
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 03 November 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.1047474
 Yusra Alyafee1
Yusra Alyafee1 Abeer Al Tuwaijri1,2
Abeer Al Tuwaijri1,2 Muhammad Umair1
Muhammad Umair1 Mashael Alharbi1Shahad Haddad1Maryam Ballow1Latifah Alayyar1
Mashael Alharbi1Shahad Haddad1Maryam Ballow1Latifah Alayyar1 Qamre Alam1Saleh Althenayyan1Nadia Al Ghilan3Aziza Al Khaldi4
Qamre Alam1Saleh Althenayyan1Nadia Al Ghilan3Aziza Al Khaldi4 Majid S. Faden5Hamad Al Sufyan6
Majid S. Faden5Hamad Al Sufyan6 Majid Alfadhel1,7*
Majid Alfadhel1,7*Background: In pregnant women at risk of autosomal recessive (AR) disorders, prenatal diagnosis of AR disorders primarily involves invasive procedures, such as chorionic villus sampling and amniocentesis.
Methods: We collected blood samples from four pregnant women in their first trimester who presented a risk of having a child with an AR disorder. Cell-free DNA (cfDNA) was extracted, amplified, and double-purified to reduce maternal DNA interference. Additionally, whole-genome amplification was performed for traces of residual purified cfDNA for utilization in subsequent applications.
Results: Based on our findings, we detected the fetal status with the family corresponding different genes, i.e., LZTR1, DVL2, HBB, RNASEH2B, and MYO7A, as homozygous affected, wild-type, and heterozygous carriers, respectively. Results were subsequently confirmed by prenatal amniocentesis. The results of AmpFLSTR™ Identifiler™ presented a distinct profile from the corresponding mother profile, thereby corroborating the result reflecting the genetic material of the fetus.
Conclusion: Herein, we detected AR disease mutations in the first trimester of pregnancy while surmounting limitations associated with maternal genetic material interference. Importantly, such detection strategies would allow the screening of pregnant women for common AR diseases, especially in highly consanguineous marriage populations. This technique would open avenues for the early detection and prevention of recessive diseases among the population.
The discovery of cell-free fetal DNA (cffDNA) in the mother’s plasma by Lo et al. (1997) in 1997 has paved the way for developing new applications in clinical practice that rely on analyzing fetal genetic material (Rather et al., 2019). cffDNA comprises approximately 3%–13% of the mother’s cell-free DNA (cfDNA) and is released into maternal circulation from placental cells undergoing apoptosis. The amount of cffDNA increases with gestational age and is cleared hours after delivery (Taglauer et al., 2014). Rapid advances in sequencing technology have facilitated the analysis of circulating cffDNA with considerable sensitivity and specificity. Since 2011, cffDNA has been employed to screen pregnant women at high risk for common fetal trisomies (Benachi et al., 2015).
Currently, the use of cffDNA for fetal screening is a rapidly developing technology, but its application remains limited. These limitations are primarily attributed to the overwhelming quantity of maternal genetic material when compared with that of fetal origin (Gerson and O'Brien, 2018). With the resolution of technical challenges and advances in genetic research, the use of cffDNA for fetal screening is expected to grow (Dhallan et al., 2007). In 2011, the UK National Health Service applied non-invasive prenatal testing (NIPT) in clinical practice for fetal sex determination (Hill et al., 2011). In 2014, the prediction of the fetal RhD type was introduced into clinical practice for the early detection and management of pregnancies in RhD-immunized women in Europe (Clausen, 2014). Cell-free fetal sex determination is currently used in NIPT services for several families with X-linked disorders, such as Duchenne muscular dystrophy and hemophilia. NIPT has also been introduced in the UK for de novo or paternal autosomal dominant mutations, such as achondroplasia. The main approach for NIPT tests is PCR, followed by digestion with a restriction enzyme (PCR-RED) (Chitty et al., 2011). The development of a new expanded next-generation sequencing panel has allowed the accurate assessment of several variants, as previously described for autosomal dominant disorders (Zhang et al., 2019). Meanwhile, identifying the most accurate, sensitive, and specific approach for NIPD of autosomal recessive (AR) disorders, in which both parents carry the same mutation as the fetus, presents considerable challenges. This can be attributed to the low level of fetal mutant alleles when compared with the high background of the maternal mutant allele in the circulating cfDNA in maternal plasma. In NIPT for AR disorders, the non-invasive droplet-digital PCR (ddPCR) assay requires the design of specific probes for familial mutations (Chang et al., 2018). Additional alternative approaches have been introduced, such as NIPT for β-thalassemia, in which haplotype dosage is assessed using semiconductor sequencing and target capture enrichment combined with specific target capture sequences (Saba et al., 2017; Koumbaris et al., 2019). These techniques have allowed the identification of haplotypes inherited by the fetus. Unfortunately, the demographic characteristics of examined samples were not provided, which is essential for detecting positivity rates. Therefore, additional improvements remain critical.
