 Anqi Chen
Anqi Chen Yukun Pan
Yukun Pan Jinzhong Chen
Jinzhong Chen- 1Department of Forensic Medicine, School of Basic Medical Sciences, Shanghai Medical College, Fudan University, Shanghai, China
- 2Barbell Therapeutics Co. Ltd., Shanghai, China
- 3State Key Laboratory of Genetic Engineering, Institute of Genetics, School of Life Sciences, Fudan University, Shanghai, China
Hyperphenylalaninemia (HPA) is the most common amino acid metabolism defect in humans. It is an autosomal-recessive disorder of the phenylalanine (Phe) metabolism, in which high Phe concentrations and low tyrosine (Tyr) concentrations in the blood cause phenylketonuria (PKU), brain dysfunction, light pigmentation and musty odor. Newborn screening data of HPA have revealed that the prevalence varies worldwide, with an average of 1:10,000. Most cases of HPA result from phenylalanine hydroxylase (PAH) deficiency, while a small number of HPA are caused by defects in the tetrahydrobiopterin (BH4) metabolism and DnaJ heat shock protein family (Hsp40) member C12 (DNAJC12) deficiency. Currently, the molecular pathophysiology of the neuropathology associated with HPA remains incompletely understood. Dietary restriction of Phe has been highly successful, although outcomes are still suboptimal and patients find it difficult to adhere to the treatment. Pharmacological treatments, such as BH4 and phenylalanine ammonia lyase, are available. Gene therapy for HPA is still in development.
Introduction
Phenylketonuria (PKU), the severe form of hyperphenylalaninemia (HPA), has been reported for near 90 years (Fölling. 1934; Grisch-Chan et al., 2019). It is the most common metabolic disorder of amino acid metabolism in humans, which is recognized by accumulated phenylalanine (Phe) in blood with an average incidence of 1:10,000 of the populations (Hillert et al., 2020). Although extensive works have been carried out to elucidate the pathological mechanism, the approach to HPA treatment has been rarely updated. Since the 1950s, a Phe-restricted diet has been the standard treatment for control the high blood Phe levels found in HPA (Sarkissian et al., 1999). Dietary management as a therapy remains a reliable and effective approach for preventing the manifestations of HPA for many (MacDonald et al., 2020). Recently, two drugs have been approved and used successfully for the correction of HPA (Keil et al., 2013; Burton et al., 2020). Several genetic diseases have been relieved using gene therapies (Welch et al., 2007; Sun, Zheng, and Simeonov 2017; Pogue et al., 2018), but the complete non-dietary treatment for HPA stays at very early-stage. This review aims to summarize the current understanding of HPA from the aspects of history, dietary treatments, pharmacological approaches, as well as the ongoing experimental gene therapies.
Milestones in PKU
In 1934, doctor Fölling (Fölling, 1934) studied two siblings with an intellectual disability and a musty odor and identified phenylpyruvic acid in their urine. The disease was named Følling’s disease and described as the inheritance of phenylpyruvic amentia (phenylketonuria, PKU) (Penrose 1935). PKU was the result of impaired Phe to Tyr conversion (Jervis 1947). Twenty years after Følling’s report, the first description of a Pherestricted diet began a new era of PKU therapy (Bickel, Gerrard, and Hickmans 1953). In 1963, the Guthrie bacterial inhibition assay (Scriver 1998) was established for detecting PKU in newborns. In 1971, the catalytic characteristics of the PAH system were determined (Kaufman 1971). A second cause of HPA, a defect in the tetrahydrobiopterin (BH4) metabolism, was identified in 1974 (Bartholome 1974). The third cause of HPA, a DnaJ heat shock protein family (Hsp40) member C12 (DNAJC12) variant, was identified in 2017 (Anikster et al., 2017). Regarding pharmacological treatment, BH4, as an adjunct treatment, decreases blood Phe concentrations in some cases (Kure et al., 1999) and phenylalanine ammonia lyase (PAL) provides an alternate treatment option (Longo et al., 2014).
Epidemiology
More than 50 years ago, measurement of the blood Phe concentrations in newborns was developed. Since then, this approach has been widely used worldwide (Guthrie and Susi 1963). Normal Phe concentrations are below 120 μmol/L, and Phe/Tyr is below 1.5. The prevalence of HPA varies worldwide (van Spronsen et al., 2021). In Europe, the prevalence ranges from 1:2,700 live births in Italy to <1:100,000 live births in Finland (Hillert et al., 2020). HPA prevalence is the lowest in Asian populations, such as Thailand, where it is 1:212,535 (Sutivijit, Banpavichit, and Wiwanitkit 2011). In China, the prevalence is 1:159,24 live births and ranges from 1:349,63 (Gansu Province) to 1: 666,667 (Guangxi Municipality) live births (Xiang et al., 2019).
Phe hydroxylation pathway and HPA
PAH, chaperone DNAJC12, and the BH4 system are necessary for metabolizing Phe to Tyr. Pathogenetic mutations in the genes synthesizing these enzymes are the potential causes of HPA, accumulating phenylpyruvate and hypotyrosinemia. The side way product phenylpyruvate is rapidly excreted in urine, but the tissue concentrations are probably too low to be of any clinical consequences (Jervis 1952). Hypotyrosinemia might be present in PKU cases and related to light pigmentation. However, there might be enough Tyr in food to support the Tyr-related neurotransmitter. Actually, supplementing the diet with Tyr does not prevent severe cognitive disability in individuals with PKU (Batshaw, Valle, and Bessman 1981). Restriction of Phe intake can prevent the major manifestations of PKU, which suggests HPA itself as the primary neurotoxin in PKU (van Vliet et al., 2018).
Cerebral effects of HPA
The most severe manifestations in PKU patients are intellectual disability and epilepsy. Similar to other aromatic amino acids and other large neutral amino acids (LNAAs), Phe is transferred to the brain by the large neutral amino acid transporter 1 (LAT1) on the blood–brain barrier (BBB) (Kanai et al., 1998). High levels of Phe mediate competition for binding to LAT1 with other LNAAs, leading to their deficiency in the brain (de Groot et al., 2010). These deficiencies are probably responsible for the impaired cerebral protein synthesis in PKU (Hoeksma et al., 2009) and contribute to brain monoamine neurotransmitter deficiencies (van Vliet et al., 2015). High Phe levels in the brain can inhibit Tyr hydroxylase (TH) and tryptophan hydroxylase 2 (TPH2), the rate-limiting steps in dopamine and serotonin synthesis, which result in disturbances of the monoamine neurotransmitters implicated as contributors to the neuropsychiatric symptoms (de Groot et al., 2010). Supplementation of LNAAs without Phe has been promoted as a treatment approach to PKU, by competitive inhibition of Phe across the BBB (van Vliet et al., 2016) (Figure 1).
