 Siddra Ijaz
Siddra Ijaz Imran Ul Haq
Imran Ul Haq Iqrar Ahmad Khan3
Iqrar Ahmad Khan3 Hayssam M. Ali
Hayssam M. Ali Sukhwinder Kaur
Sukhwinder Kaur- 1Centre of Agricultural Biochemistry and Biotechnology, University of Agriculture Faisalabad, Faisalabad, Pakistan
- 2Department of Plant Pathology, University of Agriculture Faisalabad, Faisalabad, Pakistan
- 3Institute of Horticultural Sciences, University of Agriculture Faisalabad, Faisalabad, Pakistan
- 4Botany and Microbiology Department, College of Science, King Saud University, Riyadh, Saudi Arabia
- 5Department of Plant Pathology, University of California, Davis, Davis, CA, United States
Dalbergia sissoo is an important timber tree, and dieback disease poses a dire threat to it toward extinction. The genomic record of D. sissoo is not available yet on any database; that is why it is challenging to probe the genetic elements involved in stress resistance. Hence, we attempted to unlock the genetics involved in dieback resistance through probing the NBS-LRR family, linked with mostly disease resistance in plants. We analyzed the transcriptome of D. sissoo under dieback challenge through DOP-rtPCR analysis using degenerate primers from conserved regions of NBS domain-encoded gene sequences. The differentially expressed gene sequences were sequenced and in silico characterized for predicting the expressome that contributes resistance to D. sissoo against dieback. The molecular and bioinformatic analyses predicted the presence of motifs including ATP/GTP-binding site motif A (P-loop NTPase domain), GLPL domain, casein kinase II phosphorylation site, and N-myristoylation site that are the attributes of proteins encoded by disease resistance genes. The physicochemical characteristics of identified resistance gene analogs, subcellular localization, predicted protein fingerprints, in silico functional annotation, and predicted protein structure proved their role in disease and stress resistance.
1 Introduction
Dalbergia sissoo is an important perennial tree of great economic importance due to its significance in agroforestry, forestry, and horticulture. Although it is native to the sub-Himalayan tract, it is abundantly found in other regions of Asia and the southern and central countries of America as well (Ijaz and Haq, 2021). Its high-quality timber is used in furniture making, fuel, etc. It had been severely affected by dieback disease, which was announced as an epidemic for this tree, mostly in the Punjab province (Haq et al., 2021; Ijaz and Haq, 2021). Since then, dieback losses in the D. sissoo trees have increased by 40–80%. More than 70% of plant diseases are caused by fungi (Moore et al., 2011; Ul Haq et al., 2022). Plant scientists have documented the strong involvement of phytofungi in trees’ decline compared to other ecological dynamics, including edaphic, climatic, and biotic factors. The decline is the most threatening stress on tree species, and the D. sissoo decline leads this tree to the danger of extinction (Orwa et al., 2009; Shah et al., 2010). The use of resistant germplasm to control dieback will be the most effective way to attain the disease’s sustainable management, especially for new plantations (Minocha et al., 2000). Plants possess complex defense mechanisms against phytopathogens (Sarwar et al., 2022a) that lead to biochemical and physiological alterations in plants. The first level includes pathogen recognition, anti-pathogenic protein production, interruption of pathogen infection structures, and enzymatic cell wall reinforcement (Sarwar et al., 2022b). If a pathogen overcomes it, then the second level is initiated. This level includes resistance (R) genes or their products and starts a molecular cascade of signal transductions in response to the attack (Wang et al., 2015), including kinases, phytoalexin, peroxidases, reactive oxygen species, and guanine nucleotide-binding proteins (Meng and Zhang, 2013; Zhong et al., 2019; Çetinel et al., 2022).
Resistance (R) genes encode the effector-triggered immunity system (Jones and Dangl, 2006). Most R genes encode intracellular proteins and have nucleotide-binding site (NBS) and leucine-rich repeat (LRR) domains. These proteins belong to the apoptotic ATPase (AP-ATPase or NB-ARC ATPase) family of STAND (Signal Transduction ATPase with numerous domains) P-loop NTPase. These NTPases are signal-generating bodies and act as plant and animal defense mechanisms’ switches. In the NBS-LRR class of proteins, apart from the NBS and LRR domains, a homologous region is present between these two domains, known as ARC (Riedl et al., 2005). Multiple conserved motifs, including P-loop, kinases, hydrophobic GLPL, and RNBS (resistance nucleotide-binding site), are present in the NB-ARC domain of R proteins (Baldi et al., 2004). A total of three peptide motifs in NBS are crucial for nucleotide-binding in various ATP/GTP bindings. These peptides include the P-loop (also known as a kinase-1a motif or Walker A motif), kinase-2 motif, and kinase-3a motif (Babar et al., 2021; Nasir et al., 2021).
Because of the plant defense system against pathogens and R genes’ involvement in disease resistance, it is crystal clear that identifying plant disease-resistant genes helps disease-resistant breeding in plants and provides insight into the resistance mechanism. Therefore, considering these perspectives, the D. sissoo transcriptome was probed to identify the genetic elements differentially expressed under the dieback challenge. This research study was conducted under the CAS-PARB project no. 952. In this study, we attempted to explore D. sissoo’s transcriptome through a degenerate oligonucleotide-primed polymerase chain reaction because of the unavailability of its genomic data to identify resistance genes that contribute to dieback disease resistance. Differentially expressed gene sequences (under the dieback challenge) were identified and characterized using bioinformatics tools. This study paves the way toward resistance gene identification in D. sissoo (genomic record not available yet on any database) as the contributors of disease-resistance pathways that switch on under the dieback challenge.
2 Materials and methods
2.1 Plant material collection, plant inoculation, and screening
We collected two hundred plants of Dalbergia sissoo across Pakistan. The collected plant material maintained in the greenhouse was macro propagated and screened against the pathogen of dieback diseases. Macro propagated tagged material of D. sissoo was inoculated with the fungal pathogen Ceratocystis dalbergiae (MycoBank 841380). The inoculation was done by mixing fungal isolate into the soil of growing D. sissoo plants.
2.2 Genomic DNA extraction
Before plant inoculation, we extracted their total genomic DNA using a eukaryotic genomic DNA extraction kit (Thermo Scientific, United States) and analyzed it through PCR based on a 16S DNA marker for bacteria and an ITS marker for fungi.
2.3 RNA extraction and cDNA synthesis
Total RNA was extracted from the plant samples of Dalbergia sissoo showing resistance under dieback disease challenge. A 100 mg of plant material was used for RNA isolation and purification using the GeneJET Plant RNA Purification kit and RapidOut DNA Removal kit (Thermo Scientific, United States), respectively, opting for the protocol given by the manufacturer. The dried palette of each sample was dissolved in nuclease-free water and immediately used for down stress applications; the extracted RNA samples were analyzed at 260/280 nm and 260/230 nm absorbance using UV visible NANODROP (8000 Spectrophotometer, Thermo Scientific), followed by cDNA synthesis using RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, United States). The cDNA samples were diluted (20 folds) and stored (−20°C). The quantification of synthesized cDNA was determined (at 260/280 nm absorbance).
2.4 Primer designing
The conserved regions of the NBS-LRR class of R genes were targeted for probing the genetic elements involved in the resistance against dieback in Dalbergia sissoo. The degenerate primers used in DOP-rtPCR were designed based on the NBS domain. The NBS region of the NBS-LRR domain consists of different conserved domains, like kinase P-loop, kinase-2, kinase-3A, and hydrophobic GLPL motifs, which play an essential role in plant defense mechanisms (Supplementary Table S1).