For utilizing cffDNA in AR disease screening, technological advances are particularly important in populations with high consanguineous marriages, where rare AR disorders are common (Alkuraya, 2014). Developing a screening strategy to identify family-specific AR disorders during the early stages of pregnancy remains crucial.
In the present study, we developed and validated a highly sensitive and specific whole-genome amplification approach for cffDNA purification and enrichment to screen pregnant women at high risk of having a child with family-specific AR disease. Our approach could overcome limitations such as maternal genomic interference and low fetal fractions. Furthermore, we demonstrated the feasibility of applying this strategy to pregnant women in the first trimester. The developed method could facilitate substantial advances in detecting and preventing several family-oriented AR diseases, especially in highly consanguineous marriage populations.
This study was approved by the Institutional Review Board (IRB) of the King Abdullah International Medical Research Center (KAIMRC), Riyadh, Saudi Arabia (NRC21R/083/03 approved 1 September 2022). The patients underwent a full clinical assessment for genetic and rare diseases at the Obstetrics and Gynecology Clinic of the National Guard Hospital (NGH), Riyadh, Saudi Arabia. Written informed consent for conducting the experiments and subsequent publication and storage of clinical data was provided and signed by each pregnant woman enrolled in this study.
We recruited four pregnant women and partners who were carriers of an AR disorder. All women had conceived naturally and had a singleton pregnancy with a gestational age of >10 weeks. Full and detailed family histories of any genetic disorder were obtained, along with the weight and height of each patient. An ultrasound scan was performed to confirm the number of fetuses and gestational age.
Approximately 10 ml of the peripheral venous blood sample was collected in an EDTA tube from each pregnant woman, and each tube was labeled with a unique medical record number and accession number. Extraction and library amplification of cffDNA were performed as described earlier (Alyafee et al., 2021a). Moreover, 15 ml of amniotic fluid was collected, and fetus-precipitated cells underwent direct DNA extraction using Gentra® Puregene® Tissue (Hilden, Germany) in accordance with the manufacturer’s protocol.
cffDNA libraries were size selected to ensure that fragments were within the suitable read length range of <160 bp length. First, paramagnetic beads were added to the DNA library pool, binding to the larger unwanted fragments in the sample, while the supernatant containing the desired cffDNA library was retained. Furthermore, the size-selected DNA library pool was quantified in accordance with the manufacturer’s protocol (High Sensitivity DNA Reagents & Agilent 2,100 Bioanalyzer) (Liao et al., 2014). Subsequently, the cffDNA library product was purified and cleaned twice using magnetic beads (G-Biosciences) to substantially eliminate larger DNA of maternal origin and increase the cfDNA concentration of fetal origin (Sparks et al., 2012).
The cffDNA enrichment was performed using a ReproSeq kit (Cat No. A34899, Thermo Fisher Scientific) according to the manufacturer’s protocol.
Whole-genome amplification (WGA) products of cffDNA were used to detect direct mutations using Sanger sequencing combined with short tandem repeat (STR) identifier analysis for a haplotyping-based approach (Asiri et al., 2020; Umair et al., 2020). For regions flanking mutations in DVL2, LZTR1, HBB, MYO7A, and RNASEH2B genes, the PCR reactions were performed using cffDNA WGA products in 20 μl reaction mixture consisting of 1× PCR buffer, 5 μl of 1/10 μl of the MDA product or genomic DNA, 0.5 pmol/μl of each primer set, and 10 μl AmpliTaq 360 Gold DNA Polymerases (Applied Biosystems). The initial enzyme activation and denaturation were performed at 95°C for 10 min. Thermal cycling was performed under the following conditions: 30 cycles of denaturation at 94°C for 1 min, annealing at 55°C for 1 min, and extension at 70°C for 1 min. The PCR products were subjected to cycle sequencing using a BigDye Terminator version 3.1 Ready Reaction Cycle Sequencing Kit with the forward primer. The amplified products were loaded onto an ABI Prism 3710XL Genetic Analyzer with the POP- 7 polymer (Barhoumi et al., 2019; Alhamoudi et al., 2020).
