 Junji Hu1†
Junji Hu1† Xueping Gao2†
Xueping Gao2† Longchang Chen1Yuling Kan3
Longchang Chen1Yuling Kan3 Zhaoli Du2Shuangqing Xin2Wenkai Ji2Qiang Yu2
Zhaoli Du2Shuangqing Xin2Wenkai Ji2Qiang Yu2 Lili Cao4*
Lili Cao4*- 1Department of Neurology, Zibo Changguo Hospital, Zibo, Shandong, China
- 2Yinfeng Gene Technology Co, Ltd, Jinan, Shandong, China
- 3Central Laboratory, Binzhou People’s Hospital, Binzhou, Shandong, China
- 4Department of Neurology, Cheeloo College of Medicine, Qilu Hospital, Shandong University, Jinan, China
Background: The GAP Activity Towards Rags 1 (GATOR1) complex, which includes DEPDC5, NPRL2, and NPRL3, plays a key role in epilepsy. It has been reported that focal epilepsy is associated with mutations in the NPRL3 gene in some cases. We report two rare mutations in the NPRL3 gene in two unrelated Chinese families with focal epilepsy in this study.
Methods: The proband and her brother in family E1 first experienced seizures at 1.5 and 6 years of age, respectively. Despite resection of epileptogenic foci, she still suffered recurrent seizures. The first seizure of a 20-year-old male proband in family E2 occurred when he was 2 years old. To identify pathogenic variants in these families, whole-exome sequencing (WES) was performed on genomic DNA from peripheral blood.
Results: In family E1, the trio-WES analysis of the proband and her brother without apparent structural brain abnormalities identified a heterozygous variant in the NPRL3 gene (c.954C>A, p.Y318*, NM_001077350.3). In family E2, the proband carried a heterozygous NPRL3 mutation (c.1545-1G>C, NM_001077350.3). Surprisingly, the mothers of the two probands each carried the variants, but neither had an attack. Bioinformatics analysis predicted that the mutation (c.954C>A) was in the highly conserved amino acid residues of NPRL3, which affected the α-helix of NPRL3 protein, leading to a truncated protein. The splice variant (c.1545-1G>C) resulted in the loss of the last exon of the NPRL3 gene.
Conclusion: The results of this study provide a foundation for diagnosing NPRL3-related epilepsy by enriching their genotypes and phenotypes and help us identify the genetic etiologies of epilepsy in these two families.
Introduction
Familial focal epilepsy with variable foci (FFEVF) is considered one of the most common forms of autosomal dominant epilepsy caused by mutations in the DEP domain containing 5 (DEPDC5), NPR2 like (NPRL2), and NPR3 like (NPRL3) (Dibbens et al., 2013; Ricos et al., 2016). The most common clinical feature of FFEVF is the presence of focal seizures in various cortical regions (including temporal, frontal, parietal, and occipital regions). It is reported that types of seizures usually include temporal lobe epilepsy (TLE), frontal lobe epilepsy (FLE), and nocturnal frontal lobe epilepsy (NFLE). It is interesting to note that there are notable variations in the clinical presentations of focal epilepsy in different family members that occur in various cortical regions, as well as in the features of the electroencephalogram (EEG) and magnetic resonance imaging (MRI) (Klein et al., 2012; Dibbens et al., 2013).
The NPRL3 gene (also known as C16orf35), located on 16p13.3, is a highly conserved gene widely expressed throughout development (Hughes et al., 2005). To date, 534 single nucleotide variants (SNVs) of the NPRL3 gene are included in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar), including 19 likely pathogenic and 25 pathogenic variants. There are 46 different causal mutations (SNVs and Indels) affecting the NPRL3 gene are reported in FFEVF3 (OMIM: 617118) patients in the Clinvar database (Supplementary Table S1). Nearly one-third of these mutations are truncating mutations, suggesting that the loss of function of NPRL3 may be a possible pathological mechanism. It has been widely studied that NPRL3 encodes a subunit of the GTPase-activating protein (GAP) activity toward the RAG complex 1 (GATOR1) complex, which negatively regulates the amino acid-sensing branch of the mTORC1 pathway (Bar-Peled et al., 2013). It is well known that during embryonic brain development, mTORC1 signaling plays pivotal roles in neurogenesis, synaptic transmission, and plasticity, leading to the formation of an intact cerebral cortex (Curatolo, 2015; Switon et al., 2016). Specifically, Iffland et al. confirmed that the knockdown of NPRL3 results in abnormal cell morphology in mouse neuronal cell lines, which depends on mTOR pathway (Iffland et al., 2018).
This study presents two unrelated Chinese families suffering from FFEVF3. Molecular analyses identified a rare non-sense variant and a splicing variant of the NPRL3 gene, expanding the phenotypic spectrum associated with NPRL3 variants.
Materials and methods
Patients
The present study and investigation were approved by the ethics committee of the Zibo Changguo Hospital. The peripheral blood samples were obtained from the patients and their parents, along with written informed consent. Three Chinese patients with familial focal epilepsy from two non-consanguineous families were included in this study. Detailed clinical data of patients were collected, including seizure onset, type of epilepsy, electroencephalogram (EEG), brain MRI, history of anti-epileptic treatment, and trauma history.