2.5 Degenerate oligonucleotide-primed-reverse transcription PCR
The transcriptome probing of Dalbergia sissoo under dieback challenge was performed through degenerate oligonucleotide-primed-reverse transcription PCR (DOP-rtPCR). The PCR products were analyzed on a 1.2% high-resolution agarose gel (ACTGene). The amplicons were excised from the gel, eluted using a gel purification kit (Favor-Prep), and cloned in the TA cloning vector (pTZ57R/T); the cloned products were directly sequenced through Eurofins Genomics DNA sequencing services, United States. After sequencing, the generated sequences were trimmed (BioEdit version 7.2.6.1). The high-quality trimmed sequences were BLAST (basic local alignment search tool) in NCBI (National Center for Biotechnology Information) to check their homology to stress-responsive genetic elements. Furthermore, bioinformatics-based in silico analyses were performed for the structural and functional characterization of the identified sequences.
2.6 Scanning of conserved domains and protein fingerprints
The generated sequence was subjected to Blastx to check its homology. The ExPASy translate tool (https://web.expasy.org/translate/) was used to get translated protein sequences. The translated protein was scanned for structural motifs using the ScanProsite tool (https://prosite.expasy.org/scanprosite/). The conserved domains were identified by CD-search and CDART (Geer et al., 2002). However, protein fingerprints were scanned by the PRINTS database (http://130.88.97.239/cgi-bin/dbbrowser/fingerPRINTScan/FPScan_fam.cgi).
2.7 Protein localization and physicochemical characterization
The biological function of any protein is correlated to its subcellular location. Therefore, online servers CELLO v. 2.5 (Yu et al., 2006) and SignalP 5.0 (Armenteros et al., 2019) were used to predict the subcellular localization of proteins or signal peptides. The physicochemical parameters were computed using an online ExPASy tool, ProtParam (Gasteiger et al., 2005).
2.8 Functional annotation
The web-based tools MOTIF (https://www.genome.jp/tools/motif/) and ProtoNet (Rappoport et al., 2012) were employed to identify the conserved sites or motifs. The CATH database (Orengo et al., 1997) was used to identify superfamily and functional family predictions. Functional annotation in terms of ligand binding sites, active sites prediction was estimated through the I-TASSER web server (Yang and Zhang, 2015), and Gene Ontology (GO) (molecular function, biological process, and cellular component) was determined through COFACTOR (Zhang et al., 2017) and COACH (Yang et al., 2013).
2.9 Protein modeling
The secondary structures of the translated amino acid sequences of identified RGAs were predicted and annotated using the SOPMA (self-optimized prediction from multiple alignments) (Geourjon and Deleage, 1995) online server. For homology modeling analysis, an online web server, SWISS-MODEL (Waterhouse et al., 2018), and PHYRE2 (Protein Homology/analogy Recognition Engine, Version 2.0) (Kelley et al., 2015) were used. The templates with significant score values were selected. Their PDB files were used in PyMOL v. 2.4.0 for 3-D protein imaging. The predicted protein structures were validated in ProSA-web (protein structure analysis) (Wiederstein and Sippl, 2007), getting a Z-score or energy criteria. PROCHECK (Laskowski et al., 1993) was used to check quality along with stereochemistry, and TM-align (Zhang and Skolnick, 2005) was used for model superimposition with template whose molecular graphics were visualized in RasMol v.2.7.5 (Bernstein, 2009) and RasTop v. 2.2.5.
3 Results
3.1 Screening of inoculated Dalbergia sissoo plants
Dieback is considered a mysterious disease with no well-established or well-known etiology that leads to poor disease management. However, surveying for the D. sissoo plant collection, we found almost every plant affected by this disease. We generally observed that plants were resistant but also affected by dieback disease after some time. Therefore, we tried to collect the plant material that looked healthy, particularly those that stood healthy amongst the diseased plants. The healthy collected D. sissoo material was macro propagated under controlled conditions and maintained in a greenhouse in pots with sterilized autoclaved compost. We maintained sterile conditions before inoculation to ensure that the stress had to be due to the pathogen to be used in inoculation and not due to other factors. The collected material was screened by giving pathogen stress, and those plants that showed resistance were selected for downstream applications. Before inoculation, the healthy grown macro-propagated plant material of D. sissoo was probed for bacteria and fungi presence by subjecting their isolated DNA to 16S and fungal ITS DNA marker-based PCR analysis. However, we did not get any amplification in the case of both DNA markers. It showed the absence of any hidden stress by bacteria and fungi to ensure the given stress is just due to the inoculated pathogen, and the genes activated and upregulated would be against the particular stress. It was done to make the most refined identification of resistance genes or genetic elements against dieback disease. Inoculating the macro-propagated plant material of D. sissoo, sixteen out of 200 plants were screened as resistant or tolerant sources against dieback disease (Supplementary Table S2). Apparent dieback symptoms started one month after post-inoculation. Some plants died within a few days after symptoms appeared, while some plants died within a few months from the date of the symptoms’ appearance. However, plants showing resistance against challenge were tagged as resistant sources of D. sissoo. A total of sixteen plants showing resistance or tolerance under dieback challenge were subjected to degenerate oligonucleotide-primed-reverse transcription PCR (DOP-rtPCR) analysis.
3.2 Transcriptome-based identification of resistance gene analogs against dieback disease
Based on the NBS domain, degenerate primers were designed and synthesized to amplify resistance gene analogs (RGAs) or/candidate R genes against D. sissoo’s dieback disease by degenerate oligonucleotide-primed-reverse transcriptase PCR (DOP-rtPCR) analysis (Supplementary Table S1). Among these degenerate primers, dgPL-a2F/dgGL-b1R and dgPL-a2F/dgGL-b2R primer pairs gave amplifications in JSP2, NF1, and NFP2 plants of D. sissoo. The primer pair dgPL-a1F/dgGL-b1R gave amplification in the KPK P4 plant; the primer pair dgPL-a2F/dgGL-b2R gave amplification in the HP plant; the primer pair dgPL-a1F/dgGL-b2R gave amplification in the RKP2 plants. Amplified products were sequenced. The trimmed high-quality sequences were aligned through a multiple sequence alignment program, MAFFT (multiple alignment using fast Fourier transform). The alignment displayed three identified sequences as contig; hence, their consensus sequence was obtained using DNASTAR Lasergene v. 7.1.0 SeqMan pro (SeqManTMII). The contig sequence was designated as Ds-DbRCaG-01-Rga1. The rest were designated as Ds-DbRCaG-01-Rga2p, Ds-DbRCaG-03-Rga4p, and Ds-DbRCaG-05-Rga6p.
3.3 Conserved domain scanning
The translated protein sequence of the Ds-DbRCaG-01-Rga1 was subjected to CDART and CD-search web tools. The identified polynucleotide DNA sequences predicted to have encoded a polypeptide chain or a protein sequence with a signature motif (the P-loop NTPase domain belongs to the P-loop NTPase superfamily) are the attributes of disease resistance genes encoding proteins. For structural motifs’ the scanning ScanProsite tool was used that showed ATP/GTP-binding site motif A (P-loop) signature motif (GgkgqGKS) along with post-translational modification sites or PTM sites, as N-myristoylation site (MYRISTYL), Casein kinase II phosphorylation site (CK2_PHOSPHO_SITE) with phosphoserine as an intra-domain predicted feature. The phosphate-binding loop is a highly conserved motif of the nucleotide-binding site (NBS) of R genes involved in ATP/GTP binding and is a fundamental feature of ATP/GTP binding proteins (Saraste et al., 1990). However, the translated protein sequences of identified RGAs (Ds-DbRCaG-01-Rga2p, Ds-DbRCaG-03-Rga4p, and Ds-DbRCaG-05-Rga6p) showed no conserved domains upon searching through CDART and CD programs.