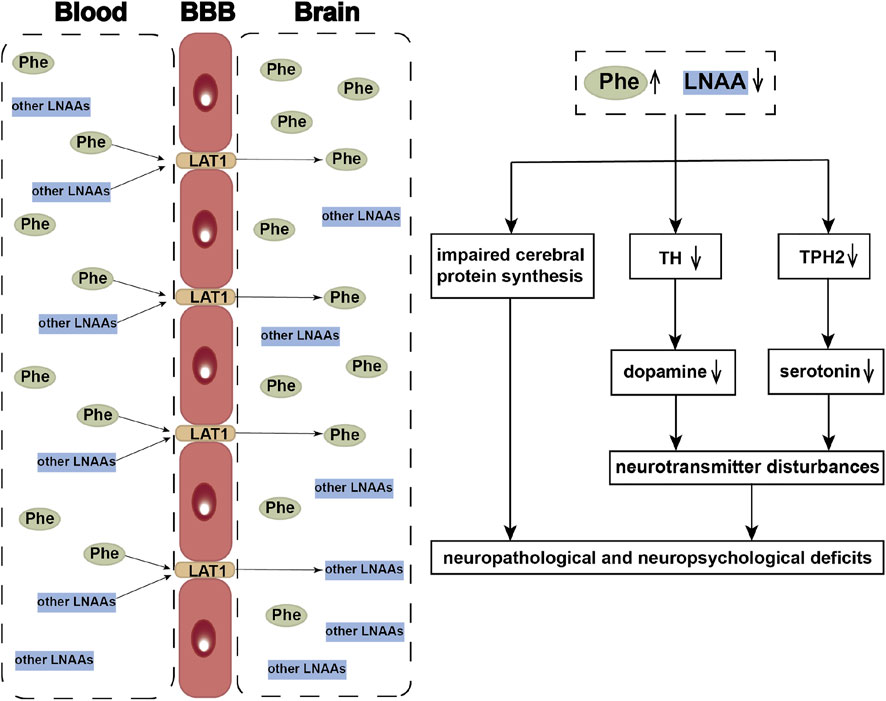
FIGURE 1. Neuropathological and neuropsychological deficits of HPA. LAT1 functions as a transporter mediating uptake of large neutral amino acids (LNAAs), such as phenylalanine (Phe), tyrosine (Tyr), and tryptophan etc., across the blood-brain barrier (BBB). When phenylalanine hydroxylase is defective, the elevated blood Phe concentrations competitively inhibit the other LNAAs transporting into the brain. The reduced LNAAs concentrations contribute to the impaired cerebral protein synthesis and neurotransmitter deficiency, which further resulted in the occurrence of neuropathological and neuropsychological deficits. LNAAs, large neutral amino acids; Phe, phenylalanine; Tyr, tyrosine; TH, Tyr hydroxylase; TPH2, tryptophan hydroxylase 2.
In vitro experiments with both cultured neurons and animal models, HPA caused disturbances of neuronal dendritic growth and synaptic connectivity (Hartwig et al., 2006; Hörster et al., 2006). HPA can impair the cholesterol or other brain lipids synthesis and thereby interfere with myelin-related structures (Shefer et al., 2000). HPA has also resulted in the forming of amyloid-like fibrils, a pathological structure similar to the amyloid plaques associated with Alzheimer’s disease (Adler-Abramovich et al., 2012).
The cerebral energy metabolism was abnormal in a PKU animal model. Glucose metabolism is reduced in the frontal cortex of hyperphenylalaninaemic Pahenu2 mice, which might be related to behavioral abnormalities (Qin and Smith 2007; Winn et al., 2016). HPA can inhibit pyruvate kinase or other enzymes involved in glycolysis or oxidative phosphorylation (Miller, Hawkins, and Veech 1973). Recently, several reports have suggested that HPA alters the methylation pattern and increases oxidative stress (Dobrowolski et al., 2016; van der Goot et al., 2019).
Diagnosis and screening
The genotypes of patients with HPA are considered deficient in PAH, BH4 and DNAJC12 (Table 1). Most cases of HPA are caused by pathogenetic mutations of the PAH gene located on human chromosome 12 (Woo et al., 1983). More than 1000 PAH pathogenetic mutations have been reported where patients might be compound heterozygous for two different PAH variants, and more than 2000 genotypes resulted in the HPA phenotype (Hillert et al., 2020).
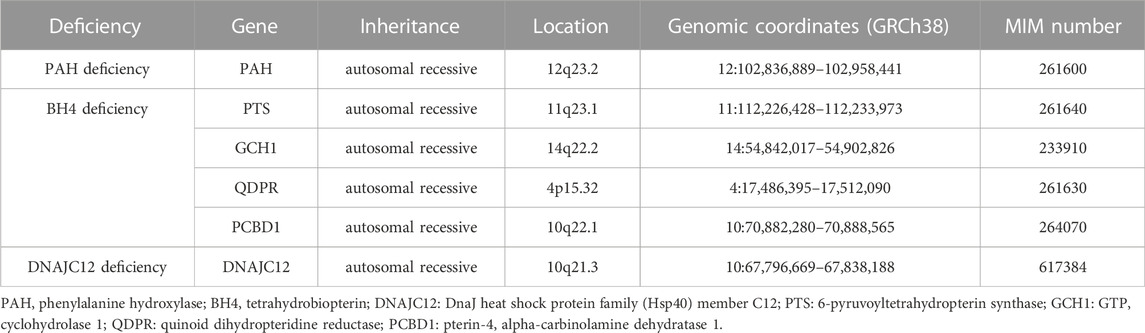
TABLE 1. The genes associated with hyperphenylalaninemia (HPA).
PAH pathogenetic mutations are inherited in an autosomal-recessive manner and result in expressing mutant protein with low/no catalytic activity, or even the absence of PAH protein expression (Garbade et al., 2019). BH4 deficiency due to inherited defects was found in the biopterin system, which consists of GTP cyclohydrolase 1 (GTPCH), 6-pyruvoyl-tetrahydropterin synthase (PTPS), dihydropteridine reductase (DHPR) or pterin-4a-carbinolamine dehydratase (PCD). PAH is disrupted in the absence of the chaperone DNAJC12, and this is described as an additional cause of inherited HPA (van Spronsen et al., 2017).
The Guthrie filter paper based newborn screening test for HPA has resulted in diagnoses in the neonatal period worldwide. To screen for PKU, the bacterial inhibition assay (BIA) and fluorimetric microassay (FMA) are employed to quantify Phe levels. A better method, tandem mass spectrometry (TMS) allows Phe and Tyr to be measured simultaneously. The PKU-positive screening result is determined by a cut-off of Phe concentrations ranging from 120 to 240 μmol/L in combination with a Phe to Tyr ratio >1.5–2 (van Wegberg et al., 2017).
Once HPA has been determined, it is necessary to distinguish PAH deficiency, disorders of the BH4 metabolism, and DNAJC12 defects. A panel comprising all genes known to cause HPA (possibly a larger panel with more than HPA-related genes) can provide final diagnostic confirmation and predict the metabolic phenotype in PAH deficiency. This is valuable for HPA diagnosis and treatment, especial for autosomal-recessive guanosine triphosphate cyclohydrolase (GTPCH) or serine racemase (SR) deficiency, which might present with normal blood Phe in the neonatal screening (van Wegberg et al., 2017). Thanks to the improvement of genotyping, most BH4 loading tests are covered for BH4 deficiencies, even in all HPA cases (Blau et al., 2014).