Whole-exome sequencing (WES) analysis
WES sequencing was performed by YinFeng Gene Technology Co., Ltd. (Jinan, China). First, genomic DNA was extracted from 2 ml peripheral blood using a Magnetic universal Genomic DNA Kit (TIANGEN, China) according to the manufacturer’s protocol. The DNA libraries were generated in accordance with the Illumina standard protocol. The exome sequences were captured using IDT xGen Exome Research Panel v 1.0 (Integrated DNA Technologies, Coralville, Iowa, United States) and sequenced on an Illumina NovaSeq 6,000 machine (Illumina, CA, United States) with an average 100-fold depth coverage.
Bcl To Fastq software (Illumina) was performed to process Raw image files. Low quality reads were filtered out, and the Burrows-Wheeler Aligner (BWA) (Heng et al., 2009) was used to align the high-quality reads to the reference human genome. Single nucleotide variants (SNVs) and indels were called by GATK (Depristo et al., 2011), and the generated VCF files were merged in a trio model. All variants were annotated using ANNOVAR (Kai et al., 2010), including minor allele frequencies from the 1000 Genomes Project, ExAC, and gnomAD databases, and deleteriousness and conservation scores predicted by MutationTaster (MT), SIFT, PolyPhen-2, GERP++, CADD, Revel score, and M-CAP databases. The pathogenicity of variants was assessed based on the American College of Medical Genetics (ACMG) guidelines (Richards et al., 2015).
Sanger sequencing
The identified variants in the NPRL3 gene were verified by Sanger sequencing. Genomic DNA extracted from each peripheral blood sample was used as template for PCR amplification. The primers used in this study were as follows: NPRL3 c.954C>A: 5′-TTAGGGAGGAAGTCTCGGGC-3′ and 5′-GGGACCTGGGTATGCTAGTGG-3'; NPRL3 c.1545-1G>C: 5′- AGAACCCTGGTCCCAACATCA-3′ and 5′-AGCACGCTGCGGAACTTGT-3'. The product was cleaned and sequenced by YinFeng Gene Technology Co., Ltd.
In silico protein structure analysis
The effect of identified mutation on the secondary structure of NPRL3 protein was predicted with the help of SOPMA (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_ automat.pl?page=/NPSA/npsa_sopma.html) (Deléage, 2017). The effect of the mutations on the 3D structure of the NPRL3 protein was investigated using SWISS-MODEL software (Andrew et al., 2018).
Extraction of variants from the literature
To analyze the correlation between NPRL3 and epilepsy, 11 original publications were collected from PubMed (https://www.ncbi.nlm.nih.gov/pubmed; accessed October 2022), and a list of the reported epilepsy-related variants in the NPRL3 gene was established and shown in Supplementary Table S2 and Supplementary Information.
Results
Clinical outcomes
In family E1, the female proband (II:1) and her affected brother (II:2), born to a healthy parent of non-consanguineous marriage, were from the Shandong province of China. The proband denied any history of infectious disease or infection. The proband had the onset of seizure at the age of 1 year and 6 months, manifested mainly as paroxysmal loss of consciousness, bilateral upward gaze or upward eyeball deviation, teeth biting, facial convulsions, left limb convulsions, or limb convulsions. After receiving poor anti-epileptic treatment in 1998, the patient (II:1) underwent epileptogenic foci resection in Beijing Yuquan Hospital in 2007, but the seizure occurred 2 months after the operation. After many adjustments to anti-epileptic drugs, there were no attacks for 2 years. The patient, however, relapsed in 2017. The medication was adjusted in time and no seizure has yet occurred so far. The EEG at age 23 showed diffuse paroxysms of spike-wave discharges in the left hemisphere, with a prominent anterior head (Figure 2C), but the brain MRI showed no abnormalities.
The younger brother (II:2) suffered his first seizure during sleep at the age of six with symptoms such as paroxysmal loss of consciousness, limb twitching, and teeth clenching. The patient (II:2) took Sodium Valproate Sustained-release tablets in the local hospital, but experienced poor effects and occurred two sleep episodes. After adjusting the anti-epileptic drugs in 2012, this patient remained seizure-free until the age of 18. EEG revealed frequent spike-slow wave complexes in the left posterior head (O1, P3) (Figure 2C). There were no abnormalities found on the brain MRI.
In family E2, the proband, a 20-year-old male, had his first focal seizure while awake at 2 years of age and was unconscious for 1–2 min, after which the seizures became intermittent. At the age of 10 years, this patient was suspected of intracranial hemorrhage caused by falling from a height (1 m) due to seizures and underwent craniocerebral surgery. This patient exhibited paroxysmal tachycardia, fear, and unrestricted behavior since the age of 18 years that lasted for approximately 1 min. It manifested as shouting in sleep, twisting of the body, dystonia of the right upper limb, waving of the left hand, pedaling of the lower limbs, and seizures occurring one to five times a night and lasting approximately 10 s to 1 min. EEG revealed persistent spike-slow wave complexes in the left central, parietal and midline areas (C3, P3, Cz, Pz) (Figure 2F). Brain MRI showed cortical thickening of the left frontal gyrus. The proband’s aunt (III:1) and grandfather’s younger brother (II:3) had a history of epilepsy.