3.4 Protein fingerprint scanning
The translated protein’s fingerprint of identified RGA (Ds-DbRCaG-01-Rga1) was scanned by the PRINTS database (http://130.88.97.239/cgi-bin/dbbrowser/fingerPRINTScan/FPScan_fam.cgi). The fingerprint with the highest hit was “CSAPPISMRASE,” with an E-value of 2.5e+03. This five-element fingerprint is the cyclophilin peptidylprolyl cis-trans isomerase signature. Cyclophilins (CYPs) are members of the peptidylprolyl cis-trans isomerase family involved in catalyzing cis-trans isomerization of the peptidylprolyl bond. In plants, cyclophilin and cyclophilin-like proteins are present across all the subcellular sections. Their exact physiological role in plants is still a matter of speculation, with few exceptions. However, they have been involved in different physiological processes, including organogenesis, transcriptional regulation, hormone-signaling pathways, photosynthetic signaling pathways, stress adaptation, and defense responses (Barbosa and Park, 2019).
The translated protein sequence of Ds-DbRCaG-01-Rga2p, Ds-DbRCaG-03-Rga4p, and Ds-DbRCaG-05-Rga6p displayed no conserved domains in CDART and CD program-based searches. However, in FPScan, these deduced protein sequences predicted with protein fingerprints provided a link to their involvement in stress response or immune response. The Ds-DbRCaG-01-Rga2p is predicted with a BOMBESINR (bombesin receptor signature) fingerprint. The BOMBESINR, a six-element fingerprint, tags a signature to bombesin receptors belonging to guanine-nucleotide-binding-coupled receptors (GPCRs). The GPCRs are transmembrane receptors that work as signal transduction in response to environmental stimuli or extracellular signals. The GPCR family contains seven hydrophobic regions spanning the membrane as a C-terminal phosphorylated site in the cytoplasm and an N-terminus glycosylated site in the extracellular space. These seven transmembrane regions (helices) are linked via three extracellular loops and three alternative cytoplasmic loops. The six-motif signature of BOMBESINR, as a derivative from initially aligned conserved segments, highlights the aligned region, characteristic of bombesin receptors. In the six-motif region, motif-1 spans at the C terminal of the transmembrane domain-2, ushering into the first external loop, motif-2 is positioned at the N-terminus of transmembrane domain-3, motif-3 spans the 2nd external loop’ segment, motif-4 occupies the third cytoplasmic loop, motif-5 spans the third external loop’ segment, ushering into transmembrane domain-7; however, motif-6 resides at the C-terminal region. Hence, the translated sequence of Ds-DbRCaG-01-Rga2p showed homology to motif-5 and motif 6 of the BOMBESINR signature. The GPCRs are well characterized in animals and yeast, but little is known about their role in plants. However, Lu and coworkers (Lu et al., 2019) explained that GPCRs are involved in plant stress tolerance besides their involvement in growth and development.
The translated sequence of Ds-DbRCaG-03-Rga4p was predicted with the HIGHMOBLTYIY (high mobility group protein) fingerprint. High mobility group (HMG) proteins are chromosomal proteins involved in transcription regulation and nuclear localization. The HMG proteins are subdivided into three families: HMG-A/T binding, HMG-box, and HMG-nucleosome binding (Grasser et al., 2007). According to the literature, HMG-box protein regulates plant immune responses (Choi et al., 2016). The HIGHMOBLTYIY is a five-element fingerprint that gives a signature for the HMG-I/HMG-Y family. These five motifs were derived from an initial alignment of seven sequences. Motif-1 encodes the DNA binding first hook domain, motif-2 and 3 span the second hook domain, motif-4 compasses the third hook domain, and motif-5 encodes the C-terminal region rich in acidic amino acids. However, the translated sequence of Ds-DbRCaG-03-Rga4p showed homology to motif-2 and motif-3 of the HIGHMOBLTYIY signature. However, no fingerprint was predicted for Ds-DbRCaG-05-Rga6p.
3.5 In silico functional characterization
For the functional characterization of proteins, molecular weight is a crucial gauge. Therefore, the translated protein sequence of the identified RGA, Ds-DbRCaG-01-Rga1 (120 amino acid residues), was subjected to the physicochemical characterization that gave the molecular weight, 13118.30 Da, molecular formula, C591H920N146O168S9, and a predicted theoretical isoelectric point (pI) of 5.53 value. The pI value revealed it as acid. The extinction coefficient provides protein–protein and protein–ligand interactions for quantitative analysis (Prabhu et al., 2020). The computed extinction coefficient was 12950M-1 cm-1 for the predicted protein concentration in water at 280 nm. The
The instability index (II) was computed to be 37.26, which classifies this protein as stable. Because the protein has an instability index >40, it is considered unstable and vice versa. However, proteins with a higher aliphatic index are considered highly thermally stable proteins. The aliphatic amino acid residues contribute to the high thermal stability in a directly proportional protein to the aliphatic index. The computed aliphatic index value of translated protein Ds-DbRCaG-01-Rga1 was 90.25, showing it is thermophilic and may resist the stressed environment. The grand average of hydropathicity (GRAVY) is also a vital physicochemical property. It illustrates the protein’s interaction with water and presents its hydrophilic or hydrophobic nature. The computed gravy score of 0.133 showed its hydrophobic nature because positively rated proteins are more hydrophobic (Kyte and Doolittle, 1982).
The physicochemical properties of the deduced amino acid sequences of Ds-DbRCaG-01-Rga2p, Ds-DbRCaG-03-Rga4p, and Ds-DbRCaG-05-Rga6p were also computed. The aliphatic index of Ds-DbRCaG-01-Rga2p and Ds-DbRCaG-03-Rga4p was 100.85 and 93.07, respectively, showing their thermophilic nature. However, the aliphatic index of Ds-DbRCaG-05-Rga6p was 71.33, which is comparatively lower than Ds-DbRCaG-01-Rga1, Ds-DbRCaG-01-Rga2p, and Ds-DbRCaG-03-Rga4p, but is toward the higher side as well.
A signal peptide predicts the target site to which that protein would be transported. Information about its localization is also vital in determining the protein’s function. We predicted the cellular localization of the Ds-DbRCaG-01-Rga1 protein using the CELLO2GO web server. It predicted the cytoplasmic localization of Ds-DbRCaG-01-Rga1 with a significant score (1.237), followed by its localization in the chloroplast, scoring a 1.084 value. The Ds-DbRCaG-01-Rga2p and Ds-DbRCaG-05-Rga6p were predicted with extracellular localization. The Pathogenesis-related proteins of the tobacco (NtPR1) and Arabidopsis-related proteins (AtPR1) were reported to be localized in extracellular space (Vigers et al., 1992; Pecenkova et al., 2017). Furthermore, the localization of pathogenesis-related protein PR10.2 of Plasmopara viticola was predicted in subcellular compartments, i.e., the nucleus, cytoplasm, and chloroplast nuclear region (He et al., 2013).
3.6 In silico functional annotation
Ds-DbRCaG-01-Rga1 showed a conserved domain in CDART and CD-search tools. Different web-based tools were used for its in silico functional annotation, including MOTIF, CATH, COFACTOR, COACH, and I-TASSER. The MOTIF search tool displayed the results based on NCBI-CDD and Pfam databases. It represented the prediction of different motifs given in Table 1. However, the significant hits based on the NCBI-CDD database search characterized the translated protein sequence of RGA as ribulose bisphosphate carboxylase/oxygenase activase-RuBisCO activase (Rca) and Torsin; however, Pfam predicted the presence of Torsin and AAA+ (ATPase family associated with various cellular activities). These results supported that the translated protein sequence of Ds-DbRCaG-01-Rga1 is a disease resistance protein because “Torsins” are essential, disease-relevant AAA+ (ATPases associated with various cellular activities) proteins (Chase et al., 2017). Similarly, Rubisco activase (Rca) is a molecular chaperone or AAA + chaperone (Hayer-Hartl and Hartl, 2020), and molecular chaperons are associated with rescuing the cell in a stressed environment and contributing to plant immunity (Park and Seo, 2015). The ProtoNet tool also predicted the similarity of the identified putative disease resistance gene(s) sequence to the Rubisco activase encoding gene (Table 1) with GO molecular function and ATP binding.