Dietary management
HPA screening aims for early diagnosis and prevention of intellectual disability by dietary management. Since the intellectual disability caused by PKU is irreversible, the prevention is more important than treatment. The dietary management of PKU should be initiated as soon as possible to prevent the cognitive and neurologic deficits (Evers et al., 2020). Unlike patients who experienced the neurocognitive consequences of late diagnosis and treatment, patients diagnosed and treated as infants may experience improved growth and development (Knox 1960). For late-diagnosed PKU patients, dietary management is also recommended, because behavior and epilepsy can be improved after dietary control (Koch et al., 1999).
Phe is an essential amino acid that cannot be produced by the body, and the blood Phe level is highly dependent on Phe intake. For this reason, dietary management should be successful. However, there is still a higher incidence of attention-deficit–hyperactivity disorder and specific learning disabilities in PKU, even with good dietary control (Arnold et al., 2004). Dietary management is comprised of limited natural protein intake, supplementation with a Phe-free amino acid mixture, and consumption of low-protein food products (MacDonald et al., 2020). Although the dietary treatment must be individualized and monitored, the protein and Phe deficiency can also cause adverse effects such as growth restriction, anorexia, alopecia, lethargy, and eczematous eruptions (Hanley et al., 1970). The basis of dietary treatment has changed little since 1953. Despite substantial efforts to improve quality, taste, and consumption methods of Phe-free amino acid mixtures, acceptance can be poor (Daly et al., 2021). Phe control during childhood is particularly important. The children themselves, as well as their family members, should understand the importance of Phe control, and sometimes it is necessary to hold the method of Phe test (Bilginsoy et al., 2005). Higher blood Phe concentrations are not always associated with poorer neurocognitive outcomes (Leuzzi et al., 2020), and the need to decrease blood Phe levels in adult PKU patients is unsure (Burlina et al., 2019). These adult patients might consider stopping dietary treatment. Consequently, they may not resume normal natural protein intake and be at risk of deficiency for some micronutrients (Lammardo et al., 2013). Regarding maternal PKU, the risk of fetal developmental abnormalities is increased if the maternal blood Phe concentrations exceed 360 μmol/L. For optimal metabolic control, the American College of Medical Genetics (ACMG) recommends lifelong maintenance of Phe concentrations within the range of 120–360 μmol/L (van Wegberg et al., 2017).
Pharmacological treatments
Sapropterin (Kuvan®, BioMarin) is a BH4 synthetic analog, an oral drug approved by FDA in 2008. Sapropterin is an exogenous synthetic BH4, and it is given as an effective replacement for endogenous BH4. The rationale for Sapropterin administration is to restore the Phe metabolism by enhancing the activity of the defective PAH. Since BH4 is a cofactor of PAH, the excess cofactor would help stimulate residual PAH to process Phe, and thereby decrease the blood Phe concentrations (Battaglia-Hsu and Feillet, 2010; Dubois and Cohen 2010). Sapropterin can increase dietary Phe tolerance (Keil et al., 2013) in some patients, thus enabling them to liberalize dietary restrictions. However, this drug does not response to all patients with PKU or BH4 deficiency (Dubois and Cohen 2010). Ten years later, a pegylated Phe ammonia lyase (pPAL, Palynziq®, BioMarin), was approved by FDA. The rationale for Palynziq® to reduce blood Phe concentrations through converting Phe to ammonia and transcinnamic acid (Thomas et al., 2018). The drug also changed the lives of PKU patients (Burton et al., 2020). Based on clinical experience and knowledge of adverse immunological events, the guidelines for pegvaliase treatment induction and maintenance in PKU patients have been proposed (Longo et al., 2019). Of all patients, 60.7% were able to achieve blood Phe concentrations less than 360 μmol/L without restricting dietary protein intake (Thomas et al., 2018). The pPAL injection for treatment of PKU patients has proved effective (Thomas et al., 2018). As a foreign protein, the immune-mediated responses remain the most important safety issue for this drug (Gupta et al., 2018).
Experimental therapies
The lifelong restricted diet supplemented with Phe-free protein substitutes has been the gold standard treatment for PKU patients (Burlina et al., 2020). However, the patient adherence to this therapy trends to be difficult after childhood, owing to the substantial time, cost and lifestyle burdens (Rose et al., 2019). Consequently, an effective, non-pathological, and long-term non-dietary restriction treatment is urgently needed. Gene therapy has been applied to cure genetic diseases such as spinal muscular atrophy (SMA) (Mendell et al., 2017; Waldrop et al., 2020) and hemophilia B (Lu et al., 2016; George et al., 2017). New approaches using gene therapy for PKU are considered promising because of the results from established PKU murine and mouse models (Isabella et al., 2018; Li et al., 2021). US and European authorities have approved injectable pegvaliase for PKU, at the same time, they have also incited other approaches to improve enzyme-based therapies to decrease the frequency and severity of this adverse effect (Hydery and Coppenrath 2019).
Rubius Therapeutics evaluated the safety and tolerability of RTX-134, the allogeneic human red blood cells (RBCs) expressing the AvPAL (which consists an Anabaena variabilis phenylalanine ammonia lyase gene inside the cell); unfortunately, this clinical trial failed to provide the expected signals of efficacy. Although the clinical trial using liver cell transplantation is in the recruiting process, the potential for transplant rejection will be unavoidable. Therefore, all participants must be treated with life-long immune suppression medications (NCT01465100), which will expose the patients to higher risks of an immune-mediated adverse reaction. Compared to the transplantation of enzyme-loaded RBCs, the enzyme substitution therapy with phenylalanine ammonialyase (PAL) appears more promising for decreasing Phe concentrations. CDX-6114 (Codexis Inc. And Nestlé) is a PAL-like enzyme that can remove Phe during digestion. To date, the results from three clinical trials (NCT03577886, NCT03797664, and NCT04085666) showed that the drug was well tolerated at different dose levels without any serious adverse events or GI-related symptoms. Unfortunately, further investigation was stopped because of the altered compositions. Other orally administered enzymes are SYNB 1618 and SYNB 1943 (Synlogic Biotic), which are engineered bacterial therapeutic drugs for oral delivery. SYNB 1618 and SYNB 1943 are expected to control Phe levels in patients with PKU. The strong positive results with these two drugs will undoubtedly initiate a phase 3 clinical trial in the near future. It has been reported that more than 10% catalytic activity is necessary for correcting PHA to below 700 μmol/L Phe (Hamman et al., 2005). However, the results observed from several PKU-related gene therapies were far from satisfactory, which suggested the treatments were still in an early stage.
For genetic disorders, genome editing using recombinant adeno-associated virus (rAAV) vectors is one of the most popular gene therapy strategies (Wang, Tai, and Gao 2019). The open read frame (ORF) of PAH is relatively small, which makes it fit well to the AAV vector. Furthermore, the native expression and residence of PAH is in liver cells, where the protein expression is easy and intracellular proteins would not be exposed to the immune system (Wang, Tai, and Gao 2019). The benefits mentioned above have laid the foundations for AAV-based gene therapy for PKU.
