén, J., et al. (2017). Cardiomyocyte regeneration: A consensus statement. Circulation 136, 680–686. doi:10.1161/CIRCULATIONAHA.117.029343
Esner, M., Meilach, S. M., Relaix, F., Nicolas, J. F., Cossu, G., and Buckingham, M. (2006). Smooth muscle of the dorsal aorta shares a common clonal origin with skeletal muscle of the myotome. Development 133, 737–749. doi:10.1242/dev.02226
Ferrari, G., Cusella-De Angelis, G., Coletta, M., Paolucci, E., Stornaiuolo, A., Cossu, G., et al. (1998). Muscle regeneration by bone marrow-derived myogenic progenitors. Science 279, 1528–1530. doi:10.1126/science.279.5356.1528
Galli, D, , Innocenzi, A., Staszewsky, L., Zanetta, L., Sampaolesi, M., Bai, A., et al. (2005). Mesoangioblasts, vessel-associated multipotent stem cells, repair the infarcted heart by multiple cellular mechanisms: A comparison with bone marrow progenitors, fibroblasts, and endothelial cells. Arterioscler. Thromb. Vasc. Biol. 25, 692–697. doi:10.1161/01.ATV.0000156402.52029.ce
Gálvez, B. G., Covarello, D., Tonlorenzi, R., Brunelli, S., Dellavalle, A., Crippa, S., et al. (2009). Human cardiac mesoangioblasts isolated from hypertrophic cardiomyopathies are greatly reduced in proliferation and differentiation potency. Cardiovasc. Res. 83, 707–716. doi:10.1093/cvr/cvp159
Galvez, B. G., Sampaolesi, M., Barbuti, A., Crespi, A., Covarello, D., Brunelli, S., et al. (2008). Cardiac mesoangioblasts are committed, self-renewable progenitors, associated with small vessels of juvenile mouse ventricle. Cell Death Differ. 15, 1417–1428. doi:10.1038/cdd.2008.75
Gerli, M. F. M., Moyle, L. A., Benedetti, S., Ferrari, G., Ucuncu, E., Ragazzi, M., et al. (2019). Combined notch and PDGF signaling enhances migration and expression of stem cell markers while inducing perivascular cell features in muscle satellite Cells.Combined notch and PDGF signalling enhances expression of stem cell markers while inducing perivascular cell features in muscle satellite cells. Stem Cell Rep. 12, 461–473. doi:10.1016/j.stemcr.2019.01.007
Giacomazzi, G., Giovannelli, G., Rotini, A., Costamagna, D., Quattrocelli, M., and Sampaolesi, M. (2021). Isolation of mammalian mesoangioblasts: A Subset of pericytes with myogenic potential. Methods Mol. Biol. 2235, 155–167. doi:10.1007/978-1-0716-1056-5_11
Giacomazzi, G., Holvoet, B., Trenson, S., Caluwé, E., Kravic, B., Grosemans, , H., et al. (2017). MicroRNAs promote skeletal muscle differentiation of mesodermal iPSC-derived progenitors. Nat. Commun. 8, 1249. doi:10.1038/s41467-017-01359-w
Giannotta, M., Benedetti, S., Tedesco, F. S., Corada, M., Trani, M., D’Antuono, R., et al. (2014). Targeting endothelial Junctional Adhesion Molecule-A/EPAC/Rap-1 axis as a novel strategy to increase stem cell engraftment in dystrophic muscles. EMBO Mol. Med. 6, 239–258. doi:10.1002/emmm.201302520
Gibson, A. J., Karasinski, J., Relvas, J., Moss, J., Sherratt, T. G., Strong, P. N., et al. (1995). Dermal fibroblasts convert to a myogenic lineage in mdx mouse muscle. J. Cell Sci. 108, 207–214. doi:10.1242/jcs.108.1.207