A panel of informative STR markers is located within a 2 MB interval around LZTR1, DVL2, and HBB. Linked markers were selected using the UCSC Genome Browser (http://genome.ucsc.edu/). The residual WGA product of cffDNA was used to confirm the haplotyping results. All forward primers were fluorescently labeled at the 5′ end by either fluorescein (Fam) or hexachlorofluorescein (HEX). The PCR was performed in 20 μl reactions, containing 5 μl of WGA product of cffDNA or 50 ng/μl genomic DNA, 10 µl of AmpliTaq 360 Gold DNA Polymerases (Applied Biosystems), and 0.5 pmol/μl of each primer set. Initial enzyme activation and denaturation were performed at 95°C for 10 min, followed by thermal cycling under the following conditions: denaturation at 94°C for 1 min, annealing at 50°C for 1 min, and extension at 70°C for 1 min for 25 cycles of amplification. Fragment analysis was performed using GeneScan software and ABI PRISM 3710 Genetic Analyzer (Applied Biosystems). Finally, to detect cfDNA of maternal origin, AmpFLSTR™ Identifiler™ Plus PCR Amplification kit was used as described by the manufacturer (Applied Biosystems).
In the present study, we recruited four pregnant women to detect fetal-related AR disorders in the maternal blood using cfDNA. All eligible participants were of Saudi Arabian ethnicity, single fetuses, and carriers with partners for at least one AR disorder and at high risk of having an affected child. The average age of the recruited pregnant women was (33.8 ± 5.6) years (range, 26–39 years), and the average gestational age at the time of NIPT was 12 weeks (range, 10–14 weeks). The average cffDNA pre-enrichment fraction of reported cases was 9%, ranging from 6 to 13%. Clinical descriptions of all pregnant women recruited are summarized in Table 1.
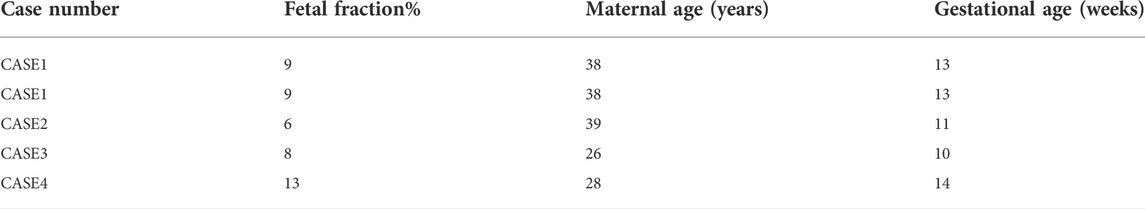
TABLE 1. General description for recruited patients.
Our first case was a 38-year-old pregnant woman with a previous history of an affected daughter with two pathogenic homozygous variants in [DVL2 (NM_004422.3): c.1690C>T; (p.Gln564Ter) and LZTR1 (NM_006767.4): c.1943–256C>T] genes. The patient was recruited for subsequent pregnancy at 13 weeks of gestation after ultrasound imaging revealed fetal hydrops. Regular NIPT results showed a high risk of Down syndrome, which was confirmed by arrayCGH. Moreover, the WGA product of cffDNA was used to detect whether the fetus was affected by the two previously mentioned mutated genes. Based on the direct point mutation detection results for both genes, the fetus was homozygous for the LZTR1 gene and wild-type for the DVL2 gene. To confirm the accuracy of obtained results, informative STR linking analysis was performed for both genes to detect the presence of the paternal allele, as well as exclude allele dropout and maternal genetic material interference. Moreover, the AmpFLSTR kit results revealed external and maternal contamination. The results of this patient are summarized in Figure 1.