WES results analysis
Trio-WES was performed to sequence the family E1 with an average depth of 100 X. The WES sequencing of this family did not reveal any significant loss of heterozygosity or potentially destructive copy number variation within affected family members. With normal individuals as negative controls, the trio-WES analysis identified 2,796 Single Nucleotide Variants (SNVs) and small Indels in this family, including 1,245 variants in autosomal dominant (AD) inheritance patterns, 53 de novo variants, 27 compound heterozygous variants, and 17 homozygous variants. Variants with less than 1% MAF in publicly available databases were screened, including dbSNP, 1000 Genomes Project, ExAC, and GnomAD databases. Through the genotype-phenotype correlation analysis, a heterozygous non-sense variant (c.954C>A, p.Y318*, NM_001077350.3) of NPRL3 at chr16: 93296 (hg38) was found to be carried in the proband and the younger brother. This variant was identified as the final candidate mutation related to seizures in this family. The c.954C>A was in exon 10 of NPRL3, which caused the 318th amino acid alteration from tyrosine to stop codon, resulting in a truncated NPRL3 protein (Figures 1A,B). The variation was located at the H1 domain of the Helix-turn Helix (Figure 1C). It is important to note that neither the gnomAD, ExAC, or 1,000 Genomes databases have documented this variant. With the use of the standards and guidelines for the interpretation of sequence variants by the American College of Medical Genetics and Genomics (ACMG), the evaluation of pathogenicity of the variant (c.954C>A, p.Y318*) in NPRL3 gene was pathogenic (PVS1+PM2_Supporting + PP4).
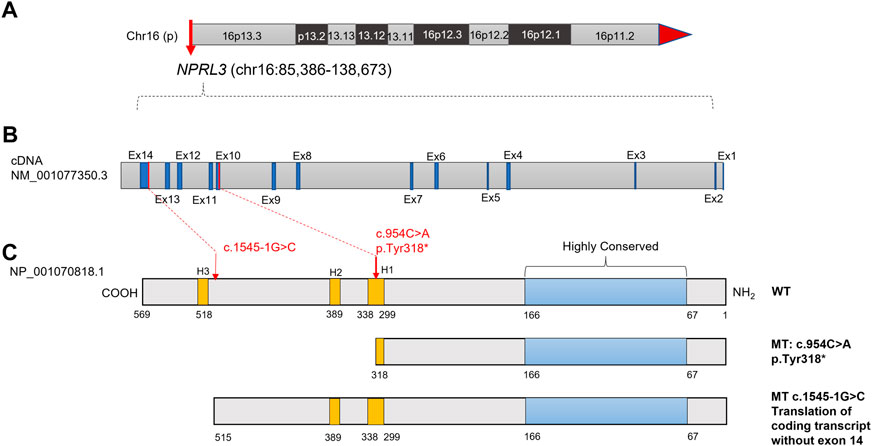
FIGURE 1. Diagram of the NPRL3 protein with the positions of the mutations found in the two families with FFEVF3. (A) Location of NPRL3 gene on chromosome 16. (B) Diagram of the NPRL3 cDNA. The exons were highlighted in dark blue, and the red lines indicated the mutation position. (C) Diagram of the NPRL3 protein. A highly conserved region at the N-terminus was highlighted in pale blue, the position of three Helix-turn-Helix domains H1, H2, and H3 was highlighted in yellow, and the red arrow indicates the mutation position.
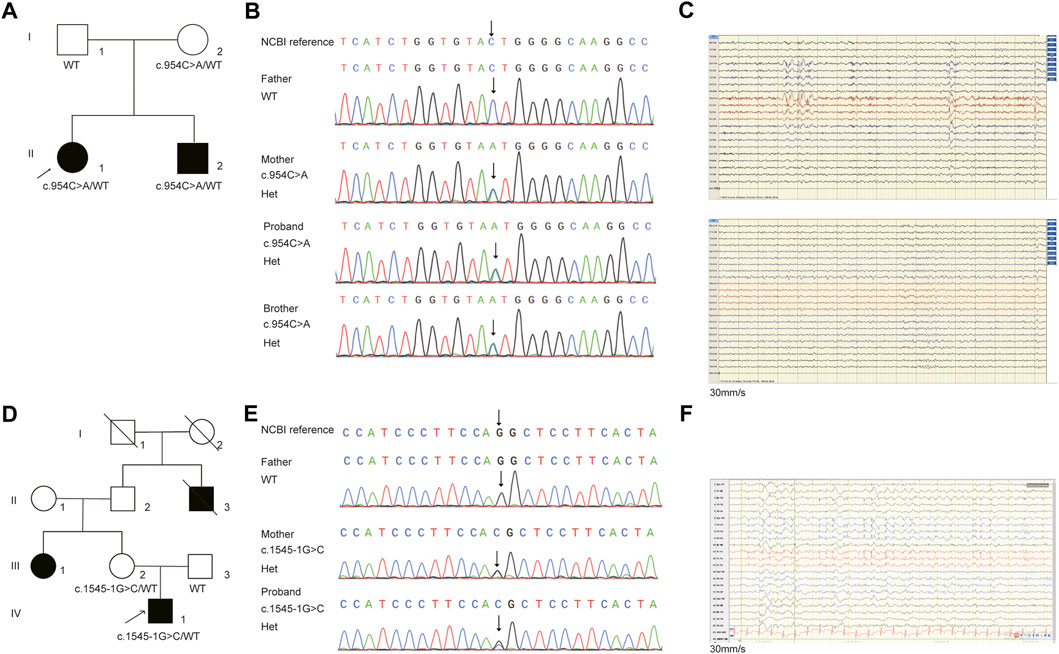
FIGURE 2. The pedigree and Sanger sequencing validation of (C)954C>A and (C)1545-1G>C in NPRL3 gene. (A) Pedigrees of affected family E1. Squares and circles indicate males and females, respectively. Arrows indicate the proband. (B) DNA sequencing data of the mutation identified in the family E1. The arrow indicates the location of the mutation. (C) EEG of the patients. Above: EEG of the patient II1 in family E1 showing diffuse paroxysms of irregular spike waves in the left hemisphere, with prominent anterior head. Below: EEG of the patient II2 in family E1 showing frequent spike-slow wave complexes in the left posterior head (O1, P3). (D) Pedigrees of affected family E2. Squares and circles indicate males and females, respectively. Arrows indicate the proband. (E) DNA sequencing data of the mutation identified in the family E2. The arrow indicates the location of the mutation. (F) EEG of the patient in family E2 showing persistent spike-slow wave complexes in left central, parietal and midline areas (C3, P3, Cz, Pz).