Since PKU is a genetic disorder owing to the mutation in the PAH gene, genome editing-based gene therapies have been developed to modify the genes. AAV-based CRISPR-Cas base editors provided more than 20% PAH activity in Pahenu2 mice and restored physiological blood Phe concentrations (Villiger et al., 2018; Richards et al., 2020). By using a dual AAV-based editing system, Zhou et al. (Zhou et al., 2022) achieved efficient correction of PKU-related mutations in vitro and in vivo. Co-delivery of SaCas9/sgRNA/donor templates with AAV receptor (AAVR) via AAV8 vectors also corrected the single mutation (PahR408W), and dramatically increased the editing efficiency in vivo (Yin et al., 2022). The advantage of this method is that the modified cells were maintained throughout the hepatocyte proliferation. Although effective in mice, no human clinical trials are reported. In addition, AAV-based gene therapies with AAV2/5-PAH were not successful; they could not correct the PAH deficiency without a high dosage of AAV (1014 vg/mouse) that might result in liver damage (Mochizuki et al., 2004; Oh et al., 2004). Alternatively, vectors pseudotyped with capsids from AAV serotype 8 were generated to explore a long-term non-dietary restriction treatment. The rAAV serotype 8 vectors exerted neither hepatic toxicity nor immunogenicity in the treated mice, and the blood Phe decreased to normal levels independent of the gender differences (Ding, Georgiev, and Thöny 2006; Rebuffat et al., 2010; Tao et al., 2020). At the same time, therapeutic ranges of Phe reverted the hair from brown to black in PAH-deficient mice. The better results came from AAV2/8-PAH (Rebuffat et al., 2010) and AAVHSC15 (Ahmed et al., 2020), which obtained a long-lasting correction of PAH activity in Pahenu2 mice. Similar observations were made using either the recombinant triplecistronic AAV2 pseudotype 1 vector (Ding et al., 2008) or the pseudotyped rAAV2/8-hPAH vector (Jung et al., 2008). Remarkably, the offspring of the treated mice were rescued from the pathologic effect (Jung et al., 2008). In a comparative study, both the rAAV2 pseudotype 1 (rAAV2/1) and rAAV2/8 vectors showed the long-term phenylalanine clearance. Although an elevated phenylalanine level was detected in female mice after 8–10 months of rAAV2/8 injection, it was corrected by either administering synthetic tetrahydrobiopterin supplementation or injecting a different AAV pseudotype vector (Rebuffat et al., 2010). Overall, the rAAV8 vectors not only corrected hyperphenylalaninemia in both males and females, but, more importantly, they exerted neither hepatic toxicity nor immunogenicity in Phe-deficient mice.
The effective outcomes and the feasibility of a single intravenous injection have paved the way to develop the clinical gene therapy procedure for PKU patients. At present, two activated clinical trials are ongoing with AAVHSC15 (NCT03952156 and NCT05222178), and one with AAV2/8 (NCT04480567). HMI-102 and HMI-103 exerted therapeutic effects by using an AAVHSC15 vector containing a functional copy of the human PAH gene. Both of them were the in vivo treatments that delivered functioning PAH genes to the liver by one-time I.V. administration. The difference is in the way the drugs work in the body. HMI-102 is designed to deliver the functional gene in the form of episomes, and HMI-103 creates the functional PAH protein by the integrated PAH gene and unintegrated PAH episomes (Table 2).
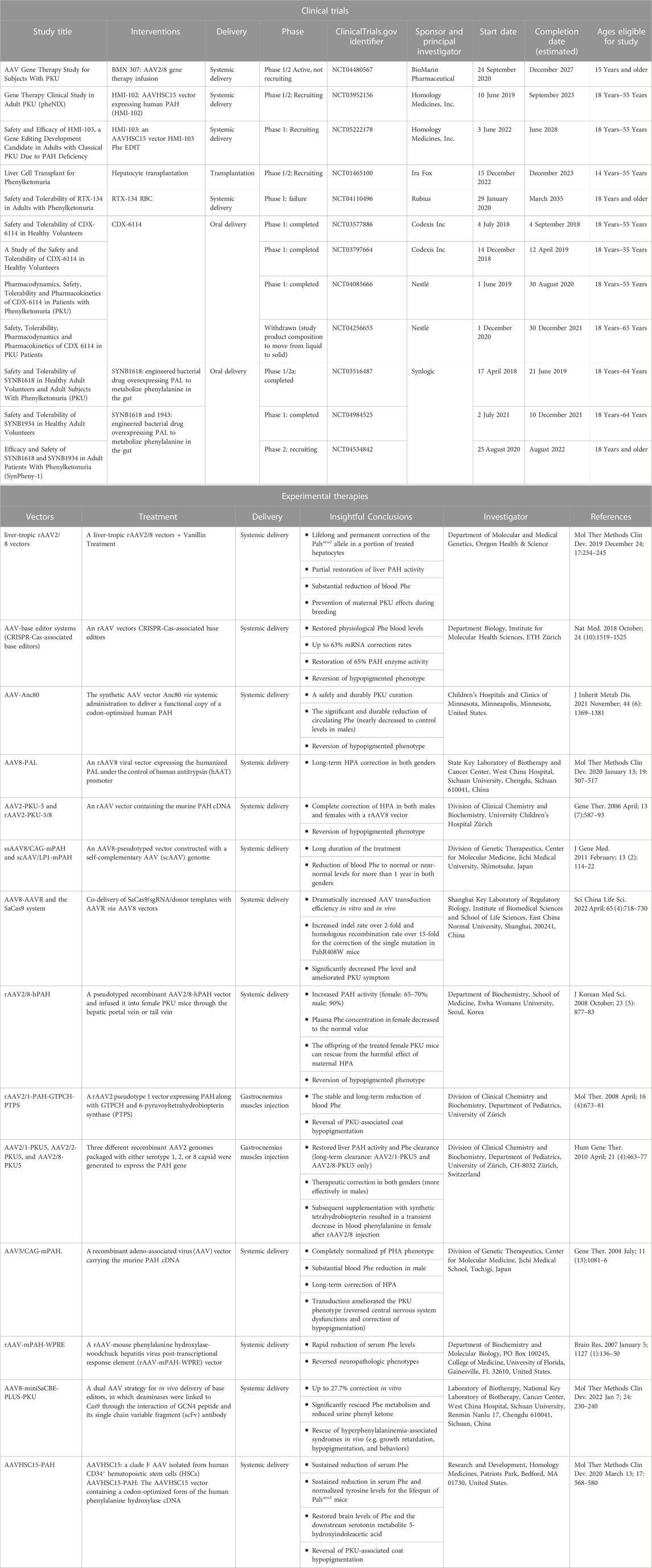
TABLE 2. Summary of the clinical trials and AAV-based experimental gene therapies.
