 Nam K. Lee1
Nam K. Lee1 Stephen P. Cass1Samuel P. Gubbels1Helen Z. Gomez1,2Melissa A. Scholes1,3†
Stephen P. Cass1Samuel P. Gubbels1Helen Z. Gomez1,2Melissa A. Scholes1,3† Herman A. Jenkins1
Herman A. Jenkins1 Regie Lyn P. Santos-Cortez1,4*
Regie Lyn P. Santos-Cortez1,4*- 1Department of Otolaryngology—Head and Neck Surgery, School of Medicine, University of Colorado Anschutz Medical Campus, Aurora, CO, United States
- 2Human Medical Genetics and Genomics Program, University of Colorado Anschutz Medical Campus, Aurora, CO, United States
- 3Department of Pediatric Otolaryngology, Children’s Hospital Colorado, Aurora, CO, United States
- 4Center for Children’s Surgery, Children’s Hospital Colorado, Aurora, CO, United States
Cholesteatoma is a rare and benign disease, but its propensity to cause erosive damage through uninhibited growth can be detrimental to hearing and health. Prior reports indicated a genetic component to pathogenesis in at least a subset of patients. In this study, we aimed to identify rare DNA variants in affected patients. The salivary DNA of six patients whose middle ear tissues were obtained during tympanoplasty/mastoidectomy surgeries were submitted for exome sequencing. Tissue samples from the same patients were previously submitted for mRNA sequencing and analyzed for differentially expressed genes (DEGs). From the generated exome sequence data, rare predicted-to-be-damaging variants were selected within previously identified DEGs, and the candidate genes within which these rare variants lie were used for network analysis. Exome sequencing of six DNA samples yielded 5,078 rare variants with minor allele frequency <.001. A total of 510 variants were predicted to be deleterious and 52 were found to lie within previously identified DEGs. After selecting variants based on quality control measures, 12 variants were identified all from one pediatric patient. Network analysis identified ten significant cellular pathways, including protein transport, viral process, regulation of catalytic activity and cell cycle, and apoptotic and rhythmic processes. We hypothesize that the candidate genes identified in this study may be part of key signaling pathways during the mucosal response to middle ear infection. The occurrence of multiple rare variants may play a role in earlier onset of cholesteatoma formation in chronic otitis media.
1 Introduction
Cholesteatoma is a rare and benign disease of the middle ear, affecting approximately one per 10,000 individuals, but due to its potential for growth, may result in a wide array of detrimental consequences (Kemppainen et al., 1999). This cyst-like mass containing keratinous debris with patchy squamous epithelial lining can grow beyond the boundaries of the middle ear leading to erosion of the temporal bone. Conductive and sensorineural hearing loss, soft tissue abscesses, facial paralysis, and meningitis, among other potential complications, may follow the destruction of the middle and inner ear, facial nerve, or skull base (Kuo et al., 2015). Existing treatment options lack any medical alternatives and are limited to surgery. The surgical treatment of cholesteatoma is often conducted in multiple stages and requires careful and frequent follow-up to assess for any recurrence or regrowth of residual disease (Kuo et al., 2015).
There are two distinct categories of cholesteatoma: Congenital, found behind an intact tympanic membrane, and acquired, often accompanying chronic infection of the middle ear. Between its two forms, acquired cholesteatoma presents more frequently with over 70% of cases in association with a history of chronic otitis media (Kemppainen et al., 1999). With an understanding of chronic infection and inflammation as the basis for disease origin, numerous theories on the pathogenesis of cholesteatoma were developed (Castle, 2018). Although emerging evidence implicates contrasting theories, syndromic and familial forms of cholesteatoma suggest a role of genetics in the disease process, thus providing an alternative approach and credence to further investigations into the genetic basis for cholesteatoma formation (Bacciu et al., 2005; Prinsley, 2009; Lim et al., 2014; Jennings et al., 2018; Collins et al., 2020).