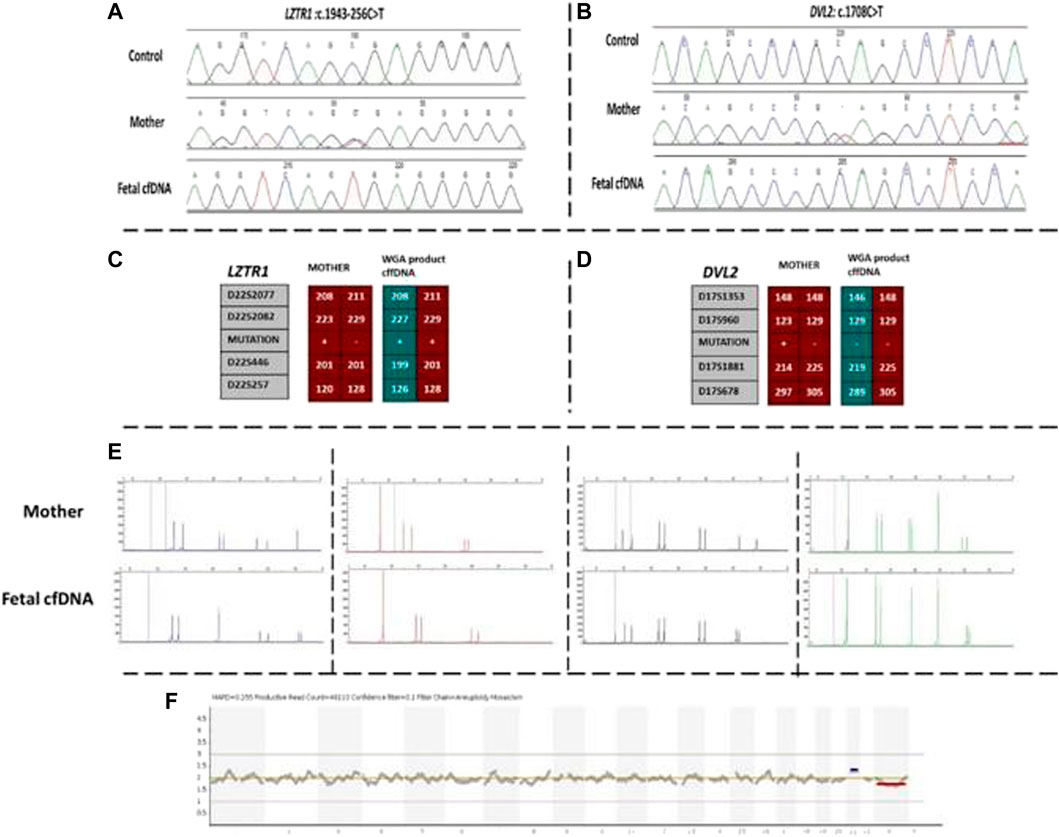
FIGURE 1. Findings of case 1. (A) Sanger results for LZTR1 gene for mother and WGA product of cffDNA. (B) Sanger results for DVL2 gene for mother and WGA product of cffDNA. (C,D) Karyomaps comparison for LZTR1 and DVL2, respectively. (E) AmpFLSTR™ Identifiler™ Plus kit results for the mother and WGA product of cffDNA to detect maternal interference. (F) Ion reporter genome viewer for the WGA product of the cffDNA showing the high risk of trisomy 21 in the male fetus. cffDNA, cell-free fetal DNA; WGA, Whole-genome amplification.
Next, we recruited additional patients to confirm that the developed method could be applied to different diseases and genomic regions. As described in Table 2, we enrolled three additional families with distinct genes involving different chromosomes. Results of direct point mutation of all genes using WGA products of cffDNA revealed the wild-type status of the fetus for cases 2 [HBB (NM_000518.5):c.17C>T; (p.Pro6Leu)] and 4 [MY O 7A (NM_001127180.2):c.1190C>T; (p.Ala397Val)], whereas the fetus of case 3 exhibited a carrier status [RNASEH2B (NM_024570.4):c.356A>G; (p.Asp119Gly)]. Similar to the first patient, allele dropout and maternal cell contamination interference were excluded by several informative STRs around related genes, and prenatal genetic amniocenteses confirmed the cffDNA results. The detailed results for all recruited patients are presented in Figures 2, 3.
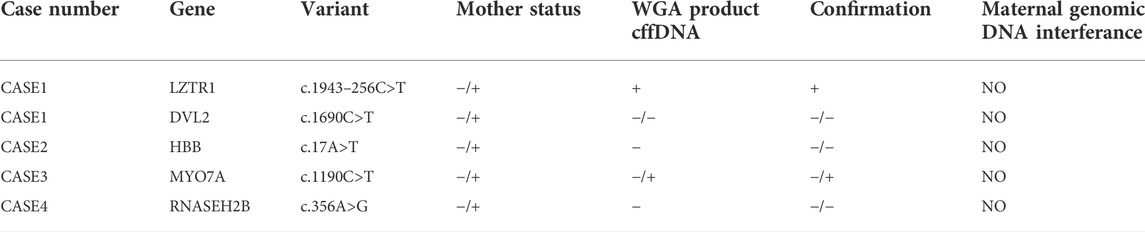
TABLE 2. Summary of results and outcomes observed.