Trio-WES analysis revealed that the mother also carried the heterozygous NPRL3 mutation (Figures 2A,B). The Sanger sequencing further confirmed that the variant c.954C>A/p.Y318* was detected in both the affected members in this family, which was inherited from the mother, and the father did not carry this mutation (Figure 2B). The mother, however, did not display any symptoms of epilepsy and refused to undergo an EEG. We reevaluated the filtering steps for further investigation by screening the de novo variants, compound heterozygous and homozygous variants. However, we did not detect any relevant pathogenic or likely pathogenic variants in this family, nor did copy number variants (CNVs) within the WES detection range. The uncertain significance (VUS) variants in other genes associated with epilepsy are shown in Supplementary Table S3.
For the patient (IV-1) in family E2, 3,509 SNVs and small Indels variants were identified by WES in proband mode. A heterozygous splicing variant (c.1545-1G>C, NM_001077350.3) of the NPRL3 gene at chr16:86871 (hg38) was identified in the proband by the same analysis pipeline. Sanger sequencing further confirmed that this variant was inherited from his mother, who had no relevant clinical phenotype (Figures 2D,E). The variant c.1545-1G>C located at intron 13 of NPRL3, as the canonical splice acceptor site, may abolish the acceptor site, resulting in the loss of the last exon of NPRL3 gene (Figures 1B,C). Furthermore, c.1545-1G>C has not yet been reported, and it was not found in the gnomAD, ExAC and 1,000 Genomes databases. According to the ACMG standards and guidelines, the variant c.1545-1G>C was classified as “Uncertain significance” (PVS1_Moderate + PM2_Supporting + PP4). Furthermore, no other significant variants were found in other epilepsy-related genes, as well as CNV within the WES detection range. The VUS variants in other genes associated with epilepsy are shown in Supplementary Table S4.
Pathogenic assessment
Pathogenicity of the variants (c.954C>A/p.Y318*, c.1545-1G>C) in the NPRL3 gene was further assessed. Variation impact prediction on protein function showed damaging/pathogenic/deleterious/disease causing automatic in different algorithms (BayesDel, EIGEN, FATHMM-MKL, LRT, MutationTaster, and CADD). The truncating mutation (p.Y318*) may cause malformations of NPRL3 protein, leading to insufficient functional haploids by prediction of different algorithms (BayesDel, EIGEN, FATHMM-MKL, MutationTaster, and Polyphen2) (Supplementary Table S5). The splicing variant (c.1545-1G>C) was predicted as damaging/pathogenic by BayesDel, EIGEN and FATHMM-MKL (Supplementary Table S6). By prediction of Splice AI (https://spliceailookup.broadinstitute.org/), c.1545-1G>C leads to acceptor loss, resulting in loss of the last exon of NPRL3 (Supplementary Table S7). Cross-species conservation analysis by multispecies alignment of the amino acids encoded by NPRL3 showed that the p.318 tyrosine was highly conserved among Homo sapiens (human), Rattus_norvegicus (Rat), Mus musculus (Mouse), Canis lupus familiaris (Dog), Macaca mulatta (Rhesus macaque), and other different species (Figure 3A). It was predicted by SOPMA that the p.Y318* mainly affected the α-helix of the NPRL3 protein and caused the protein to be truncated, and c.1545-1G>C resulting in deletion of the last exon may affect the α-helix of the NPRL3 protein (Figure 3B).
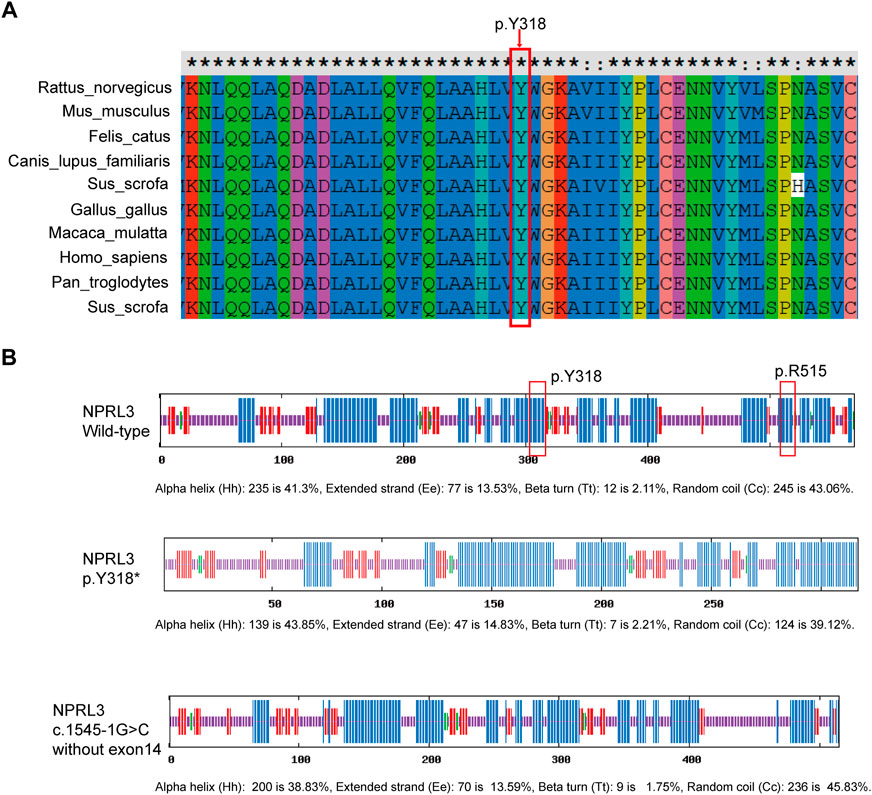
FIGURE 3. Conservation of p.Y318 in NPRL3. (A) Conservation of p.Y318 in NPRL3 across several species: Homo sapiens (human), Rattus_norvegicus (Rat), Mus musculus (Mouse), Canis lupus familiaris (Dog), Macaca mulatta (Rhesus macaque), and other different species. The p.Y318 is highly conserved among different species. The red box indicates the location of the mutation change. (B) Effects of the p.Y318* and (C)1545-1G>C variants on the secondary structure of the NPRL3 protein predicted by SOPMA (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html). The blue bar indicates the α-helix, the red bar indicates the extended strand, the green bar indicates the ß-turn, the yellow bar indicates the random coil. The red box indicates the location of the mutation change.