Gilbert, G., Kadur Nagaraju, C., Duelen, R., Amoni, M., Bobin, P., Eschenhagen, T., et al. (2021). Incomplete assembly of the dystrophin-associated protein complex in 2D and 3D-cultured human induced pluripotent stem cell-derived cardiomyocytes. Front. Cell Dev. Biol. 9, 737840. doi:10.3389/fcell.2021.737840
Giordani, L., He, G. J., Negroni, E., Sakai, H., Law, J. Y. C., Siu, M. M., et al. (2019). High-dimensional single-cell cartography reveals novel skeletal muscle-resident cell populations. Mol. Cell 74, 609–621. doi:10.1016/j.molcel.2019.02.026
Jalal, S., Dastidar, S., and Tedesco, F. S. (2021). Advanced models of human skeletal muscle differentiation, development and disease: Three-dimensional cultures, organoids and beyond. Curr. Opin. Cell Biol. 73, 92–104. doi:10.1016/j.ceb.2021.06.004
Joe, A. W., Yi, L., Natarajan, A., Le Grand, F., So, L., Wang, J., et al. (2010). Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 12, 153–163. doi:10.1038/ncb2015
Kawai, Y., Tohyama, S., Arai, K., Tamura, T., Soma, Y., Fukuda, K., et al. (2002). Scaffold-free tubular engineered heart tissue from human induced pluripotent stem cells using bio-3D printing technology in vivo. Front. Cardiovasc Med. 8, 806215. doi:10.3389/fcvm.2021.806215,
Keefe, A. C., Lawson, J. A., Flygare, S. D., Fox, Z. D., Colasando, M. P., Mathew, S. J., et al. (2015). Muscle stem cells contribute to myofibres in sedentary adult mice. Nat. Commun. 6, 7087. doi:10.1038/ncomms8087
Kelly, R., Alonso, S., Tajbakhsh, S., Cossu, G., and Buckingham, M. (1995). Myosin light chain 3F regulatory sequences confer regionalized cardiac and skeletal muscle expression in transgenic mice. J. Cell Biol. 129, 383–396. doi:10.1083/jcb.129.2.383
Kim, K., Doi, A., Wen, B., Ng, K., Zhao, R., Cahan, P., et al. (2010). Epigenetic memory in induced pluripotent stem cells. Nature 467, 285–290. doi:10.1038/nature09342
Ku, Y. H., Cho, B. J., Kim, M. J., Lim, S., Park, Y. J., Jang, H. C., et al. (2017). Rosiglitazone increases endothelial cell migration and vascular permeability through Akt phosphorylation. BMC Pharmacol. Toxicol. 18, 62. doi:10.1186/s40360-017-0169-y
Kuroiwa, Y., Kaneko-Ishino, T., Kagitani, F., Kohda, T., Li, L. L., TadaM., , et al. (1996). Peg3 imprinted gene on proximal chromosome 7 encodes for a zinc finger protein. Nat. Genet. 12, 186–190. doi:10.1038/ng0296-186
Lancrin, C., Sroczynska, P., Stephenson, C., Allen, T., Kouskoff, V., and Lacaud, G. (2009). The haemangioblast generates haematopoietic cells through a haemogenic endothelium stage. Nature 457, 892–895. doi:10.1038/nature07679
Liu, N., Garry, G. A., Li, S., Bezprozvannaya, S., Sanchez-Ortiz, E., Chen, B., et al. (2017). A Twist2-dependent progenitor cell contributes to adult skeletal muscle. Nat. Cell Biol. 19, 202–213. doi:10.1038/ncb3477
Lolmede, K., Campana, L., Vezzoli, M., Bosurgi, L., Tonlorenzi, R., Clementi, E., et al. (2009). Inflammatory and alternatively activated human macrophages attract vessel-associated stem cells, relying on separate HMGB1- and MMP-9-dependent pathways. J. Leukoc. Biol. 85, 779–787. doi:10.1189/jlb.0908579
Marini, V., Marino, F., Aliberti, F., Giarratana, N., Pozzo, E., Duelen, R., et al. (2022). Long-term culture of patient-derived cardiac organoids recapitulated Duchenne Duchenne muscular dystrophy cardiomyopathy and disease progression. Front. Cell Dev. Biol. 10, 878311. doi:10.3389/fcell.2022.878311