TABLE 1. Functional motifs of Ds-DbRCaG-01-Rga1 predicted through Motif prediction tools, ProtoNet, and MOTIF.
The superfamily and functional family of translated protein Ds-DbRCaG-01-Rga1 were identified through the CATH database, revealing that it belongs to the superfamily P-loop containing nucleotide triphosphate hydrolases and the functional family Ribulose bisphosphate carboxylase/oxygenase activase (Rubisco activase, Rca). These results also supported it as a putative disease resistance protein because the P-loop is the signature motif of disease-resistant proteins in plants. Hence, the identified differentially expressed gene sequence of Dalbergia sissoo under dieback challenge was submitted to NCBI with the GenBank accession number MW533149.
Function annotation is a crucial criterion for the characterization of a protein. The COACH Meta server, based on I-TASSER structure prediction, determined the functional annotation of the translated protein of an identified disease resistance gene sequence or RGA. These databases predicted 41Phe (F), 42Tyr (Y), 44Ala (A), 47Phe (F), 77Lys (K), 78Gly (G), 80Gly (G), 81Lys (K), 82Ser (S), and 83Phe (F), as protein-ligand binding sites (Supplementary Figure S1) based on a high confidence score (C-score) that relates to more reliable prediction. The ligand-binding site residues, 77Lys (K), 78Gly (G), 80Gly (G), 81Lys (K), and 82Ser (S), are amino acid residues of the P-loop domain. The active site residue predicted using I-TASSER was 86Glu (E), as glutamate (E) residue is a characteristic feature of active kinases (Huse and Kuriyan, 2002). The Gene Ontology (GO) reconnoitered through the COFACTOR server represented its predominant role in response to stimuli and stresses (Table 2).
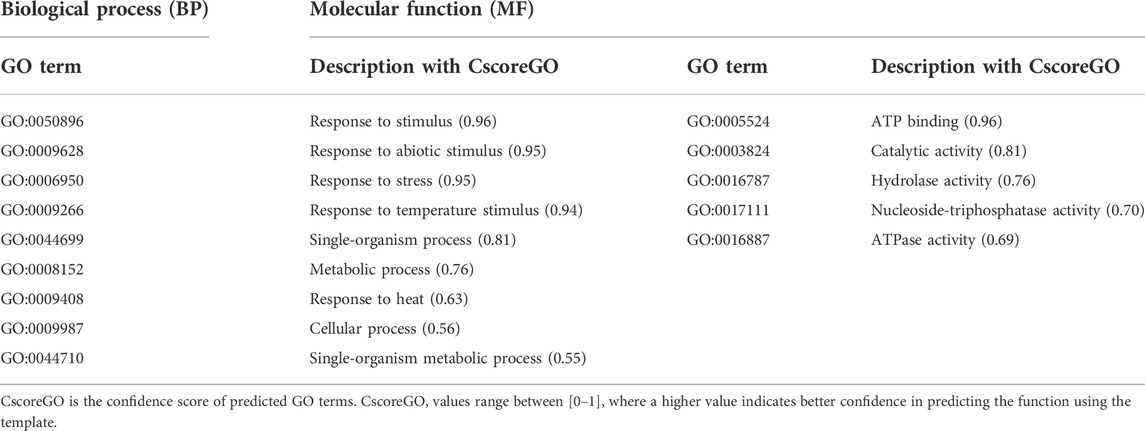
TABLE 2. In silico functional annotation of Ds-DbRCaG-01-Rga1 predicted through COFACTOR and COACH web-based tools.
3.7 Annotation of protein secondary structure
As no conserved domains were predicted in Ds-DbRCaG-01-Rga2p, Ds-DbRCaG-03-Rga4p, and Ds-DbRCaG-05-Rga6p, through CD-search and CDART; therefore, these RGAs were subjected to structure prediction to unravel their role in disease resistance. The SOPMA program was executed to predict their secondary structure. The α-helix (Hh), random coil (Cc), β-turn (Tt), and extended strand (Ee) percentiles were observed in the translated protein sequences of these RGAs. The 20.34% Hh and 20.34% Ee with a significant share of Cc at 50.50% were predicted in Ds-DbRCaG-01-Rga2p′s translated protein sequence. The Ds-DbRCaG-03-Rga4p′s translated protein sequence was predicted with 17.54% Hh, 25.44% Ee, and 7.02% Tt, with a significant share of Cc at 50%. The Ds-DbRCaG-05-Rga6p′s translated protein sequence was also predicted with 18.75% Ee and 6.25% Tt with a significant share of Cc at 75% secondary features.
3.8 Homology modeling and 3D imaging
The 3D model of Ds-DbRCaG-01-Rga2p displayed homology to the template 1X6I (crystal structure of ygfY from Escherichia coli). A total of thirty-one amino acid residues of Ds-DbRCaG-01-Rga2p (9–39) have been modeled with template 1X6I residues (50–80) with 7.26% confidence and 13% sequence identity. The aligned region of the template confirmed the succinate dehydrogenase_SDH5 superfamily domain. Succinate dehydrogenase (SDH) is an enzyme involved in mitochondrial reactive oxygen species (ROS), while SDH is involved in enhancing the production of ROS in any plant stress. ROS production is a crucial indicator of the plant stress response. Likewise, the FAD (flavin adenine dinucleotide) cofactor attachment to SDH1 through the delivery vehicles, SDH5 or SDHAF2 (Eletsky et al., 2012), mediates the production of fumarate from succinate in the tricarboxylic acid cycle (Krebs cycle). Moreover, if the plant suffers from stress, SDH upregulates the expression of stress-related genes (Jardim-Messeder et al., 2015). The enhanced production of salicylic acid also correlates with mitochondrial ROS, ultimately SDH (Belt et al., 2017). SDH has subunits, SDH1-SDH5, though SDH5 has been newly identified in prokaryotes and eukaryotes. Even its homologous, SDHAF2 (succinate dehydrogenase assembly factor 2), has also been identified in Arabidopsis thaliana (Huang et al., 2013). SDH5 was identified to be involved in the flavinylation process.
Ds-DbRCaG-03-Rga4p showed maximum homology with a significant hit to template 1GC6 (crystal structure of the radixin FERM domain complexed with inositol-(1,4,5)-triphosphate). A total of fifteen amino acid residues of Ds-DbRCaG-03-Rga4p (28–42) have been modeled with template 1GC6 residues (275–289) with 28% confidence and 13.33% sequence identity. The aligned region of the template revealed the pleckstrin homology (PH) like domain. Several proteins involved in cellular signal transmission, membrane transport, phospholipid modification, and cytoskeletal regulation are characterized as having a PH domain (Blomberg and Nilges, 1997; Rebecchi and Scarlata, 1998; Lemmon et al., 2002; Huang et al., 2013). The PH domains are lipid or phospholipid-binding domains that facilitate membrane localization (Maffucci and Falasca, 2001). The PH domain association in lipid binding provides the association of PH domain-containing proteins with lipid signaling that enhances plant disease resistance (Tang et al., 2005).