Furthermore, SWISS-MODEL was used to predict the molecular effects of the variants on NPRL3 protein. As shown in Figure 4A, once the 318th amino acid altered from tyrosine to a stop codon, the NPRL3 protein was forced to be truncated, and its conformation changed subsequently. The alternation may affect the function of NPRL3 protein, leading to damage. As shown in Figure 4B, when the last exon was lost, the conformation of the protein changed, which may lead to a change in protein function.
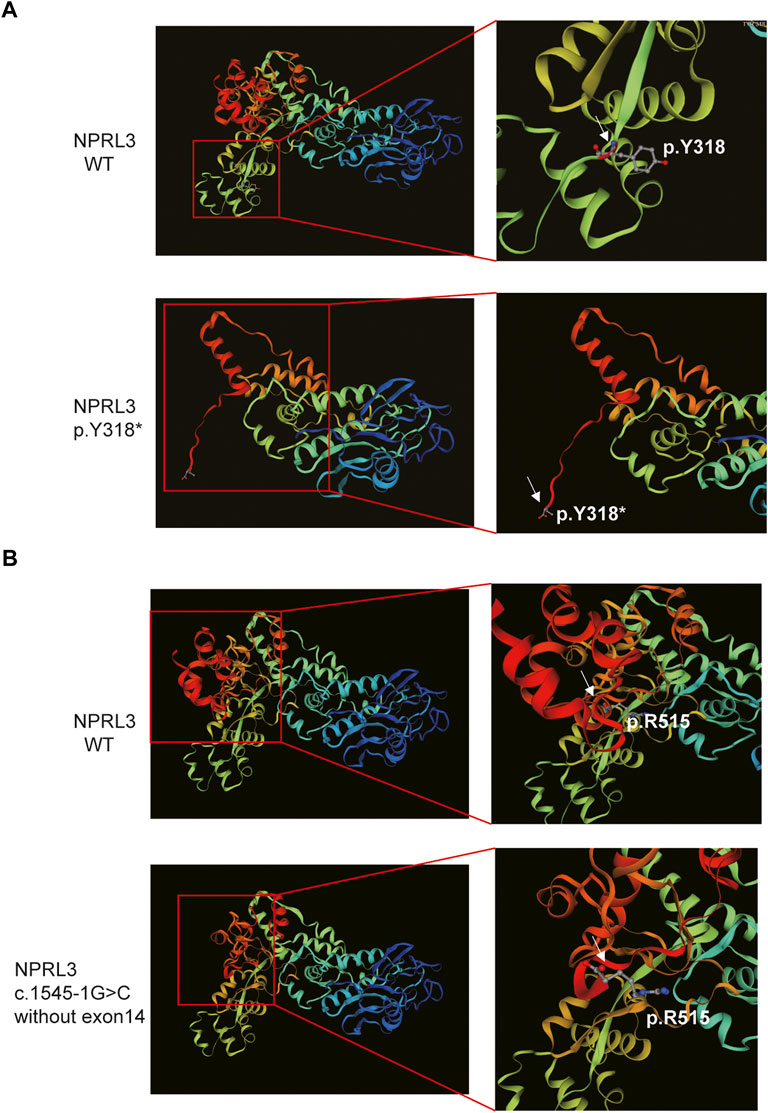
FIGURE 4. The 3D modeling of the wild-type and variant protein indicated different structures (visualized by SWISS-MODEL). (A) Comparison of wild-type and p.Y318* predicted structure. Left panel: Overall 3D modeling of wild-type and variant proteins. Right panel: Partial magnification of 3D models of wild-type and variant proteins. White arrow indicated the position of the p.Y318. (B) The 3D modeling of the wild-type and the mutant protein without exon 14. Left panel: Overall 3D modeling of wild-type and variant proteins. Right panel: Partial magnification of 3D models of wild-type and variant proteins. White arrow indicated the position of the last amino acid of the mutant protein.
Korenke et al. reported a multiplex family with nocturnal frontal lobe epilepsy caused by NPRL3 mutation and showed incomplete penetrance in the family. Therefore, we speculated that the rare truncated mutation of NPRL3 p.Y318* was identified as the potential pathogenic variant of patients in the family E1. The splicing variant (c.1545-1G>C) may be the potential causative gene in the family E2.