Mauro, A. (1961). Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 9, 493–495. doi:10.1083/jcb.9.2.493
Messina, G., Sirabella, D., Monteverde, S., Galvez, B. G., Tonlorenzi, R., Schnapp, E., et al. (2009). Skeletal muscle differentiation of embryonic mesoangioblasts requires Pax3 activity. Stem Cells 27, 157–164. doi:10.1634/stemcells.2008-0503
Minasi, M. G., Riminucci, M., De Angelis, L., Borello, U., Berarducci, B., Innocenzi, A., et al. (2002). The meso-angioblast: A multipotent, self-renewing cell that originates from the dorsal aorta and differentiates into most mesodermal tissues. Development 129, 2773–2783. doi:10.1242/dev.129.11.2773
Mitchell, K. J., Pannérec, A., Cadot, B., Parlakian, A., Besson, V., Gomes, E. R., et al. (2010). Identification and characterization of a non-satellite cell muscle resident progenitor during postnatal development. Nat. Cell Biol. 12, 257–266. doi:10.1038/ncb2025
Monsoro-Burq, A. H., Bontoux, M., Teillet, M. A., and Le Douarin, N. (1994). MHeterogeneity in the development of the vertebra. Proc. Natl. Acad. Sci. U. S. A. 91, 10435–10439. doi:10.1073/pnas.91.22.10435
Palumbo, R., Sampaolesi, M., Marchis, F. D., Tonlorenzi, R., Colombetti, S., Mondino, A., et al. (2004). Extracellular HMGB1, a signal of tissue damage, induces mesoangioblast migration and proliferation. J. Cell Biol. 164, 441–449. doi:10.1083/jcb.200304135
Pannérec, A., Formicola, L., Besson, V., Marazzi, G., and Sassoon, D. A. (2013). Defining skeletal muscle resident progenitors and their cell fate potentials. Development 140, 2879–2891. doi:10.1242/dev.089326
Pawlikowski, B., Pulliam, C., Dalla Betta, N., Kardon, G., and Olwin, B. (2015). Pervasive satellite cell contribution to uninjured adult muscle fibers. Skelet. Muscle 5, 42. doi:10.1186/s13395-015-0067-1
Périé, S., Trollet, C., Mouly, V., Vanneaux, V., Mamchaoui, K., Bouazza, B., et al. (2014). Autologous myoblast transplantation for oculopharyngeal muscular dystrophy: A phase I/IIa clinical study. Mol. Ther. 22, 219–225. doi:10.1038/mt.2013.155
Quattrocelli, M., Costamagna, D., Giacomazzi, G., Camps, J., and Sampaolesi, M. (2014). Notch signaling regulates myogenic regenerative capacity of murine and human mesoangioblasts. Cell Death Dis. 5, e1448. doi:10.1038/cddis.2014.401
Quattrocelli, M., Giacomazzi, G., Broeckx, S. Y., Ceelen, L., Bolca, S., Spaas, J. H., et al. (2016). Equine-induced pluripotent stem cells retain lineage commitment toward myogenic and chondrogenic fates. Stem Cell Rep. 6, 55–63. doi:10.1016/j.stemcr.2015.12.005
Quattrocelli, M., Palazzolo, G., Perini, I., Crippa, S., Cassano, M., and Sampaolesi, M. (2012). Mouse and human mesoangioblasts: Isolation and characterization from adult skeletal muscles. Methods Mol. Biol. 798, 65–76. doi:10.1007/978-1-61779-343-1_4
Quattrocelli, M., Swinnen, M., Giacomazzi, G., Camps, J., Barthélemy, I., Ceccarelli, G., et al. (2015). Mesodermal iPSC-derived progenitor cells functionally regenerate cardiac and skeletal muscle. J. Clin. Invest. 125, 4463–4482. doi:10.1172/JCI82735