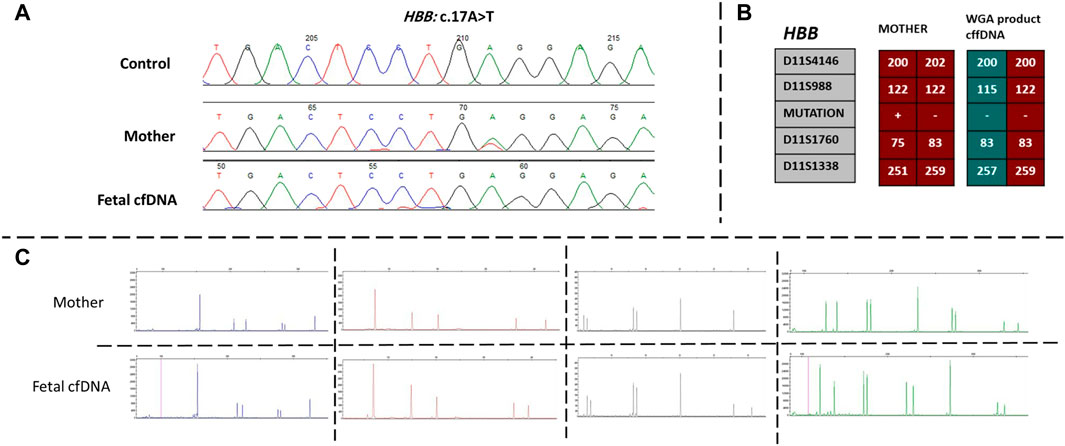
FIGURE 2. Findings of case 2. (A) Sanger results for HBB gene for mother and WGA product of cffDNA. (B) Karyomaps comparison for HBB, gene between the mother and WGA product of cffDNA to detect the paternal allele and exclude ADO probability. (C) AmpFLSTR™ Identifiler™ Plus kit results for the mother and WGA product of cffDNA to detect maternal interference. ADO, allele dropout; cffDNA, cell-free fetal DNA; WGA, Whole-genome amplification.
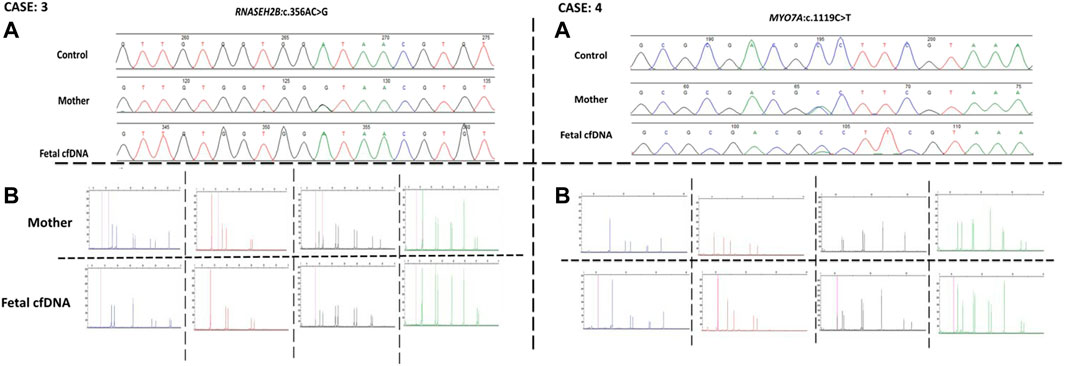
FIGURE 3. Case 3 and case 4 findings. (A) Sanger results for RNASEH2B, and MYOA7A genes respectively, for the mothers and WGA products of cffDNA. (B) AmpFLSTR™ Identifiler™ Plus kit results for the mothers and WGA product of cffDNA to detect maternal interference.
Following the discovery of cffDNA by Lo et al. (1997) in pregnant women, NIPT with high-throughput sequencing has been widely adopted for the clinical detection of chromosomal abnormalities. This high-throughput DNA sequencing technique can rapidly and effectively detect large-scale genetic alterations with high accuracy and specificity for trisomies T21, T18, and T13 (Gregg et al., 2016).
Currently, NIPT for aneuploidy is commonly used worldwide as a preliminary screening test for the most common fetal trisomies (T21, T18, and T13) (Scotchman et al., 2020). NIPT is employed either exclusively as an initial test or confirmed by regular invasive prenatal testing. For non-invasive prenatal diagnosis of monogenic disorders, several technical and analytical obstacles still need to be resolved. Herein, we aimed to develop a new approach using cfDNA in fetal screening for AR diseases. First-trimester screening for AR diseases can be vital, specifically for populations with high numbers of consanguineous marriages. Considering these populations, a significant percentage of individuals might be carriers of one or more AR disorders, potentially inducing a high burden on families and respective communities. Currently, the only preventive measure for AR in these populations involves premarital screening or carrier screening and subsequent preimplantation genetic testing for monogenetic disorders if both partners are found to be carriers (Alfadhel et al., 2021, 2022).