An ideal strategy for gene therapy is expected to be non-pathogenic, transduction efficient, as well as sustained long-term expressed. PAH-deficient mice achieved nearly complete restoration of liver PAH activity and reversed symptoms without dietary supervision using AAV-mediated gene therapy. Despite the conventional AAV-pseudotype vectors, a series of newly developed AAV vectors also achieved the satisfactory results. Harmful changes in the brains of Pahenu2 mice were reversed after the portal vein delivery of an rAAV-mouse PAH-woodchuck hepatitis virus post-transcriptional response element (rAAV-mPAH-WPRE) vector (Embury et al., 2007). Kaiser et al. (2021) synthesized the AAV vector Anc80, a synthetic serotype using in silico techniques, to deliver a functional copy of a codon-optimized human PAH gene to the Pahenu2 mice. They observed the circulating Phe was reduced nearly to the control levels in males, but the clinic curative effect still needs to be proved in human trials.
Conclusion
PKU results from the severe form of HPA, a syndrome recognized by high concentrations of blood Phe. Consequently, blood Phe levels is the most important marker for diagnosis and treatment. Over the past 50 years, the classical lifelong dietary treatment has been regarded as the most effective approach to prevent disease consequences. Recently, the pharmaceutical therapies have provided alternative options for treatment. Several studies have suggested that AAV-based gene therapy might be another promising approach for HPA curation using only one-time administration without dietary restriction, while its security and efficacy await the results of ongoing clinical trials. Considering the uncertainties around the capacity and long-term durability of gene therapy, more optimizations are needed in the near future.
Author contributions
Conceptualization, JC and YP; writing—original draft preparation, AC; writing—review and editing, AC; supervision, JC and YP; project administration, JC and YP; funding acquisition, JC and YP. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Key Research and Development Project of China (grant number: 2021YFC2301500) and Shanghai Pujiang Program (grant number: No. 2020PJD019).
Conflict of interest
Author YP is employed by Barbell Therapeutics Co. Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adler-Abramovich, L., Vaks, L., Carny, O., Trudler, D., Magno, A., Caflisch, A., et al. (2012). 'Phenylalanine assembly into toxic fibrils suggests amyloid etiology in phenylketonuria. Nat. Chem. Biol. 8, 701–706. doi:10.1038/nchembio.1002
Ahmed, S. S., Rubin, H., Wang, M., Faulkner, D., Sengooba, A., Dollive, S. N., et al. (2020). 'Sustained correction of a murine model of phenylketonuria following a single intravenous administration of AAVHSC15-PAH. Mol. Ther. Methods Clin. Dev. 17, 568–580. doi:10.1016/j.omtm.2020.03.009
Anikster, Y., Haack, T. B., Vilboux, T., Pode-Shakked, B., Thöny, B., Shen, N., et al. (2017). 'Biallelic mutations in DNAJC12 cause hyperphenylalaninemia, dystonia, and intellectual disability. Am. J. Hum. Genet. 100, 257–266. doi:10.1016/j.ajhg.2017.01.002
Arnold, G. L., Vladutiu, C. J., Orlowski, C. C., Blakely, E. M., and DeLuca, J. (2004). 'Prevalence of stimulant use for attentional dysfunction in children with phenylketonuria. J. Inherit. Metab. Dis. 27, 137–143. doi:10.1023/B:BOLI.0000028725.37345.62
Bartholome, K. (1974). 'Letter: A new molecular defect in phenylketonuria. Lancet 2, 1580. doi:10.1016/s0140-6736(74)90337-7
Batshaw, M. L., Valle, D., and Bessman, S. P. (1981). 'Unsuccessful treatment of phenylketonuria with tyrosine. J. Pediatr. 99, 159–160. doi:10.1016/s0022-3476(81)80985-7
Battaglia-Hsu, S. F., and Feillet, F. (2010). Sapropterin in the treatment of phenylketonuria. Clin. Med. Insights Ther. 2, CMT.S2721–31. doi:10.4137/cmt.s2721
Bickel, H., Gerrard, J., and Hickmans, E. M. (1953). 'Influence of phenylalanine intake on phenylketonuria. Lancet 265, 812–813. doi:10.1016/s0140-6736(53)90473-5
Bilginsoy, C., Waitzman, N., Leonard, C. O., and Ernst, S. L. (2005). 'Living with phenylketonuria: Perspectives of patients and their families. J. Inherit. Metab. Dis. 28, 639–649. doi:10.1007/s10545-005-4478-8
Blau, N., Duran, M., Gibson, K. M., and Dionisi, C. V. (2014). 'Physician’s guide to the diagnosis, treatment, and follow-up of inherited metabolic diseases. Congenital Disorders of Glycosylation. Switzerland: Springer Nature.
Burlina, A. P., Cazzorla, C., Massa, P., Loro, C., Gueraldi, D., and Burlina, A. B. (2020). The impact of a slow-release large neutral amino acids supplement on treatment adherence in adult patients with phenylketonuria. Nutrients 12, 2078. doi:10.3390/nu12072078
Burlina, A. P., Lachmann, R. H., Manara, R., Cazzorla, C., Celato, A., van Spronsen, F. J., et al. (2019). 'The neurological and psychological phenotype of adult patients with early-treated phenylketonuria: A systematic review. J. Inherit. Metab. Dis. 42, 209–219. doi:10.1002/jimd.12065
Burton, B. K., Longo, N., Vockley, J., Grange, D. K., Harding, C. O., Decker, C., et al. (2020). 'Pegvaliase for the treatment of phenylketonuria: Results of the phase 2 dose-finding studies with long-term follow-up. Mol. Genet. Metab. 130, 239–246. doi:10.1016/j.ymgme.2020.06.006
Daly, A., Evans, S., Pinto, A., Ashmore, C., and MacDonald, A. (2021). Protein substitutes in PKU; their historical evolution. Nutrients 13, 484. doi:10.3390/nu13020484
de Groot, M. J., Hoeksma, M., Blau, N., Reijngoud, D. J., and van Spronsen, F. J. (2010). 'Pathogenesis of cognitive dysfunction in phenylketonuria: Review of hypotheses. Mol. Genet. Metab. 99, S86–S89. doi:10.1016/j.ymgme.2009.10.016