Relaix, F., Polimeni, M., Rocancourt, D., Ponzetto, C., Schäfer, B. W., and Buckingham, M. (2003). The transcriptional activator PAX3-FKHR rescues the defects of Pax3 mutant mice but induces a myogenic gain-of-function phenotype with ligand-independent activation of Met signaling in vivo. Genes Dev. 17, 2950–2965. doi:10.1101/gad.281203
Relaix, F., Weng, X., Marazzi, G., Yang, E., Copeland, N., JeNkiNsN., , et al. (1996). Pw1, a novel zinc finger gene implicated in the myogenic and neuronal lineages. Dev. Biol. 177, 383–396. doi:10.1006/dbio.1996.0172
Rigamonti, E., Touvier, T., Clementi, E., Manfredi, A. A., Brunelli, S., and Rovere-Querini, P. (2013). Requirement of inducible nitric oxide synthase for skeletal muscle regeneration after acute damage. J. Immunol. 190, 1767–1777. doi:10.4049/jimmunol.1202903
Ronzoni, F. L., Giarratana, N., Crippa, S., Quattrocelli, M., Cassano, M., Ceccarelli, G., et al. (2021). Guide cells support muscle regeneration and affect neuro-muscular junction organization. Int. J. Mol. Sci. 22 (4), 1939. doi:10.3390/ijms22041939
Roston, R. A., Lickorish, D., and Buchholtz, E. A. (2013). Anatomy and age estimation of an early blue whale (balaenoptera musculus) fetus. Anat. Rec. 296, 709–722. doi:10.1002/ar.22678
Rotini, A., Martínez-Sarrà, E., Duelen, R., Costamagna, D., Di Filippo, E. S., Giacomazzi, G., et al. (2018). Aging affects the in vivo regenerative potential of human mesoangioblasts. Aging Cell 17 (2), e12714. doi:10.1111/acel.12714
Sacchetti, B., Funari, A., Remoli, C., Giannicola, G., Kogler, G., Liedtke, S., et al. (2016). No identical “mesenchymal stem cells” at different times and sites: Human committed progenitors of distinct origin and differentiation potential are incorporated as adventitial cells in microvessels. Stem Cell Rep. 6, 897–913. doi:10.1016/j.stemcr.2016.05.011
Saccone, V., Consalvi, S., Giordani, L., Mozzetta, C., Barozzi, I., Sandona, M., et al. (2014). HDAC-regulated myomiRs control BAF60 variant exchange and direct the functional phenotype of fibro-adipogenic progenitors in dystrophic muscles. Genes Dev. 28, 841–857. doi:10.1101/gad.234468.113
Safadi, A., Livne, E., Silbermann, M., and Reznick, A. Z. (1991). Activity of alkaline phosphatase in rat skeletal muscle localized along the sarcolemma and endothelial cell membranes. J. Histochem. Cytochem. 39, 199–203. doi:10.1177/39.2.1987264
Salerno, N., Salerno, L., Marino, F., Scalise, M., Chiefalo, A., Panuccio, G., et al. (2022). Myocardial regeneration protocols towards the routine clinical scenario: An unseemly path from bench to bedside. EClinicalMedicine 50, 101530. doi:10.1016/j.eclinm.2022.101530
Salvatori, G., Lattanzi, L., Coletta, M., Aguanno, S., Vivarelli, E., Kelly, R., et al. (1995). Myogenic conversion of mammalian fibroblasts induced by differentiating muscle cells. J. Cell Sci. 108, 2733–2739. doi:10.1242/jcs.108.8.2733
Sampaolesi, M., Torrente, Y., Innocenzi, A., Tonlorenzi, R., D'Antona, G., Pellegrino, M. A., et al. (2003). Cell therapy of alpha-sarcoglycan null dystrophic mice through intra-arterial delivery of mesoangioblasts. Science 301, 487–492. doi:10.1126/science.1082254