Therefore, the availability of suitable screening methods for a wide range of AR disorders within a specific community is a promising strategy for early preventive measures (Alfadhel et al., 2019; Alyafee et al., 2021b). The probability of a fetus being affected or healthy has been successfully reported in previous reports; however, limitations such as maternal genetic DNA interference and/or low fetal fraction were found to persist. In the present study, we enriched the percentage of fetal cfDNA by double-size selection purification, as previously described (Liang et al., 2018; Hu et al., 2019). Subsequently, to overcome the effect of double purification of markedly low levels of cfDNA starting material, we used a WGA approach using an Ampliseq library preparation kit. This facilitated the amplification of our target cffDNA to reach detectable levels, similar to the direct detection of point mutations. To confirm the applicability of our developed approach, we attempted to detect the fetal status of four pregnant women carriers and their partners with different AR diseases. The first case was a couple who were carriers of an extremely rare and likely pathogenic mutation with a previous history of an affected child. Our approach enabled the detection of high-risk fetal aneuploidy, mosaic trisomy 21, as well as allowed the accurate identification of the fetal status of two different genes within two different chromosomes. Moreover, reducing maternal genetic interference allowed the detection of an affected homozygous fetus for LZTR1 gene variants causing schwannomatosis disease and wild-type status for the DVL2 gene. Case 2 presented similar findings, where we detected the wild-type status of the sickle cell disease mutation in a normal healthy fetus. In addition, we detected non-maternal alleles to rule out the likelihood of allele dropout in determining the wild-type status.
We recruited two additional patients for further confirmation. The two couples were carriers of pathogenic variants in the MYO7A gene (Usher syndrome) and RNASEH2B (Aicardi-Goutières syndrome). The results demonstrate the ability of our approach to minimize maternal DNA interference while detecting the single-point mutation status of the fetus. Furthermore, all cffDNA results were confirmed by Sanger sequencing or arrayCGH of fetal cells obtained by amniocenteses.
In the present study, we present a successful approach for isolating and enriching fetal cfDNA to detect AR diseases. The ability to detect high-risk fetuses in the first trimester of pregnancy, especially in populations with AR diseases, has great value. The mutant allele carrier status in adults is approximately 1%, and among newborns, mutant alleles account for nearly 10% of pediatric hospitalizations and ∼20% of infant mortalities. New and effective methods, such as the method presented in the present study, that can aid in rapid prenatal diagnosis are needed to prevent genetic disorders and improve the quality of life.
In conclusion, this feasibility study examined a novel approach for NIPT of AR disorders. In this approach, cffDNA underwent double-size selection and purification, followed by WGA for enrichment. Using this approach, we could precisely detect the fetal status considering the corresponding family gene. Therefore, this study offers solutions to reduce maternal genetic interference and low levels of cffDNA, aiding in the more accurate detection of high-risk fetuses with AR disorders. Further confirmation with additional cases involving different genes and locations is required for optimum results to evaluate the percentage of false-positive and false-negative cases. Moreover, additional studies assessing factors that may impact the final cffDNA yield and quality should be considered to further validate this new approach.
The data presented in the study are deposited in the LOVD online repository: https://databases.lovd.nl/. Individual ID # 00418812, 00418890, 00418891.
The studies involving human participants were reviewed and approved by King Abdullah International Medical Research Center (KAIMRC), Riyadh, Saudi Arabia (NRC21R/083/03 approved 1 September 2022). The patients/participants provided their written informed consent to participate in this study.
YA: Designed and wrote the project; MA: Project Management, editing, and PI; MU: Manuscript writing, editing, and submission; SH, MA, MB, and QA: Performed the experiment; AAT, MA, SH, MB, LA, QA, and SA: counselors; AAK: logistic support; MF, NA, and HA: patient recruitment.
This work was funded by the King Abdullah International Medical Research Center (Project No. NRC21R/083/03).