Ding, Z., Georgiev, P., and Thöny, B. (2006). 'Administration-route and gender-independent long-term therapeutic correction of phenylketonuria (PKU) in a mouse model by recombinant adeno-associated virus 8 pseudotyped vector-mediated gene transfer. Gene Ther. 13, 587–593. doi:10.1038/sj.gt.3302684
Ding, Z., Harding, C. O., Rebuffat, A., Elzaouk, L., Wolff, J. A., and Thöny, B. (2008). 'Correction of murine PKU following AAV-mediated intramuscular expression of a complete phenylalanine hydroxylating system. Mol. Ther. 16, 673–681. doi:10.1038/mt.2008.17
Dobrowolski, S. F., Lyons-Weiler, J., Spridik, K., Vockley, J., Skvorak, K., and Biery, A. (2016). 'DNA methylation in the pathophysiology of hyperphenylalaninemia in the PAH(enu2) mouse model of phenylketonuria. Mol. Genet. Metab. 119, 1–7. doi:10.1016/j.ymgme.2016.01.001
Dubois, E. A., and Cohen, A. F. (2010). Sapropterin. Br. J. Clin. Pharmacol. 69, 576–577. doi:10.1111/j.1365-2125.2010.03643.x
Embury, J. E., Charron, C. E., Martynyuk, A., Zori, A. G., Liu, B., Ali, S. F., et al. (2007). 'PKU is a reversible neurodegenerative process within the nigrostriatum that begins as early as 4 weeks of age in Pah(enu2) mice. Brain Res. 1127, 136–150. doi:10.1016/j.brainres.2006.09.101
Evers, R. A. F., van Wegberg, A. M. J., Anjema, K., Lubout, C. M. A., van Dam, E., van Vliet, D., et al. (2020). 'The first European guidelines on phenylketonuria: Usefulness and implications for BH(4) responsiveness testing. J. Inherit. Metab. Dis. 43, 244–250. doi:10.1002/jimd.12173
Fölling, A. (1934). 'Über Ausscheidung von Phenylbrenztraubensäure in den Harn als Stoffwechselanomalie in Verbindung mit Imbezillität. Hoppe-Seyler´s. Z. fur Physiol. Chem. 227, 169–181. doi:10.1515/bchm2.1934.227.1-4.169
Garbade, S. F., Shen, N., Himmelreich, N., Haas, D., Trefz, F. K., Hoffmann, G. F., et al. (2019). 'Allelic phenotype values: A model for genotype-based phenotype prediction in phenylketonuria. Genet. Med. 21, 580–590. doi:10.1038/s41436-018-0081-x
George, L. A., Sullivan, S. K., Giermasz, A., Rasko, J. E. J., Samelson-Jones, B. J., Ducore, J., et al. (2017). 'Hemophilia B gene therapy with a high-specific-activity factor IX variant. N. Engl. J. Med. 377, 2215–2227. doi:10.1056/NEJMoa1708538
Grisch-Chan, H. M., Schwank, G., Harding, C. O., and Thöny, B. (2019). 'State-of-the-Art 2019 on gene therapy for phenylketonuria. Hum. Gene Ther. 30, 1274–1283. doi:10.1089/hum.2019.111
Gupta, S., Lau, K., Harding, C. O., Shepherd, G., Boyer, R., Atkinson, J. P., et al. (2018). 'Association of immune response with efficacy and safety outcomes in adults with phenylketonuria administered pegvaliase in phase 3 clinical trials. EBioMedicine 37, 366–373. doi:10.1016/j.ebiom.2018.10.038
Guthrie, R., and Susi, A. (1963). A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics 32, 338–343. doi:10.1542/peds.32.3.338
Hamman, K., Clark, H., Montini, E., Al-Dhalimy, M., Grompe, M., Finegold, M., et al. (2005). 'Low therapeutic threshold for hepatocyte replacement in murine phenylketonuria. Mol. Ther. 12, 337–344. doi:10.1016/j.ymthe.2005.03.025
Hanley, W. B., Linsao, L., Davidson, W., and Moes, C. A. (1970). 'Malnutrition with early treatment of phenylketonuria. Pediatr. Res. 4, 318–327. doi:10.1203/00006450-197007000-00002
Hartwig, C., Gal, A., Santer, R., Ullrich, K., Finckh, U., and Kreienkamp, H. J. (2006). 'Elevated phenylalanine levels interfere with neurite outgrowth stimulated by the neuronal cell adhesion molecule L1 in vitro. FEBS Lett. 580, 3489–3492. doi:10.1016/j.febslet.2006.05.026
Hillert, A., Anikster, Y., Belanger-Quintana, A., Burlina, A., Burton, B. K., Carducci, C., et al. (2020). 'The genetic landscape and epidemiology of phenylketonuria. Am. J. Hum. Genet. 107, 234–250. doi:10.1016/j.ajhg.2020.06.006
Hoeksma, M., Reijngoud, D. J., Pruim, J., de Valk, H. W., Paans, A. M., and van Spronsen, F. J. (2009). 'Phenylketonuria: High plasma phenylalanine decreases cerebral protein synthesis. Mol. Genet. Metab. 96, 177–182. doi:10.1016/j.ymgme.2008.12.019
Hörster, F., Schwab, M. A., Sauer, S. W., Pietz, J., Hoffmann, G. F., Okun, J. G., et al. (2006). 'Phenylalanine reduces synaptic density in mixed cortical cultures from mice. Pediatr. Res. 59, 544–548. doi:10.1203/01.pdr.0000203091.45988.8d
Hydery, T., and Coppenrath, V. A. (2019). A comprehensive review of pegvaliase, an enzyme substitution therapy for the treatment of phenylketonuria. Drug Target Insights 13, 1177392819857089. doi:10.1177/1177392819857089
Isabella, V. M., Ha, B. N., Castillo, M. J., Lubkowicz, D. J., Rowe, S. E., Millet, Y. A., et al. (2018). 'Development of a synthetic live bacterial therapeutic for the human metabolic disease phenylketonuria. Nat. Biotechnol. 36, 857–864. doi:10.1038/nbt.4222
Jervis, G. A. (1952). Studies on phenylpyruvic oligophrenia; phenylpyruvic acid content on blood. Proc. Soc. Exp. Biol. Med. 81, 715–720. doi:10.3181/00379727-81-19998
Jervis, G. A. (1947). 'Studies on phenylpyruvic oligophrenia; the position of the metabolic error. J. Biol. Chem. 169, 651–656. doi:10.1016/s0021-9258(17)30882-7
Jung, S. C., Park, J. W., Oh, H. J., Choi, J. O., Seo, K. I., Park, E. S., et al. (2008). 'Protective effect of recombinant adeno-associated virus 2/8-mediated gene therapy from the maternal hyperphenylalaninemia in offsprings of a mouse model of phenylketonuria. J. Korean Med. Sci. 23, 877–883. doi:10.3346/jkms.2008.23.5.877
Kaiser, R. A., Weber, N. D., Trigueros-Motos, L., Allen, K. L., Martinez, M., Cao, W., et al. (2021). 'Use of an adeno-associated virus serotype Anc80 to provide durable cure of phenylketonuria in a mouse model. J. Inherit. Metab. Dis. 44, 1369–1381. doi:10.1002/jimd.12392
Kanai, Y., Segawa, H., Miyamoto, K. I., Uchino, H., Takeda, E., and Endou, H. (1998). Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98). J. Biol. Chem. 273, 23629–23632. doi:10.1074/jbc.273.37.23629