Ds-DbRCaG-05-Rga6p showed homology by significantly hitting the template 6I4H (crystal structure of plasmodium falciparum actin I (F54Y mutant) in the Ca-ATP state). A total of twenty-nine amino acid residues of Ds-DbRCaG-05-Rga6 (1–29) have been modeled with template 6I4H residues (7–35) with 31% sequence similarity and 17.24% sequence identity. However, the template’s aligned region confirmed the nucleotide-binding domain of the sugar kinase/HSP70/actin superfamily. As the superfamily members of the sugar kinases, actin, and heat shock-related proteins, they showed structural homology in 3D folding but differed in their function. However, these proteins are characterized by ATP hydrolysis or phosphotransferase (Hurley, 1996). The sugar kinases are involved in metabolic regulation, as does the ATP-dependent phosphorylation of sugars. HSP70 is a well-known cellular protein folding chaperone with a specialized ATP-binding domain. Actin is a cellular filament that plays a central role in the cytoskeleton structure. All three proteins have a specialized nucleotide-binding region in a similar position and conformation.
The crystalline structure of the template 1X6I chain A showed the SDH5 domain. The model 1X6I template is displayed as a helical bundle of multiple helices joined by loops. The predicted model of Ds-DbRCaG-01-Rga2p’s translated protein displayed structural homology to the part of the SDH5 domain of the template model, as shown in Figure 1A; the homology zone between the template model and the predicted translated protein of RGA share the same color. The crystalline structure of template 1GC6 showed two domains: (1) the N-terminal B41 domain and (2) a C-terminal FERM or PH-like domain connected with the loop. The predicted model of Ds-DbRCaG-03-Rga4p’s translated protein showed structural homology to a PH-like domain possessing perpendicular antiparallel β strands and one helix (Figure 1B). The crystalline structure of the template 6I4H chain A displayed the NBD domain of sugar kinase/HSP70/actin with a conserved core structure, βββαβαβα. The turns between β1 and β2 majorly contribute to the nucleotide-binding site. The predicted model of Ds-DbRCaG-05-Rga6p′s translated protein showed structural homology at different regions of the NBD domain for three β-pleated sheets (predicted model of Ds-DbRCaG-05-Rga6p) joined via loops, as shown in Figure 1C.
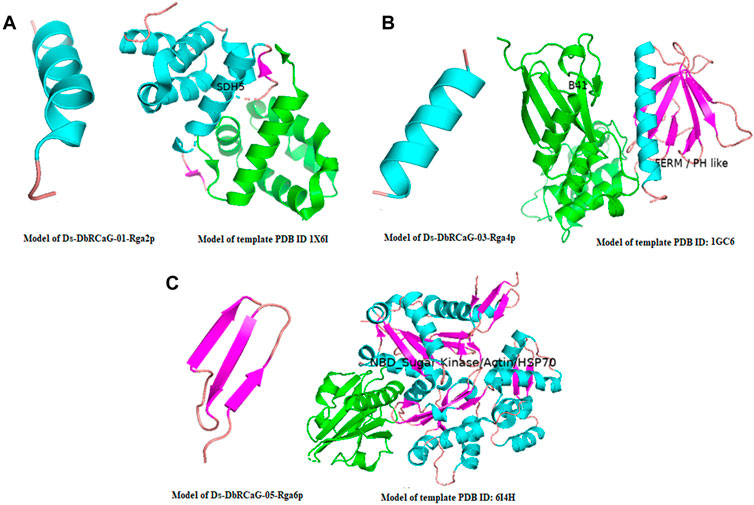
FIGURE 1. 3-D imaging of Ds-DbRCaG-01-Rga2p, Ds-DbRCaG-03-Rga4p, and Ds-DbRCaG-05-Rga6p. The homology zone of the template model and the model of predicated protein structure of each identified RGA showing with same colors: (A) predicted protein structure of Ds-DbRCaG-01-Rga2p, (B) predicted protein structure of Ds-DbRCaG-03-Rga4p, and (C) predicted protein structure of Ds-DbRCaG-05-Rga6p. The secondary models of template proteins were retrieved from the RCSB Protein Data Base, https://www.rcsb.org/
3.9 3D model validation
The quality of 3D models was determined by generating Ramachandran plots. The plot consisted of amino acid residues found in Phi (φ) and Psi (ψ) angles based on the graphical data representation. For Ds-DbRCaG-01-Rga2p and Ds-DbRCaG-03-Rga4p, the highest value model was observed with 100% residues in the most favored region. The model validation study of Ds-DbRCaG-05-Rga6p indicated 90.9% residues in the most favored region, 4.5% in the additional allowed region, and 4.5% in the generously allowed region. In general, the value ≥ 90% of residues in the most favored region is crucial to predict that the model is of good quality (Figure 2). The calculated Z-scores for predicted models of Ds-DbRCaG-01-Rga2p, Ds-DbRCaG-03-Rga4p, and Ds-DbRCaG-05-Rga6p were -1.43, -0.63, and -0.18, respectively, validating the models to be satisfactory. The superimposition of the models with the templates was analyzed by computing TM scores and RMSD (root mean square deviation) values. The TM score of Ds-DbRCaG-01-Rga2p, Ds-DbRCaG-03-Rga4p, and Ds-DbRCaG-05-Rga6p was 0.15016, 0.05050, and 0.08237, respectively, unraveling the random structural similarity of superimposed models with their templates. In comparison, the RMSD values of Ds-DbRCaG-01-Rga2p, Ds-DbRCaG-03-Rga4p, and Ds-DbRCaG-05-Rga6p with their respected templates were 0.97 Å, 0.06 Å, and 0.11 Å, respectively. The RMSD value scales the similarity between two atomic coordinates in a superimposed position. The lower the RMSD value (< 2), the higher the accuracy of the results (Kirchner and Guntert, 2011). The estimated RMSD values of models imparted confidence that residues were well superimposed. The predicted models were folded exactly in the same manner as their respective templates. The superposed full atom structures of the whole chain are given in Figure 3. The in silico prediction of the expressome of Dalbergia sissoo under dieback disease stress revealed the role of identified RGAs in disease and stress resistance.
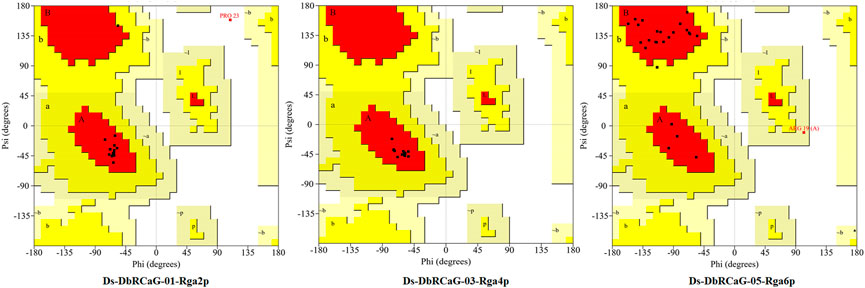
FIGURE 2. Ramachandran plot of Ds-DbRCaG-01-Rga2p, Ds-DbRCaG-03-Rga4p, and Ds-DbRCaG-05-Rga6p. Their most favored region is red, additionally allowed in yellow, generously allowed in light yellow, and disallowed regions are indicated in white fields.
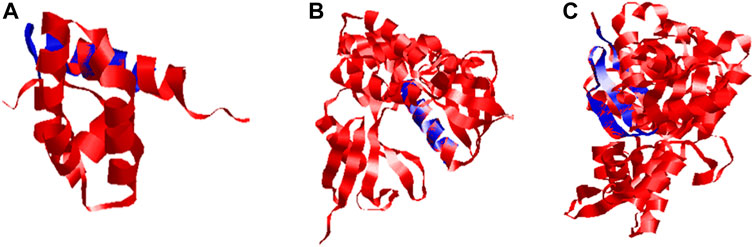
FIGURE 3. Superposed ribbon structure of predicted proteins models of Ds-DbRCaG-01-Rga2p, Ds-DbRCaG-03-Rga4p, and Ds-DbRCaG-05-Rga6p with their respective template regions. Blue indicates identified RGAs predicted proteins, while red indicates the template protein. (A) Ds-DbRCaG-02-Rga2 superposed to model 1X6I, (B) Ds-DbRCaG-03-Rga4p superposed to model 1GC6, and (C) Ds-DbRCaG-05-Rga6p superposed to model 6I4H.