The authors thank the participants and their families for participating in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AR, autosomal recessive disorder; CGH, comparative genomic hybridization; CFF DNA, cell-free fetal DNA; IRB, Institutional Review Board; KAIMRC, King Abdullah International Medical Research Centre; NGH, National Guard Hospital; NSG, next-generation sequencing; NIPT, non-invasive prenatal testing; WGA, whole-genome amplification
Alfadhel, M., Abadel, B., Almaghathawi, H., Umair, M., Rahbeeni, Z., Faqeih, E. A., et al. (2022). HMG-CoA lyase deficiency: A retrospective study of 62 Saudi patients. Front. Genet. 13, 880464. doi:10.3389/fgene.2022.880464
Alfadhel, M., Almuqbil, M., Mutairi, F. A., Umair, M., Almannai, M., Alghamdi, M., et al. (2021). The leukodystrophy Spectrum in Saudi Arabia: Epidemiological, clinical, Radiological and genetic data. Front. Pediatr. 9, 633385. doi:10.3389/fped.2021.633385
Alfadhel, M., Umair, M., Almuzzaini, B., Alsaif, S., AlMohaimeed, S. A., Almashary, M. A., et al. (2019). Targeted SLC19A3 gene sequencing of 3000 Saudi newborn: A pilot study toward newborn screening. Ann. Clin. Transl. Neurol. 6 (10), 2097–2103. doi:10.1002/acn3.50898
Alhamoudi, K. M., Bhat, J., Nashabat, M., Alharbi, M., Alyafee, Y., Asiri, A., et al. (2020). A Missense mutation in the UGDH gene is associated with developmental delay and Axial Hypotonia. Front. Pediatr. 8, 71. doi:10.3389/fped.2020.00071
Alkuraya, F. S. (2014). Genetics and genomic medicine in Saudi Arabia. Mol. Genet. genomic Med. 2 (5), 369–378. doi:10.1002/mgg3.97
Alyafee, Y., Al Tuwaijri, A., Alam, Q., Umair, M., Haddad, S., Alharbi, M., et al. (2021a). Next generation sequencing based non-invasive prenatal testing (NIPT): First report from Saudi Arabia. Front. Genet. 12, 630787. doi:10.3389/fgene.2021.630787
Alyafee, Y., Alam, Q., Altuwaijri, A., Umair, M., Haddad, S., Alharbi, M., et al. (2021b). Next-generation sequencing-based pre-Implantation genetic testing for aneuploidy (PGT-A): First report from Saudi Arabia. Genes (Basel) 12 (4), 461. doi:10.3390/genes12040461
Asiri, A., Aloyouni, E., Umair, M., Alyafee, Y., Tuwaijri, A. A., Alhamoudi, K. M., et al. (2020). Mutated RAP1GDS1 causes a new syndrome of dysmorphic feature, intellectual disability & speech delay. Ann. Clin. Transl. Neurol. 7 (6), 956–964. doi:10.1002/acn3.51059
Barhoumi, T., Nashabat, M., Ghanem, B., Al-hallaj, A. S., Boudjellal, M., Umair, M., et al. (2019). Delta like-1 gene mutation: A novel Cause of Congenital Vertebral Malformation. Front. Genet. 10, 534. doi:10.3389/fgene.2019.00534
Benachi, A., Letourneau, A., Kleinfinger, P., Senat, M. V., Gautier, E., Favre, R., et al. (2015). Cell-free DNA analysis in maternal plasma in cases of fetal abnormalities detected on ultrasound examination. Obstet. Gynecol. 125 (6), 1330–1337. doi:10.1097/AOG.0000000000000874
Chang, M. Y., Ahn, S., Kim, M. Y., Han, J. H., Park, H. R., Seo, H. K., et al. (2018). One-step noninvasive prenatal testing (NIPT) for autosomal recessive homozygous point mutations using digital PCR. Sci. Rep. 8 (1), 2877. doi:10.1038/s41598-018-21236-w
Chitty, L. S., Griffin, D. R., Meaney, C., Barrett, A., Khalil, A., Pajkrt, E., et al. (2011). New aids for the non-invasive prenatal diagnosis of achondroplasia: Dysmorphic features, charts of fetal size and molecular confirmation using cell-free fetal DNA in maternal plasma. Ultrasound Obstet. Gynecol. 37 (3), 283–289. doi:10.1002/uog.8893
Clausen, F. B. (2014). Integration of noninvasive prenatal prediction of fetal blood group into clinical prenatal care. Prenat. Diagn. 34 (5), 409–415. doi:10.1002/pd.4326
Dhallan, R., Guo, X., Emche, S., Damewood, M., Bayliss, P., Cronin, M., et al. (2007). A non-invasive test for prenatal diagnosis based on fetal DNA present in maternal blood: A preliminary study. Lancet (London, Engl. 369 (9560), 474–481. doi:10.1016/S0140-6736(07)60115-9
Gerson, K. D., and O'Brien, B. M. (2018). Cell-free DNA: Screening for single-gene disorders and determination of fetal rhesus D Genotype. Obstet. Gynecol. Clin. North Am. 45 (1), 27–39. doi:10.1016/j.ogc.2017.11.001