Treatment of patients
The proband in family E1, had adjusted the oral medication several times after epilepsy recurrence after epileptogenic focus resection. The proband is currently taking Nanxing Quanxie capsules at 1.4 g twice daily, Sodium valproate sustained release pills at 0.5 g per night, Lamotrigine tablets at 100 mg twice daily, and Clonazepam tablets at 2 mg per night, and no seizures have yet occurred during the proband’s 3 years of treatment. The younger brother takes the Nanxing Quanxie capsule at 1.05 g twice a day, Oxcarbazepine tablets at 0.3 g in the morning and 0.6 g at night, and Sodium valproate sustained release tablets at 0.5 g per night. The drugs have kept him seizure-free since 2012. As a result, the EEGs of the sister and brother have returned to normal satisfactorily.
Discussion
This study identified two variants in the NPRL3 gene. Two rare NPRL3 variants (c.954C>A/p.Y318*, c.1545-1G>C) were identified in two Chinese families experiencing FFEVF3. Each of these variants exposes a greater spectrum of mutations in NPRL3. The c.954C>G/p.Y318* mutation in the NPRL3 gene has been documented in the ClinVar database and dbSNP database (rs1596500172), which is interpreted as pathogenic (https://www.ncbi.nlm.nih.gov/clinvar/variation/652768/). The c.1545-1G>C (#0000691935) variant has been reported in the Leiden Open Variation Database (LOVD) database (https://databases.lovd.nl/shared/variants/0000691935#00025406). The c.1545-1G>C variant may produce abnormal splicing, resulting in the loss of the last exon of NPRL3. Sim et al. reported a frameshift variant in NPRL3 (c.1375_1376dupAC, p.S460Pfs*20) in a family with familial cortical dysplasia (Sim et al., 2016). Five heterozygous mutations in the NPRL3 gene (c.275G>A/p.R92Q; c.745G>A/p.E249K; c.835_836insT/p.S279Ffs*52; c.954_955insCCCA/p.W319Pfs*1; and c.1376_1377insAC/p.S460Pfs*20) was identified in 404 unrelated patients with focal epilepsy using WES analysis (Ricos et al., 2016). A novel mutation of NPRL3 (c.1522delG/p.E508Rfs*46) has been reported in a family of multiplex epilepsy (Korenke et al., 2016). Canavati et al. identified a de novo non-sense variant c.1063C>T/p.Q355* in NPRL3 in a Palestinian family with familial focal epilepsy with variable foci (Canavati et al., 2019). We reviewed the literature and this study, and found that twenty-two of the 25 NPRL3 variants associated with epilepsy were LoF, 15/25 patients were affected with focal epilepsy, and 12/17 with normal brain MRI (Supplementary Table S2). These studies reveal that NPRL3 is a causative gene for epilepsy.
NPRL3, NPRL2, and DEPDC5 form the GATOR1 complex, which controls the activity of the mTORC1 signaling pathway (Bar-Peled et al., 2013). It is generally known that the mTOR signaling pathway is a master regulator of cell growth and numerous diseases are characterized by loss of control in cell division and differentiation. Therefore, mutations within the genes involved in the mTORC1 signaling have been recognized as causative for different genetic diseases, including epilepsy. As a key regulatory of mTORC1 signaling, loss-of-function mutation of the GATOR1 subunit results in the activation of constitutive mTORC1 signaling, leading to morphological changes, cell size enhancement and abnormal cortical lamination (Marsan et al., 2016). Iffland et al. further confirmed that brain tissue of patients with NPRL3 c1375_1376dupAC showed increased cell size and indicated that NPRL3 knockdown led to the cell morphology of mouse neuronal cell lines in a mTOR-dependent manner and altered the subcellular localization of mTOR in neural precursor cells in vitro, further suggesting the critical role of NPRL3 in neurons (Iffland et al., 2018). Collectively, NPRL3 variants might cause seizures by interfering with mTORC1 signaling. Mutations truncated in the NPRL3 gene lead to a 50% reduction in its transcription levels, suggesting that haploinsufficiency may be the pathogenic mechanism. Therefore, this may also be the underlying mechanism of the NPRL3 p.Y318* variant for seizures in the family E1 in the present study.
Emerging evidence reveals that mutations within genes involved in the mTOR pathway cause several brain abnormalities, including focal cortical dysplasia (FCD). Sim et al. reported that most (4/6) individuals with epilepsy carried a frameshift mutation of the NPRL3 gene (c.1375_ 1,376 dupAC) and showed dysplastic brain lesions (Sim et al., 2016). There were, however, no significant brain abnormalities in two epilepsy patients carrying this mutation (Sim et al., 2016). Subsequently, Korenke et al. identified a truncating mutation of NPRL3 (c.1522delG/p.E508Rfs*46) in a multiplex family with neural frontal lobe epilepsy without any brain structural abnormalities (Korenke et al., 2016). As shown in Supplementary Table S2, even in the same family with the NPRL3 variant, some patients showed structural brain abnormalities while others showed no brain abnormalities. The NPRL3 pedigree has been reported to exhibit heterogeneous clinical phenotypes with abnormal or normal imaging and EEG findings (Iffland et al., 2022). In the present study, two patients with truncated NPRL3 variants in family E1 also showed no abnormalities in brain MRIs. However, in family E2, the patient had cortical thickening of the left frontal gyrus. Epilepsy caused by NPRL3 may not necessarily manifest structural abnormalities in the brain. There is a need to further study the different clinical phenotypes caused by the NPRL3 variants.