Sampaolesi, M., Blot, S., D’Antona, G., Granger, N., Tonlorenzi, R., Innocenzi, A., et al. (2006). Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature 444, 574–579. doi:10.1038/nature05282
Sanes, J. R., Rubenstein, J. L., and Nicolas, J. F. (1986). Use of a recombinant retrovirus to study post-implantation cell lineage in mouse embryos. EMBO J. 5, 3133–3142. doi:10.1002/j.1460-2075.1986.tb04620.x
Santoni de Sio, F. R., Gritti, A., Cascio, P., Neri, M., Sampaolesi, M., Galli, C., et al. (2008). Lentiviral vector gene transfer is limited by the proteasome at postentry steps in various types of stem cells. Stem Cells 26 (8), 2142–2152. doi:10.1634/stemcells.2007-0705
Scaffidi, P., Misteli, T., and Bianchi, M. E. (2002). Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195. doi:10.1038/nature00858
Sciorati, C., Galvez, B. G., Brunelli, S., Tagliafico, E., Ferrari, S., Cossu, G., et al. (2006). Ex vivo treatment with nitric oxide increases mesoangioblast therapeutic efficacy in muscular dystrophy. J. Cell Sci. 119, 5114–5123. doi:10.1242/jcs.03300
Stallcup, W. B. (2013). Bidirectional myoblast-pericyte plasticity. Dev. Cell 24, 563–564. doi:10.1016/j.devcel.2013.02.018
Stoklund Dittlau, K., Krasnow, E. N., Fumagalli, L., Vandoorne, T., Baatsen, P., Kerstens, A., et al. (2021). Human motor units in microfluidic devices are impaired by FUS mutations and improved by HDAC6 inhibition. Stem Cell Rep. 16, 2213–2227. doi:10.1016/j.stemcr.2021.03.029
Swiers, G., Rode, C., Azzoni, E., and de Bruijn, M. F. (2013). A short history of hemogenic endothelium. Blood Cells Mol. Dis. 51, 206–212. doi:10.1016/j.bcmd.2013.09.005
Tagliafico, E., Brunelli, S., Bergamaschi, A., De Angelis, L., Scardigli, R., Galli, D., et al. (2004). TGFbeta/BMP activate the smooth muscle/bone differentiation programs in mesoangioblasts. J. Cell Sci. 117, 4377–4388. doi:10.1242/jcs.01291
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi:10.1016/j.cell.2007.11.019
Tamaki, T., Okada, Y., Uchiyama, Y., Tono, K., Masuda, M., Wada, M., et al. (2007). Synchronized reconstitution of muscle fibers, peripheral nerves and blood vessels by murine skeletal muscle-derived CD34(-)/45 (-) cells. Histochem. Cell Biol. 128, 349–360. doi:10.1007/s00418-007-0331-5
Tanaka, K. K., Hall, J. K., Troy, A. A., Cornelison, D. D., Majka, S. M., and Olwin, B. B. (2009). Syndecan-4-expressing muscle progenitor cells in the SP engraft as satellite cells during muscle regeneration. Cell Stem Cell 4, 217–225. doi:10.1016/j.stem.2009.01.016
Tedesco, F. S., Gerli, M. F., Perani, L., Benedetti, S., Ungaro, F., Cassano, M., et al. (2012). Transplantation of genetically corrected human iPSC-derived progenitors in mice with limb-girdle muscular dystrophy. Sci. Transl. Med. 4, 140ra89. doi:10.1126/scitranslmed.3003541
Tedesco, F. S., Hoshiya, H., D’Antona, G., Gerli, M. F. M., Messina, G., Antonini, S., et al. (2011). Stem cell-mediated transfer of a human artificial chromosome ameliorates muscular dystrophy. Sci. Transl. Med. 3, 96ra78. doi:10.1126/scitranslmed.3002342
Tirone, M., Tran, N. L., Ceriotti, C., Gorzanelli, A., Canepari, M., Bottinelli, R., et al. (2018). High mobility group box 1 orchestrates tissue regeneration via CXCR4. J. Exp. Med. 215, 303–318. doi:10.1084/jem.20160217