4 Discussion
Plants have an innate immune system against pathogens. Upon infecting plants, pathogens produce pathogen-associated molecular patterns (PAMPs), and plants detect them by transmembrane pattern recognition receptors, which leads to the activation of signaling pathways and is known as PAMP-triggered immunity (Dodds and Rathjen, 2010). The strong invaders suppress this immunity by secreting effector proteins or effectors into plant cells to arrest signaling pathways (Kamoun, 2006). In response to effectors, plants generate intracellular immune receptors to recognize the effectors and develop effector-triggered immunity. This immunity establishes a hypertensive response resulting from programmed activation of localized cell death (Jones and Dangl, 2006). The immune receptors encoded by R genes are particular. These intracellular R proteins have N-terminal nucleotide-binding sites (NBS) and a C-terminal leucine-rich repeat (LRR). The R proteins recognize and interact with their corresponding effectors through the N-terminal NBS domain (Dangl and Jones, 2001; Mucyn et al., 2006; Burch-Smith et al., 2007), which indicates the recognition specificity of specific R genes to their specific effectors (Luck et al., 2000). The N-terminal NBS domain of the NBS-LRR class of R proteins is projected for effector recognition (Ade et al., 2007).
Therefore, in this study, to identify resistance gene analogs (candidate resistance gene sequences) in D. sissoo against dieback disease, the nucleotide-binding site (NBS) of the NBS-LRR class of R genes was used to probe the transcriptome of D. sissoo. The NBS region of the NBS-LRR domain consists of different conserved domains, like kinase P-loop, kinase-2, kinase-3A, and hydrophobic GLPL motifs (McHale et al., 2006), which play an essential role in plant defense mechanisms. The Blast tool revealed the differentially expressed identified DNA sequence upregulated under dieback stress has homology to the P-loop NTPase superfamily with a significant hit. The P-loop domain has been reported to be a part of NBS-LRR containing RGAs in different plant species like rice, sugar beet, maize, and coconut and is considered a characteristic domain of disease resistance proteins in plants (Tian et al., 2004; Wenkai et al., 2006; Rachana et al., 2016).
The MOTIF web-based tool characterized the RGA translated protein sequence as ribulose bisphosphate carboxylase/oxygenase activase-RuBisCO activase (Rca) and Torsin; however, Pfam predicted the presence of Torsin and AAA+ (ATPase family associated with various cellular activities). This characterization revealed the translated protein sequence of the identified RGA as a disease resistance protein because “Torsins” are essential, disease-relevant AAA+ (ATPases associated with various cellular activities) proteins (Chase et al., 2017). Correspondingly, Rubisco activase (Rca), as a molecular chaperone (AAA + chaperone), is associated with extricating the cell in a stressed environment and contributing to plant immunity (Park and Seo, 2015; Hayer-Hartl and Hartl, 2020). Similarly, the functional annotation of the translated protein sequence of the identified RGA using COFACTOR, COACH, and I-TASSER bioinformatics tools reconnoitered its predominant role in response to stresses.
The translated sequences of Ds-DbRCaG-01-Rga2p, Ds-DbRCaG-03-Rga4p, and Ds-DbRCaG-05-Rga6p were found to have no-match of conserved domains from databases for conserved domains. Predicted protein fingerprints search displayed bombesin receptor signature (BOMBESINR) for Ds-DbRCaG-01-Rga2p that showed it as transmembrane receptors; guanine-nucleotide-binding protein-coupled receptors (GPCRs) reported to be involved in stress tolerance in plants (Lu et al., 2019). Ds-DbRCaG-03-Rga4p is predicted to possess a fingerprint of a high mobility group protein (HIGHMOBLTYIY) that contributes to regulating plant immune response (Choi et al., 2016).
Using online web servers, we performed the structural characterization of identified RGAs. The Ds-DbRCaG-01-Rga2p showed homology to the template PDB ID: 1X6I. The alignment region confirmed the succinate dehydrogenase_SDH5 domain, involved in producing reactive oxygen species (ROS) and upregulating stress-responsive genes (Jardim-Messeder et al., 2015; Belt et al., 2017). The Ds-DbRCaG-03-Rga4p showed significant homology to template PDB ID: 1GC6. The alignment region between the template model and the Ds-DbRCaG-03-Rga4p predicted protein model validated the homology with lipid or-binding pleckstrin homology (PH), strongly associated with disease resistance (Tang et al., 2005). The structural homology of the predicted protein model of Ds-DbRCaG-05-Rga6p with the nucleotide-binding domain of sugar kinase/HSP70/actin domain of template model PDB ID: 6I4H revealed strong support for its putative role in stress resistance (Hurley, 1996). The molecular and bioinformatics-based analyses predicted the presence of motifs including ATP/GTP-binding site motif A (P-loop NTPase domain), GLPL domain, casein kinase II phosphorylation site, and N-myristoylation site, etc. that are the attributes of proteins encoded by disease resistance genes. The physicochemical attributes of identified resistance gene analogs, subcellular localization, predicted protein fingerprints, in silico functional annotation, and predicted protein structure proved their role in disease and stress resistance.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, MW533149.
Author contributions
SI, IU, and IK conceived, designed, and developed the experiment. SI, IU, and HR performed experiments. SI, IU, IK, HA, and SK analyzed and interpreted the data and drafted and revised the manuscript.
Funding
The Researchers Supporting Project (RSP-2021/123), King Saud University, Riyadh, Saudi Arabia, provided funding support for publication.
Acknowledgments
We highly acknowledge Researchers Supporting Project number (RSP-2021/123) King Saud University, Riyadh, Saudi Arabia.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1036029/full#supplementary-material
References
Ade, J., DeYoung, B. J., Golstein, C., and Innes, R. W. (2007). Indirect activation of a plant nucleotide binding site–leucine-rich repeat protein by a bacterial protease. Proc. Natl. Acad. Sci. U. S. A. 104, 2531–2536. doi:10.1073/pnas.0608779104
Armenteros, J. J., Tsirigos, K. D., Sonderby, C. K., Petersen, T. N., Winther, O., Brunak, S., et al. (2019). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423. doi:10.1038/s41587-019-0036-z
Babar, M., Ijaz, S., Khan, M. S., and Ul Haq, I. (2021). Computational genomics based probing of resistance gene analogs (RGAs) in mungbean under cercospora leaf spot disease challenge. Pak. J. Agri. Sci. 58, 1523–1536. doi:10.21162/PAKJAS/21.439
Baldi, P., Patocchi, A., Zini, E., Toller, C., Velasco, R., and Komjanc, M. (2004). Cloning and linkage mapping of resistance gene homologues in apple. Theor. Appl. Genet. 109, 231–239. doi:10.1007/s00122-004-1624-x
Barbosa, D. S. I., and Park, S. W. (2019). Versatility of cyclophilins in plant growth and survival: A case study in Arabidopsis. Biomolecules 9, 1–14.
Belt, K., Huang, S., Thatcher, L. F., Casarotto, H., Singh, K. B., Van Aken, O., et al. (2017). Salicylic acid-dependent plant stress signaling via mitochondrial succinate dehydrogenase. Plant Physiol. 173, 2029–2040. doi:10.1104/pp.16.00060
Blomberg, N., and Nilges, M. (1997). Functional diversity of PH domains: An exhaustive modelling study. Fold. Des. 2, 343–355. doi:10.1016/S1359-0278(97)00048-5
Burch-Smith, T. M., Schiff, M., Caplan, J. L., Tsao, J., Czymmek, K., and Dinesh-Kumar, S. P. (2007). A novel role for the TIR domain in association with pathogen-derived elicitors. PLoS Biol. 14 (1), e68. doi:10.1371/journal.pbio.0050068
Çetinel, A. H. S., Çetinel, B., Gokce, A., Tatli, C., and Erdik, E. (2022). “Molecular mechanisms–relayed plant defense responses against fungal pathogens,” in Phytomycology and molecular biology of plant–pathogen interactions. Editor B. S. Parkways (Boca Raton, FL: CRC Press), 87–110.