Kaufman, S. (1971). 'The phenylalanine hydroxylating system from mammalian liver. Adv. Enzymol. Relat. Areas Mol. Biol. 35, 245–319. doi:10.1002/9780470122808.ch6
Keil, S., Anjema, K., van Spronsen, F. J., Lambruschini, N., Burlina, A., Bélanger-Quintana, A., et al. (2013). 'Long-term follow-up and outcome of phenylketonuria patients on sapropterin: A retrospective study. Pediatrics 131, e1881–e1888. doi:10.1542/peds.2012-3291
Knox, W. E. (1960). An evaluation of the treatment of phenylketonuria with diets low in phenylalanine. Pediatrics 26, 1–11. doi:10.1542/peds.26.1.1
Koch, R., Moseley, K., Ning, J., Romstad, A., Guldberg, P., and Guttler, F. (1999). 'Long-term beneficial effects of the phenylalanine-restricted diet in late-diagnosed individuals with phenylketonuria. Mol. Genet. Metab. 67, 148–155. doi:10.1006/mgme.1999.2863
Kure, S., Hou, D. C., Ohura, T., Iwamoto, H., Suzuki, S., Sugiyama, N., et al. (1999). 'Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. J. Pediatr. 135, 375–378. doi:10.1016/s0022-3476(99)70138-1
Lammardo, A. M., Robert, M., Rocha, J. C., van Rijn, M., Ahring, K., Bélanger-Quintana, A., et al. (2013). 'Main issues in micronutrient supplementation in phenylketonuria. Mol. Genet. Metab. 110, S1–S5. doi:10.1016/j.ymgme.2013.08.008
Leuzzi, V., Chiarotti, F., Nardecchia, F., van Vliet, D., and van Spronsen, F. J. (2020). 'Predictability and inconsistencies of cognitive outcome in patients with phenylketonuria and personalised therapy: The challenge for the future guidelines. J. Med. Genet. 57, 145–150. doi:10.1136/jmedgenet-2019-106278
Li, Y., Tan, Z., Zhang, Y., Zhang, Z., Hu, Q., Liang, K., et al. (2021). 'A noncoding RNA modulator potentiates phenylalanine metabolism in mice. Science 373, 662–673. doi:10.1126/science.aba4991
Longo, N., Dimmock, D., Levy, H., Viau, K., Bausell, H., Bilder, D. A., et al. (2019). 'Evidence- and consensus-based recommendations for the use of pegvaliase in adults with phenylketonuria. Genet. Med. 21, 1851–1867. doi:10.1038/s41436-018-0403-z
Longo, N., Harding, C. O., Burton, B. K., Grange, D. K., Vockley, J., Wasserstein, M., et al. (2014). 'Single-dose, subcutaneous recombinant phenylalanine ammonia lyase conjugated with polyethylene glycol in adult patients with phenylketonuria: An open-label, multicentre, phase 1 dose-escalation trial. Lancet 384, 37–44. doi:10.1016/S0140-6736(13)61841-3
Lu, W., Zhou, Q., Yang, H., Wang, H., Gu, Y., Shen, Q., et al. (2016). 'Gene therapy for hemophilia B mice with scAAV8-LP1-hFIX. Front. Med. 10, 212–218. doi:10.1007/s11684-016-0438-y
MacDonald, A., van Wegberg, A. M. J., Ahring, K., Beblo, S., Bélanger-Quintana, A., Burlina, A., et al. (2020). 'PKU dietary handbook to accompany PKU guidelines. Orphanet J. Rare Dis. 15, 171. doi:10.1186/s13023-020-01391-y
Mendell, J. R., Al-Zaidy, S., Shell, R., Arnold, W. D., Rodino-Klapac, L. R., Prior, T. W., et al. (2017). 'Single-Dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 377, 1713–1722. doi:10.1056/NEJMoa1706198
Miller, A. L., Hawkins, R. A., and Veech, R. L. (1973). 'Phenylketonuria: Phenylalanine inhibits brain pyruvate kinase in vivo. Science 179, 904–906. doi:10.1126/science.179.4076.904
Mochizuki, S., Mizukami, H., Ogura, T., Kure, S., Ichinohe, A., Kojima, K., et al. (2004). 'Long-term correction of hyperphenylalaninemia by AAV-mediated gene transfer leads to behavioral recovery in phenylketonuria mice. Gene Ther. 11, 1081–1086. doi:10.1038/sj.gt.3302262
Oh, H. J., Park, E. S., Kang, S., Jo, I., and Jung, S. C. (2004). 'Long-term enzymatic and phenotypic correction in the phenylketonuria mouse model by adeno-associated virus vector-mediated gene transfer. Pediatr. Res. 56, 278–284. doi:10.1203/01.PDR.0000132837.29067.0E
Penrose, L. S. (1935). Inheritance of phenylpyruvic amentia (phenylketonuria). Lancet 226, 192–194. doi:10.1016/s0140-6736(01)04897-8
Pogue, R. E., Cavalcanti, D. P., Shanker, S., Andrade, R. V., Aguiar, L. R., de Carvalho, J. L., et al. (2018). 'Rare genetic diseases: Update on diagnosis, treatment and online resources. Drug Discov. Today 23, 187–195. doi:10.1016/j.drudis.2017.11.002
Qin, M., and Smith, C. B. (2007). 'Regionally selective decreases in cerebral glucose metabolism in a mouse model of phenylketonuria. J. Inherit. Metab. Dis. 30, 318–325. doi:10.1007/s10545-007-0583-1
Rebuffat, A., Harding, C. O., Ding, Z., and Thöny, B. (2010). 'Comparison of adeno-associated virus pseudotype 1, 2, and 8 vectors administered by intramuscular injection in the treatment of murine phenylketonuria. Hum. Gene Ther. 21, 463–477. doi:10.1089/hum.2009.127
Richards, D. Y., Winn, S. R., Dudley, S., Nygaard, S., Mighell, T. L., Grompe, M., et al. (2020). 'AAV-Mediated CRISPR/Cas9 gene editing in murine phenylketonuria. Mol. Ther. Methods Clin. Dev. 17, 234–245. doi:10.1016/j.omtm.2019.12.004
Rose, A. M., Grosse, S. D., Garcia, S. P., Bach, J., Kleyn, M., Simon, N. E., et al. (2019). The financial and time burden associated with phenylketonuria treatment in the United States. Mol. Genet. Metab. Rep. 21, 100523. doi:10.1016/j.ymgmr.2019.100523
Sarkissian, C. N., Shao, Z., Blain, F., Peevers, R., Su, H., Heft, R., et al. (1999). 'A different approach to treatment of phenylketonuria: Phenylalanine degradation with recombinant phenylalanine ammonia lyase. Proc. Natl. Acad. Sci. U. S. A. 96, 2339–2344. doi:10.1073/pnas.96.5.2339
Scriver, C. C. (1998). A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants, by Robert Guthrie and Ada Susi, Pediatrics. Pediatrics 32, 318–343. doi:10.1542/peds.102.s1.236