The NPRL3 pedigree exhibited incomplete epilepsy penetrance. In a four-generation family with six patients with nocturnal frontal lobe epilepsy, the epilepsy was only present in the proband’s generation and the next-generation (Korenke et al., 2016). It was interesting to find that individuals with heterozygous NPRL3 mutations in the parents’ generations did not exhibit epilepsy phenotypes, and the calculated overall penetration did not exceed 50% (Korenke et al., 2016). Sim et al. (Sim et al., 2016) found that all members of generation III carrying the NPRL3 variant c.1375_1376dupAC exhibited focal epilepsy, but the three individuals carrying the variant in generation II and their grandmothers were asymptomatic, which confirmed that the epilepsy penetrance in individuals with NPRL3 was 50% (4/8) in this family. Iffland et al. reported that the NPRL3 pedigree had an epilepsy penetrance of 28% and exhibited a distinct clinical phenotype with different epileptic symptoms (Iffland et al., 2022). In a family reported by Canavati et al. (Canavati et al., 2019), five individuals with FFEVF carried an NPRL3 variant c.1063C>T, the individual III-5 also carried the variant but none of the family members had epilepsy. Li et al. (Li et al., 2021) reported a six-generation Chinese family with eight FFEVF patients with mutation segregation in four generations, and the penetrance of the identified NPRL3 variant (c.316C>T) was 50% or less. In the present study, the mothers were both revealed to carry a heterozygous NPRL3 variant without any epilepsy phenotypes in these two families, which further confirms the incomplete penetrance of NPRL3 in FFEVF3. We found 12/25 NPRL3 variants currently reported to exhibit incomplete epilepsy penetrance (Supplementary Table S2).
Recently, germline and somatic mutations causing focal cortical dysplasia (FCD) were identified in patients carried with DEPDC5, and proposing a 2-hit- brain somatic and germline–mutational model (Ribierre et al., 2018; Lee WS et al., 2019). Bennett MF et al. (Bennett et al., 2022) reported a maternally inherited pathogenic germline variant (c.48delG) in NPRL3 gene in two brothers with FCD, and found a somatic variant (c.338C>T) in WNT2 gene in the brain-derived DNA from the elder brother at an allele fraction estimated at 0.3% by ddPCR, further confirming the 2-hit model in FCD caused by NPRL3. Therefore, the 2-hit model of germline and somatic mutations might cause the different clinical phenotypes and penetrance insufficiency in NPRL3 families. However, we cannot obtain frozen brain tissue samples from the patients in this study, which prevented us from identifying somatic variants in NPRL3 and other epilepsy-related genes.
Additionally, there are still several limitations to this study. Our study did not confirm the damaging effect of the splicing variant on NPRL3 transcription in vivo or in vitro. The direct functional effects of these two variants were not examined. Furthermore, more cases are needed to establish the relationship between different variants and heterogeneous clinical phenotypes, expanding the whole spectrum of the phenotype of NPRL3 variants.
Conclusion
Our study identified two rare NPRL3 variants in two unrelated families through WES analysis, including a truncating variant and a splicing variant, further emphasizing the role of NPRL3 in epileptogenesis. Furthermore, the present study confirmed the role of NPRL3 as a cause of familial focal epilepsy. Our findings expand the clinical and molecular spectrum of NPRL3.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories can be found below: Sequence Read Archive (SRA) data (www.ncbi.nlm.nih.gov ›sra), the accession number is PRJNA893418.
Ethics statement
The studies involving human participants were reviewed and approved by the ethics committee of the Zibo Changguo Hospital. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
JH and LC managed the patient and provided clinical information. JH and XG wrote the manuscript. YK, ZD, SX, WJ, QY, and LC contributed to WES sequencing and analysis. LC contributed to critical revision of the manuscript for important intellectual content.