Chase, A. R., Laudermilch, E., and Schlieker, C. (2017). Torsin ATPases: Harnessing dynamic instability for function. Front. Mol. Biosci. 4, 29–37. doi:10.3389/fmolb.2017.00029
Choi, H. W., Manohar, M., Manosalva, P., Tian, M., Moreau, M., and Klessig, D. F. (2016). Activation of plant innate immunity by extracellular high mobility group box 3 and its inhibition by salicylic acid. PLoS Pathog. 12, e1005518. doi:10.1371/journal.ppat.1005518
Dangl, J. L., and Jones, J. D. (2001). Plant pathogens and integrated defence responses to infection. Nature 411, 826–833. doi:10.1038/35081161
Dodds, P. N., and Rathjen, J. P. (2010). Plant immunity: Towards an integrated view of plant– pathogen interactions. Nat. Rev. Genet. 11, 539–548. doi:10.1038/nrg2812
Eletsky, A., Jeong, M. Y., Kim, H., Lee, H. W., Xiao, R., Pagliarini, D. J., et al. (2012). Solution NMR structure of yeast succinate dehydrogenase flavinylation factor Sdh5 reveals a putative Sdh1 binding site. Biochemistry 51, 8475–8477. doi:10.1021/bi301171u
Gasteiger, E., Hoogland, C., Gattiker, A., Wilkins, M. R., Appel, R. D., and Bairoch, A. (2005). Protein identification and analysis tools on the ExPASy server. The proteomics protocols handbook, 571–607.
Geer, L. Y., Domrachev, M., Lipman, D. J., and Bryant, S. H. (2002). Cdart: Protein homology by domain architecture. Genome Res. 12, 1619–1623. doi:10.1101/gr.278202
Geourjon, C., and Deleage, G. (1995). Sopma: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. 11, 681–684. doi:10.1093/bioinformatics/11.6.681
Grasser, K. D., Launholt, D., and Grasser, M. (2007). High mobility group proteins of the plant HMGB family: Dynamic chromatin modulators. Biochim. Biophys. Acta 1769, 346–357. doi:10.1016/j.bbaexp.2006.12.004
Haq, I. U., Ijaz, S., and Latif, M. Z. (2021). “Diseases of Dalbergia sissoo: Etiology and integrated management of economically important diseases,” in Dalbergia sissoo: Biology, ecology and sustainable agroforestry. Editors S. Ijaz, and I. U. Haq (Boca Raton, FL: CRC Press), 63–87.
Hayer-Hartl, M., and Hartl, F. U. (2020). Chaperone machineries of rubisco–the most abundant enzyme. Trends biochem. Sci. 45, 748–763. doi:10.1016/j.tibs.2020.05.001
He, M., Xu, Y., Cao, J., Zhu, Z., Jiao, Y., Wang, Y., et al. (2013). Subcellular localization and functional analyses of a PR10 protein gene from Vitis pseudoreticulata in response to Plasmopara viticola infection. Protoplasma 250, 129–140. doi:10.1007/s00709-012-0384-8
Huang, S., Taylor, N. L., Stroher, E., Fenske, E., and Millar, A. H. (2013). Succinate dehydrogenase assembly factor 2 is needed for assembly and activity of mitochondrial complex II and for normal root elongation in Arabidopsis. Plant J. 73, 429–441. doi:10.1111/tpj.12041
Hurley, J. H. (1996). The sugar kinase/heat shock protein 70/actin superfamily: Implications of conserved structure for mechanism. Annu. Rev. Biophys. Biomol. Struct. 25, 137–162. doi:10.1146/annurev.bb.25.060196.001033
Huse, M., and Kuriyan, J. (2002). The conformational plasticity of protein kinases. Cell 109 (3), 275–282. doi:10.1016/s0092-8674(02)00741-9
Ijaz, S., and Haq, I. U. (2021). Dalbergia sissoo: Biology, ecology and sustainable agroforestry. United States: CRC Press.
Jardim-Messeder, D., Caverzan, A., Rauber, R., de Souza, F. E., Margis‐Pinheiro, M., and Galina, A. (2015). Succinate dehydrogenase (mitochondrial complex II) is a source of reactive oxygen species in plants and regulates development and stress responses. New Phytol. 208, 776–789. doi:10.1111/nph.13515
Jones, J. D., and Dangl, J. L. (2006). The plant immune system. Nature 444, 323–329. doi:10.1038/nature05286
Kamoun, S. (2006). A catalogue of the effector secretome of plant pathogenic oomycetes. Annu. Rev. Phytopathol. 44, 41–60. doi:10.1146/annurev.phyto.44.070505.143436
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi:10.1038/nprot.2015.053
Kirchner, D. K., and Guntert, P. (2011). Objective identification of residue ranges for the superposition of protein structures. BMC Bioinforma. 12, 170–211. doi:10.1186/1471-2105-12-170
Kyte, J., and Doolittle, R. F. (1982). A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132. doi:10.1016/0022-2836(82)90515-0
Laskowski, R. A., MacArthur, M. W., Moss, D. S., and Thornton, J. M. (1993). Procheck - a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291. doi:10.1107/s0021889892009944
Lemmon, M. A., Ferguson, K. M., and Abrams, C. S. (2002). Pleckstrin homology domains and the cytoskeleton. FEBS Lett. 513, 71–76. doi:10.1016/s0014-5793(01)03243-4
Lu, P., Magwang, R. O., Kirungu, J. N., Dong, Q., Cai, X., Zhou, Z., et al. (2019). Genome-wide analysis of the cotton G-coupled receptor proteins (GPCR) and functional analysis of GTOM1, a novel cotton GPCR gene under drought and cold stress. BMC genomics 20, 651–717. doi:10.1186/s12864-019-5972-y
Luck, J. E., Lawrence, G. J., Dodds, P. N., Shepherd, K. W., and Ellis, J. E. (2000). Regions outside of the leucine-rich repeats of flax rust resistance proteins play a role in specificity determination. Plant Cell 12, 1367–1377. doi:10.1105/tpc.12.8.1367
Maffucci, T., and Falasca, M. (2001). Specificity in pleckstrin homology (PH) domain membrane targeting: A role for a phosphoinositide-protein co-operative mechanism. FEBS Lett. 506, 173–179. doi:10.1016/s0014-5793(01)02909-x
McHale, L., Tan, X., Koehl, P., and Michelmore, R. W. (2006). Plant NBS-LRR proteins: Adaptable guards. Genome Biol. 7, 212–221. doi:10.1186/gb-2006-7-4-212
Meng, X., and Zhang, S. (2013). MAPK cascades in plant disease resistance signaling. Annu. Rev. Phytopathol. 51, 245–266. doi:10.1146/annurev-phyto-082712-102314
Minocha, R., and Jain, S. M. (2000). “Tissue culture of woody plants and its relevance to molecular biology,” in Molecular biology of woody plants. Editors S. M. Jain, and S. C. Minocha (Dordrecht: Springer), 315–339.
Moore, D., Robson, G. D., and Trinci, A. P. (2011). 21st century guidebook to fungi. U.K: Cambridge University Press.