Shefer, S., Tint, G. S., Jean-Guillaume, D., Daikhin, E., Kendler, A., Nguyen, L. B., et al. (2000). Is there a relationship between 3-hydroxy-3-methylglutaryl coenzyme a reductase activity and forebrain pathology in the PKU mouse? J. Neurosci. Res. 61, 549–563. doi:10.1002/1097-4547(20000901)61:5<549:AID-JNR10>3.0.CO;2-0
Sun, W., Zheng, W., and Simeonov, A. (2017). 'Drug discovery and development for rare genetic disorders. Am. J. Med. Genet. A 173, 2307–2322. doi:10.1002/ajmg.a.38326
Sutivijit, Y., Banpavichit, A., and Wiwanitkit, V. (2011). 'Prevalence of neonatal hypothyroidism and phenylketonuria in southern Thailand: A 10-year report. Indian J. Endocrinol. Metab. 15, 115–117. doi:10.4103/2230-8210.81941
Tao, R., Xiao, L., Zhou, L., Zheng, Z., Long, J., Zhou, L., et al. (2020). 'Long-Term metabolic correction of phenylketonuria by AAV-delivered phenylalanine amino lyase. Mol. Ther. Methods Clin. Dev. 19, 507–517. doi:10.1016/j.omtm.2019.12.014
Thomas, J., Levy, H., Amato, S., Vockley, J., Zori, R., Dimmock, D., et al. (2018). 'Pegvaliase for the treatment of phenylketonuria: Results of a long-term phase 3 clinical trial program (PRISM). Mol. Genet. Metab. 124, 27–38. doi:10.1016/j.ymgme.2018.03.006
van der Goot, E., Bruinenberg, V. M., Hormann, F. M., Eisel, U. L. M., van Spronsen, F. J., and Van der Zee, E. A. (2019). 'Hippocampal microglia modifications in C57Bl/6 Pah(enu2) and BTBR Pah(enu2) phenylketonuria (PKU) mice depend on the genetic background, irrespective of disturbed sleep patterns. Neurobiol. Learn. Mem. 160, 139–143. doi:10.1016/j.nlm.2018.05.002
van Spronsen, F. J., Blau, N., Harding, C., Burlina, A., Longo, N., and Bosch, A. M. (2021). Phenylketonuria. Nat. Rev. Dis. Prim. 7, 36. doi:10.1038/s41572-021-00267-0
van Spronsen, F. J., Himmelreich, N., Rüfenacht, V., Shen, N., Vliet, D. V., Al-Owain, M., et al. (2017). 'Heterogeneous clinical spectrum of DNAJC12-deficient hyperphenylalaninemia: From attention deficit to severe dystonia and intellectual disability. J. Med. Genet. 55, 249–253. doi:10.1136/jmedgenet-2017-104875
van Vliet, D., Bruinenberg, V. M., Mazzola, P. N., van Faassen, M. H., de Blaauw, P., Kema, I. P., et al. (2015). 'Large neutral amino acid supplementation exerts its effect through three synergistic mechanisms: Proof of principle in phenylketonuria mice. PLoS One 10, e0143833. doi:10.1371/journal.pone.0143833
van Vliet, D., Bruinenberg, V. M., Mazzola, P. N., van Faassen, M. H., de Blaauw, P., Pascucci, T., et al. (2016). 'Therapeutic brain modulation with targeted large neutral amino acid supplements in the Pah-enu2 phenylketonuria mouse model. Am. J. Clin. Nutr. 104, 1292–1300. doi:10.3945/ajcn.116.135996
van Vliet, D., van Wegberg, A. M. J., Ahring, K., Bik-Multanowski, M., Blau, N., Bulut, F. D., et al. (2018). 'Can untreated PKU patients escape from intellectual disability? A systematic review. Orphanet J. Rare Dis. 13, 149. doi:10.1186/s13023-018-0890-7
van Wegberg, A. M. J., MacDonald, A., Ahring, K., Bélanger-Quintana, A., Blau, N., Bosch, A. M., et al. (2017). 'The complete European guidelines on phenylketonuria: Diagnosis and treatment. Orphanet J. Rare Dis. 12, 162. doi:10.1186/s13023-017-0685-2
Villiger, L., Grisch-Chan, H. M., Lindsay, H., Ringnalda, F., Pogliano, C. B., Allegri, G., et al. (2018). 'Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med. 24, 1519–1525. doi:10.1038/s41591-018-0209-1
Waldrop, M. A., Karingada, C., Storey, M. A., Powers, B., Iammarino, M. A., Miller, N. F., et al. (2020). 'Gene therapy for spinal muscular atrophy: Safety and early outcomes. Pediatrics 146, e20200729. doi:10.1542/peds.2020-0729
Wang, D., Tai, P. W. L., and Gao, G. (2019). 'Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 18, 358–378. doi:10.1038/s41573-019-0012-9
Welch, E. M., Barton, E. R., Zhuo, J., Tomizawa, Y., Friesen, W. J., Trifillis, P., et al. (2007). 'PTC124 targets genetic disorders caused by nonsense mutations. Nature 447, 87–91. doi:10.1038/nature05756
Winn, S. R., Scherer, T., Thöny, B., and Harding, C. O. (2016). 'High dose sapropterin dihydrochloride therapy improves monoamine neurotransmitter turnover in murine phenylketonuria (PKU). Mol. Genet. Metab. 117, 5–11. doi:10.1016/j.ymgme.2015.11.012
Woo, S. L., Lidsky, A. S., Güttler, F., Chandra, T., and Robson, K. J. (1983). 'Cloned human phenylalanine hydroxylase gene allows prenatal diagnosis and carrier detection of classical phenylketonuria. Nature 306, 151–155. doi:10.1038/306151a0
Xiang, L., Tao, J., Deng, K., Li, X., Li, Q., Yuan, X., et al. (2019). 'Phenylketonuria incidence in China between 2013 and 2017 based on data from the Chinese newborn screening information system: A descriptive study. BMJ Open 9, e031474. doi:10.1136/bmjopen-2019-031474
Yin, S., Ma, L., Shao, T., Zhang, M., Guan, Y., Wang, L., et al. (2022). 'Enhanced genome editing to ameliorate a genetic metabolic liver disease through co-delivery of adeno-associated virus receptor. Sci. China. Life Sci. 65, 718–730. doi:10.1007/s11427-020-1744-6
Keywords: phenylalanine (Phe), hyperphenylalaninemia (HPA), phenylketonuria (PKU), phenylalanine hydroxylase (PAH), tetrahydrobiopterin (BH4), DnaJ heat shock protein family (Hsp40) member C12 (DNAJC12), dietary restriction, gene therapy
Citation: Chen A, Pan Y and Chen J (2023) Clinical, genetic, and experimental research of hyperphenylalaninemia. Front. Genet. 13:1051153. doi: 10.3389/fgene.2022.1051153
Received: 22 September 2022; Accepted: 06 December 2022;
Published: 04 January 2023.
Edited by:
Tieliu Shi, Hunan University of Arts and Science, ChinaReviewed by:
Siqi Hu, Seventh Medical Center of PLA General Hospital, ChinaYu Leng Phua, Mount Sinai Genomics, Inc., United States
Copyright © 2023 Chen, Pan and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yukun Pan, cGFueXVrdW5AYmFyYmVsbHR4LmNvbQ==; Jinzhong Chen, a2luZ2JlbGxjaGVuQGZ1ZGFuLmVkdS5jbg==