Tonlorenzi, R., Dellavalle, A., Schnapp, E., Cossu, G., and Sampaolesi, M. (2007). Isolation and characterization of mesoangioblasts from mouse, dog and human tissues. Curr. Protoc. Stem Cell Biol. 3, 2B. doi:10.1002/9780470151808.sc02b01s3
Tonlorenzi, R., Rossi, G., and Messina, G. (2017). Isolation and characterization of vessel-associated stem/progenitor cells from skeletal muscle. Methods Mol. Biol. 1556, 149–177. doi:10.1007/978-1-4939-6771-1_8
Uezumi, A., Fukada, S., Yamamoto, N., Takeda, S., and Tsuchida, K. (2010). Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol. 12, 143–152. doi:10.1038/ncb2014
Ugarte, G., Cappellari, O., Perani, L., Pistocchi, A., and Cossu, G. (2012). Noggin recruits mesoderm progenitors from the dorsal aorta to a skeletal; myogenic fate. Dev. Biol. 365, 91–100. doi:10.1016/j.ydbio.2012.02.015
Venereau, E., Casalgrandi, M., Schiraldi, M., Antoine, D. J., Cattaneo, A., De Marchis, F., et al. (2012). Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 209, 1519–1528. doi:10.1084/jem.20120189
Vivarelli, E., and Cossu, G. (1986). Neural control of early myogenic differentiation in cultures of mouse somites. Dev. Biol. 117, 319–325. doi:10.1016/0012-1606(86)90374-x
Wang, Y., Nakayama, M., Pitulescu, M. E., Schmidt, T. S., Bochenek, M. L., Sakakibara, A., et al. (2010). Ephrin-B2 controls VEGF-induced angiogenesis 1and lymphangiogenesis. Nature 465, 483–486. doi:10.1038/nature09002
Keywords: mesoderm, myogenic stem cells, pericyte, muscular dystrophy, muscle development
Citation: Cossu G, Tonlorenzi R, Brunelli S, Sampaolesi M, Messina G, Azzoni E, Benedetti S, Biressi S, Bonfanti C, Bragg L, Camps J, Cappellari O, Cassano M, Ciceri F, Coletta M, Covarello D, Crippa S, Cusella-De Angelis MG, De Angelis L, Dellavalle A, Diaz-Manera J, Galli D, Galli F, Gargioli C, Gerli MFM, Giacomazzi G, Galvez BG, Hoshiya H, Guttinger M, Innocenzi A, Minasi MG, Perani L, Previtali SC, Quattrocelli M, Ragazzi M, Roostalu U, Rossi G, Scardigli R, Sirabella D, Tedesco FS, Torrente Y and Ugarte G (2023) Mesoangioblasts at 20: From the embryonic aorta to the patient bed. Front. Genet. 13:1056114. doi: 10.3389/fgene.2022.1056114
Received: 28 September 2022; Accepted: 31 October 2022;
Published: 04 January 2023.
Edited by:
Feng Yue, University of Florida, United StatesReviewed by:
Katherine Vest, University of Cincinnati, United StatesCopyright © 2023 Cossu, Tonlorenzi, Brunelli, Sampaolesi, Messina, Azzoni, Benedetti, Biressi, Bonfanti, Bragg, Camps, Cappellari, Cassano, Ciceri, Coletta, Covarello, Crippa, Cusella-De Angelis, De Angelis, Dellavalle, Diaz-Manera, Galli, Galli, Gargioli, Gerli, Giacomazzi, Galvez, Hoshiya, Guttinger, Innocenzi, Minasi, Perani, Previtali, Quattrocelli, Ragazzi, Roostalu, Rossi, Scardigli, Sirabella, Tedesco, Torrente and Ugarte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giulio Cossu, Y29zc3UuZ2l1bGlvQGhzci5pdA==; Rossana Tonlorenzi, dG9ubG9yZW56aS5yb3NzYW5hQGhzci5pdA==; Silvia Brunelli, c2lsdmlhLmJydW5lbGxpQHVuaW1pYi5pdA==; Maurilio Sampaolesi, bWF1cmlsaW8uc2FtcGFvbGVzaUBrdWxldXZlbi5iZQ==; Graziella Messina, Z3JhemllbGxhLm1lc3NpbmFAdW5pbWkuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.