Mucyn, T. S., Clemente, A., Andriotis, V. M., Balmuth, A. L., Oldroyd, G. E., Staskawicz, B. J., et al. (2006). The tomato NBARC-LRR protein Prf interacts with Pto kinase in vivo to regulate specific plant immunity. Plant Cell 18, 2792–2806. doi:10.1105/tpc.106.044016
Nasir, B., Ijaz, S., Awan, F. S., and Ul Haq, I. (2021). Genome-wide probing of NBS-LRR encoding genes in red clover (Trifolium pratense L) for the identification of resistance gene analogs in Trifolium alexandrinum L. Sci. Asia. 47, 425. doi:10.2306/scienceasia1513-1874.2021.054
Orengo, C. A., Michie, A. D., Jones, S., Jones, D. T., Swindells, M. B., and Thornton, J. M. (1997). CATH–a hierarchic classification of protein domain structures. Structure 5, 1093–1108. doi:10.1016/s0969-2126(97)00260-8
Orwa, C., Mutua, A., Kindt, R., Jamnadass, R., and Simons, A. (2009). Agroforestree database: A tree reference and selection guide version 4. Narobi, KE: World Agroforestry Centre ICRAF.
Park, C. J., and Seo, Y. S. (2015). Heat shock proteins: A review of the molecular chaperones for plant immunity. Plant Pathol. J. 31, 323–333. doi:10.5423/PPJ.RW.08.2015.0150
Pecenkova, T., Pleskot, R., and Zarsky, V. (2017). Subcellular localization of Arabidopsis pathogenesis-related 1 (PR1) protein. Int. J. Mol. Sci. 18, 825. doi:10.3390/ijms18040825
Prabhu, N., Dai, L., and Nordlund, P. (2020). CETSA in integrated proteomics studies of cellular processes. Curr. Opin. Chem. Biol. 54, 54–62. doi:10.1016/j.cbpa.2019.11.004
Rachana, K. E., Naganeeswaran, S. A., Fayas, T. P., Thomas, R. J., and Rajesh, M. K. (2016). Cloning, characterization and expression analysis of NBS-LRR-type resistance gene analogues (RGAs) in coconut. Acta Bot. Croat. 75, 1–10. doi:10.1515/botcro-2016-0003
Rappoport, N., Karsenty, S., Stern, A., Linial, N., and Linial, M. (2012). ProtoNet 6.0: Organizing 10 million protein sequences in a compact hierarchical family tree. Nucleic Acids Res. 40, D313–D320. doi:10.1093/nar/gkr1027
Rebecchi, M. J., and Scarlata, S. (1998). Pleckstrin homology domains: A common fold with diverse functions. Annu. Rev. Biophys. Biomol. Struct. 27, 503–528. doi:10.1146/annurev.biophys.27.1.503
Riedl, S. J., Li, W., Chao, Y., Schwarzenbacher, R., and Shi, Y. (2005). Structure of the apoptotic protease-activating factor 1 bound to ADP. Nature 434, 926–933. doi:10.1038/nature03465
Saraste, M., Sibbald, P. R., and Wittinghofer, A. (1990). The P-loop—A common motif in ATPand GTP-binding proteins. Trends biochem. Sci. 15, 430–434. doi:10.1016/0968-0004(90)90281-f
Sarwar, M. K., Payum, A., Ijaz, S., Shakeel, Q., and Issam, G. (2022). “Fungal gene expression and interaction with host plants,” in Phytomycology and molecular biology of plant–pathogen interactions. Editor I. U. Haq, S. Ijaz, and I. A. Khan (Boca Raton, FL: CRC Press), 111–124.
Sarwar, M. K., Ul Haq, I., Ijaz, S., Khan, I. A., and Çetinel, A. H. S. (2022). “Pathogenicity genes,” in Phytomycology and molecular biology of plant–pathogen interactions. Editor I. U. Haq, S. Ijaz, and I. A. Khan (Boca Raton, FL: CRC Press), 75–86.
Shah, M. H., Mukhtar, I., and Khan, S. N. (2010). Medicinal importance and association of pathological constraints with Dalbergia sissoo. Pak J. Phytopath 22, 135–138.
Tang, D., Ade, J., Frye, C. Z., and Innes, R. W. (2005). Regulation of plant defense responses in Arabidopsis by EDR2, a PH and START domain‐containing protein. Plant J 44: 245- 257 hurley JH (1996) the sugar kinase/heat shock protein 70/actin superfamily: Implications of conserved structure for mechanism. Annu. Rev. Biophys. Biomol. Struct. 25, 137–162.
Tian, Y., Fan, L., Thurau, T., Jung, C., and Cai, D. (2004). The absence of TIR-type resistance gene analogues in the sugar beet (Beta vulgaris L.) genome. J. Mol. Evol. 58, 40–53. doi:10.1007/s00239-003-2524-4
Ul Haq, I., Ijaz, S., and Khan, I. A. (2022). Phytomycology and molecular biology of plant pathogen interactions. Boca Raton, FL: CRC Press.
Vigers, A. J., Wiedemann, S., Roberts, W. K., Legrand, M., Selitrennikoff, C. P., and Fritig, B. (1992). Thaumatin-like pathogenesis-related proteins are antifungal. Plant Sci. 83, 155–161. doi:10.1016/0168-9452(92)90074-v
Wang, H., Meng, J., Peng, X., Tang, X., Zhou, P., Xiang, J., et al. (2015). Rice WRKY4 acts as a transcriptional activator mediating defense responses toward Rhizoctonia solani, the causing agent of rice sheath blight. Plant Mol. Biol. 89, 157–171. doi:10.1007/s11103-015-0360-8
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., et al. (2018). SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303. doi:10.1093/nar/gky427
Wenkai, X., Mingliang, X., Jiuren, Z., Fengge, W., Jiansheng, L., and Jingrui, D. (2006). Genome-wide isolation of resistance gene analogs in maize (Zea mays L.) Theor. Appl. Genet. 113, 63–72. doi:10.1007/s00122-006-0272-8
Wiederstein, M., and Sippl, M. J. (2007). ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 35, W407–W410. doi:10.1093/nar/gkm290
Yang, J., Roy, A., and Zhang, Y. (2013). Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 29, 2588–2595. doi:10.1093/bioinformatics/btt447
Yang, J., and Zhang, Y. (2015). I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 43, W174–W181. doi:10.1093/nar/gkv342
Yu, C. S., Chen, Y. C., Lu, C. H., and Hwang, J. K. (2006). Prediction of protein subcellular localization. Proteins 64, 643–651. doi:10.1002/prot.21018
Zhang, C., Freddolino, P. L., and Zhang, Y. (2017). Cofactor: Improved protein function prediction by combining structure, sequence and protein–protein interaction information. Nucleic Acids Res. 45, W291–W299. doi:10.1093/nar/gkx366
Zhang, Y., and Skolnick, J. (2005). TM-Align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309. doi:10.1093/nar/gki524
Keywords: Dalbergia sissoo, resistance gene analogs, NBS-LRR, functional annotation, computational biology
Citation: Ijaz S, Haq IU, Khan IA, Ali HM, Kaur S and Razzaq HA (2022) Identification of resistance gene analogs of the NBS-LRR family through transcriptome probing and in silico prediction of the expressome of Dalbergia sissoo under dieback disease stress. Front. Genet. 13:1036029. doi: 10.3389/fgene.2022.1036029
Received: 03 September 2022; Accepted: 21 September 2022;
Published: 07 October 2022.
Edited by:
Feng Zhang, Chinese Academy of Agricultural Sciences (CAAS), ChinaReviewed by:
Yasir Iftikhar, University of Sargodha, PakistanYoulian Pan, National Research Council Canada (NRC-CNRC), Canada
Copyright © 2022 Ijaz, Haq, Khan, Ali, Kaur and Razzaq. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Imran Ul Haq, aW1yYW5fMTYxNEB5YWhvby5jb20=