Gregg, A. R., Skotko, B. G., Benkendorf, J. L., Monaghan, K. G., Bajaj, K., Best, R. G., et al. (2016). Noninvasive prenatal screening for fetal aneuploidy, 2016 update: A position statement of the American College of medical genetics and genomics. Genet. Med. 18 (10), 1056–1065. doi:10.1038/gim.2016.97
Hill, M., Finning, K., Martin, P., Hogg, J., Meaney, C., Norbury, G., et al. (2011). Non-invasive prenatal determination of fetal sex: Translating research into clinical practice. Clin. Genet. 80 (1), 68–75. doi:10.1111/j.1399-0004.2010.01533.x
Hu, P., Liang, D., Chen, Y., Lin, Y., Qiao, F., Li, H., et al. (2019). An enrichment method to increase cell-free fetal DNA fraction and significantly reduce false negatives and test failures for non-invasive prenatal screening: A feasibility study. J. Transl. Med. 17 (1), 124. doi:10.1186/s12967-019-1871-x
Koumbaris, G., Achilleos, A., Nicolaou, M., Loizides, C., Tsangaras, K., Kypri, E., et al. (2019). Targeted capture enrichment followed by NGS: Development and validation of a single comprehensive NIPT for chromosomal aneuploidies, microdeletion syndromes and monogenic diseases. Mol. Cytogenet. 12, 48. doi:10.1186/s13039-019-0459-8
Liang, B., Li, H., He, Q., Li, H., Kong, L., Xuan, L., et al. (2018). Enrichment of the fetal fraction in non-invasive prenatal screening reduces maternal background interference. Sci. Rep. 8 (1), 17675. doi:10.1038/s41598-018-35738-0
Liao, C., Yin, A. H., Peng, C. F., Fu, F., Yang, J. X., Li, R., et al. (2014). Noninvasive prenatal diagnosis of common aneuploidies by semiconductor sequencing. Proc. Natl. Acad. Sci. U. S. A. 111 (20), 7415–7420. doi:10.1073/pnas.1321997111
Lo, Y. M., Corbetta, N., Chamberlain, P. F., Rai, V., Sargent, I. L., Redman, C. W., et al. (1997). Presence of fetal DNA in maternal plasma and serum. Lancet (London, Engl. 350 (9076), 485–487. doi:10.1016/S0140-6736(97)02174-0
Rather, R. A., Dhawan, V., and Saha, S. C. (2019). Non-invasive prenatal rhesus D genotyping using cell-free foetal DNA. Indian J. Med. Res. 150 (1), 62–66. doi:10.4103/ijmr.IJMR_1787_17
Saba, L., Masala, M., Capponi, V., Marceddu, G., Massidda, M., and Rosatelli, M. C. (2017). Non-invasive prenatal diagnosis of beta-thalassemia by semiconductor sequencing: A feasibility study in the Sardinian population. Eur. J. Hum. Genet. 25 (5), 600–607. doi:10.1038/ejhg.2017.26
Scotchman, E., Chandler, N. J., Mellis, R., and Chitty, L. S. (2020). Noninvasive prenatal diagnosis of single-gene diseases: The next Frontier. Clin. Chem. 66 (1), 53–60. doi:10.1373/clinchem.2019.304238
Sparks, A. B., Struble, C. A., Wang, E. T., Song, K., and Oliphant, A. (2012). Noninvasive prenatal detection and selective analysis of cell-free DNA obtained from maternal blood: Evaluation for trisomy 21 and trisomy 18. Am. J. Obstet. Gynecol. 206 (4), 319.e311–e9. doi:10.1016/j.ajog.2012.01.030
Taglauer, E. S., Wilkins-Haug, L., and Bianchic, D. W. (2014). Review: Cell-free fetal DNA in the maternal circulation as an indication of placental health and disease. Placenta 35, S64–S68. doi:10.1016/j.placenta.2013.11.014
Umair, M., Ballow, M., Asiri, A., Alyafee, Y., Tuwaijriet, A. A., Alhamoudi, K. M., et al. (2020). EMC10 homozygous variant identified in a family with Global developmental delay, Mild intellectual disability, and speech delay. Clin. Genet. 98 (6), 555–561. doi:10.1111/cge.13842
Keywords: autosomal recessive, AR, fetal DNA, non-invasive prenatal testing for monogenic disorders (NIPTM), Single gene, whole-genome amplification
Citation: Alyafee Y, Al Tuwaijri A, Umair M, Alharbi M, Haddad S, Ballow M, Alayyar L, Alam Q, Althenayyan S, Al Ghilan N, Al Khaldi A, Faden MS, Al Sufyan H and Alfadhel M (2022) Non-invasive prenatal testing for autosomal recessive disorders: A new promising approach. Front. Genet. 13:1047474. doi: 10.3389/fgene.2022.1047474
Received: 18 September 2022; Accepted: 19 October 2022;
Published: 03 November 2022.
Edited by:
Musharraf Jelani, Islamia College University, PakistanReviewed by:
Waleed Hashmi, Ohio University, United StatesCopyright © 2022 Alyafee, Al Tuwaijri, Umair, Alharbi, Haddad, Ballow, Alayyar, Alam, Althenayyan, Al Ghilan, Al Khaldi, Faden, Al Sufyan and Alfadhel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Majid Alfadhel, ZHJhbGZhZGhlbG1AZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.