Conflict of interest
XG, ZD, SX, WJ, and QY were employed by Yinfeng Gene Technology Co, Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1054567/full#supplementary-material
References
Andrew, W., Martino, B., Stefan, B., Gabriel, S., Gerardo, T., Rafal, G., et al. (2018). SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303. doi:10.1093/nar/gky427
Bar-Peled, L., Chantranupong, L., Cherniack, A. D., Chen, W. W., Sabatini, D. M., Grabiner, B. C., et al. (2013). A tumor suppressor complex with GAP activity for the RAG GTPases that signal amino acid sufficiency to mTORC1. Science 340 (6136), 1100–1106. doi:10.1126/science.1232044
Bennett, M. F., Hildebrand, M. S., Kayumi, S., Corbett, M. A., Gupta, S., Ye, Z., et al. (2022). Evidence for a dual-pathway, 2-hit genetic model for focal cortical dysplasia and epilepsy. Neurol. Genet. 8 (1), e652. doi:10.1212/NXG.0000000000000652
Canavati, C., Klein, K. M., Afawi, Z., Pendziwiat, M., Rayyan, A. A., Kamal, L., et al. (2019). Inclusion of hemimegalencephaly into the phenotypic spectrum of NPRL3 pathogenic variants in familial focal epilepsy with variable foci. Epilepsia 60, e67–e73. doi:10.1111/epi.15665
Curatolo, P. (2015). Mechanistic target of rapamycin (mTOR) in tuberous sclerosis complex-associated epilepsy. Pediatr. Neurol. 52 (3), 281–289. doi:10.1016/j.pediatrneurol.2014.10.028
Deléage, G. (2017). Alignsec: Viewing protein secondary structure predictions within large multiple sequence alignments. Bioinformatics 33, 3991–3992. doi:10.1093/bioinformatics/btx521
Depristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Daly, M. J., Hartl, C., et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43 (5), 491–498. doi:10.1038/ng.806
Dibbens, L. M., Donatello, S., Heron, S. E., Hodgson, B. L., Chintawar, S., Crompton, D. E., et al. (2013). Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat. Genet. 45 (5), 546–551. doi:10.1038/ng.2599
Hughes, J., Cheng, J., Ventress, N., Prabhakar, S., Clark, K., Anguita, E., et al. (2005). Annotation of cis-regulatory elements by identification, subclassification, and functional assessment of multispecies conserved sequences. Proc. Natl. Acad. Sci. U. S. A. 102 (28), 9830–9835. doi:10.1073/pnas.0503401102
Iffland, P. H., Baybis, M., Barnes, A. E., Leventer, R. J., and Crino, P. B. (2018). DEPDC5 and NPRL3 modulate cell size, filopodial outgrowth, and localization of mTOR in neural progenitor cells and neurons. Neurobiol. Dis. 114, 184–193. doi:10.1016/j.nbd.2018.02.013
Iffland, P. H., Everett, M. E., Cobb-Pitstick, K. M., Bowser, L. E., Barnes, A. E., Babus, J. K., et al. (2022). NPRL3 loss alters neuronal morphology, mTOR localization, cortical lamination, and seizure threshold. Brain 145, 3872–3885. doi:10.1093/brain/awac044
Kai, W., Li, M., and Hakon, H. (2010). Annovar: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 16, e164. doi:10.1093/nar/gkq603
Klein, K. M., O’Brien, T. J., Praveen, K., Heron, S. E., Mulley, J. C., Foote, S., et al. (2012). Familial focal epilepsy with variable foci mapped to chromosome 22q12: Expansion of the phenotypic spectrum. Epilepsia 53 (8), e151–e155. doi:10.1111/j.1528-1167.2012.03585.x
Korenke, G., Eggert, M., Thiele, H., Nürnberg, P., Sander, T., and Steinlein, O. K. (2016). Nocturnal frontal lobe epilepsy caused by a mutation in the GATOR1 complex gene NPRL3. Epilepsia 57 (3), e60–e63. doi:10.1111/epi.13307
Lee Ws, S. S., Howell, K. B., Pope, K., Gillies, G., Wray, A., Maixner, W., et al. (2019). Second-hit DEPDC5 mutation is limited to dysmorphic neurons in cortical dysplasia type IIA. Ann. Clin. Transl. Neurol. 6 (7), 1338–1344. doi:10.1002/acn3.50815
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi:10.1093/bioinformatics/btp324
Li, Y., Zhao, X., Wang, S., Xu, K., Zhao, X., Huang, S., et al. (2021). A novel loss-of-function mutation in the NPRL3 gene identified in Chinese familial focal epilepsy with variable foci. Front. Genet. 12, 766354. doi:10.3389/fgene.2021.766354
Marsan, E., Ishida, S., Schramm, A., Weckhuysen, S., Baulac, S., Lecas, S., et al. (2016). Depdc5 knockout rat: A novel model of mTORopathy. Neurobiol. Dis. 89, 180–189. doi:10.1016/j.nbd.2016.02.010
Ribierre, T., Deleuze, C., Bacq, A., Baldassari, S., Marsan, E., Chipaux, M., et al. (2018). Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia-associated epilepsy. J. Clin. Invest. 128 (6), 2452–2458. doi:10.1172/JCI99384
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. Official J. Am. Coll. Med. Genet. 17 (5), 405–424. doi:10.1038/gim.2015.30
Ricos, M. G., Hodgson, B. L., Pippucci, T., Saidin, A., Ong, Y. S., Heron, S. E., et al. (2016). Mutations in the mammalian target of rapamycin pathway regulators NPRL2 and NPRL3 cause focal epilepsy. Ann. Neurology 79 (1), 120–131. doi:10.1002/ana.24547
Sim, J. C., Scerri, T., Fanjul-Fernandez, M., Riseley, J. R., Gillies, G., Pope, K., et al. (2016). Familial cortical dysplasia caused by mutation in the mammalian target of rapamycin regulator NPRL3. Ann. Neurol. 79 (1), 132–137. doi:10.1002/ana.24502
Keywords: epilepsy, Nprl3, familial focal epilepsy with variable foci, GATOR1, mTORC1 signaling
Citation: Hu J, Gao X, Chen L, Kan Y, Du Z, Xin S, Ji W, Yu Q and Cao L (2023) Identification of two rare NPRL3 variants in two Chinese families with familial focal epilepsy with variable foci 3: NGS analysis with literature review. Front. Genet. 13:1054567. doi: 10.3389/fgene.2022.1054567
Received: 27 September 2022; Accepted: 19 December 2022;
Published: 06 January 2023.
Edited by:
Mohiuddin Mohammed Taher, Umm al-Qura University, Saudi ArabiaReviewed by:
Mohammed Faruq, Institute of Genomics and Integrative Biology, IndiaSamira Kalayinia, Shaheed Rajaei Cardiovascular Medical and Research Center, Iran
Copyright © 2023 Hu, Gao, Chen, Kan, Du, Xin, Ji, Yu and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lili Cao, cWx5eWNsbEAxNjMuY29t
†These authors have contributed equally to this work