Genomic investigation can uncover key insights into rare diseases through the identification of rare but high-penetrance variants with complex inheritance patterns. Cholesteatoma has been associated with syndromes with chromosomal abnormalities or genetic variants, such as Turner, Down, and Branchio-oto-renal syndromes (Lipkin et al., 1986; Bacciu et al., 2005; Bergamaschi et al., 2008; Vaglio et al., 2008; Lim et al., 2014; Diego et al., 2018). Familial patterns of cholesteatoma have also been described in siblings, twins, and intergenerational families. In a recent study of 857 individuals, 10.4% reported a family history of cholesteatoma, which in turn was a risk factor for bilateral disease (OR 2.15; 95% CI: 1.35, 3.43; p = .001) (Collins et al., 2020). In a follow-up study by Prinsley et al. (2019), who identified three familial cases of cholesteatoma in a rural region of the United Kingdom, two rare loss-of-function variants in EGFL8 and BTNL9 were identified in one of the families (Prinsley, 2009). Based on these associations and patterns of disease presentation, there is the potential to identify genetic drivers of cholesteatoma pathogenesis and pathways that are potential targets for effective medical solutions.
In a previous study that profiled the expression of cholesteatoma, differentially expressed genes (DEGs) were identified using three cholesteatoma and three middle ear mucosal samples of six unrelated patients with cholesteatoma (Baschal et al., 2020). Contrarily, in this report, DNA extracted from saliva samples of the same six patients with cholesteatoma were submitted for exome sequencing to identify rare variants that are predicted to be damaging. We hypothesized that at least some of the DEGs identified in our previous mRNA-seq study were potentially due to rare genetic variants in the patients who provided both mRNA-seq and exome data. Identified candidate genes were then submitted for network analysis to identify protein-protein interactions and cellular pathways that are potentially involved in cholesteatoma pathogenesis.
2 Materials and methods
2.1 Ethical approval, recruitment, and sample collection
The study was approved by the Colorado Multiple Institutional Review Board (protocol 16-2673) prior to initiation. Informed consent was obtained from all study participants. The relevant clinical data from these patients were collected using the Research Electronic Data Capture (REDCap), a HIPAA-compliant electronic data capture tool (Supplementary Table S1).
In a prior study, cholesteatoma or middle ear mucosal tissues were collected in six unrelated patients with cholesteatoma (Baschal et al., 2020). From the three cholesteatoma and three middle ear mucosal tissues of the six patients, RNA was isolated and submitted for bulk mRNA-sequencing. Then, using the resulting mRNA-seq data, 1,806 DEGs were identified for cholesteatoma.
For this exome filtering study, saliva samples of the same six patients with mRNA-seq and DEG data were collected using Oragene OGR-500 kits (DNAgenotek, Ottawa, Ontario, Canada) and used to extract DNA per the manufacturer’s protocol.
2.2 Exome sequencing and analysis
The DNA extracted from six saliva samples were submitted for exome sequencing at the University of Washington Northwest Genomics Center. The Twist Bioscience Human Core Exome Kit (South San Francisco, CA, United States of America) was used for sequence capture, and sequencing was performed using an Illumina HiSeq (San Diego, CA, United States of America) to an average depth of 30×. Fastq files were aligned to the hg38 human reference sequence using Burrows-Wheeler Aligner, generating demultiplexed .bam files (Li and Durbin, 2009). The Genome Analysis Tool Kit was used for variant detection and calling, as well as generation of standard metrics used for quality control (QC) during exome analyses (McKenna et al., 2010). Low-quality and likely false-positive variants were flagged. The generated .vcf file was annotated using ANNOVAR (Wang et al., 2010).
Annotated variants were selected based on the following criteria: 1) minor allele frequency (MAF) ≤.001 in any known population in public genome databases including gnomAD, 1,000 Genomes, or Greater Middle East (GME) Variome (Lek et al., 2016; Scott et al., 2016; Azaiez et al., 2018); 2) scaled Combined Annotation Dependent Depletion score of ≥3 and/or had dbscSNV score ≥.7 (Kircher et al., 2014); (3a) predicted to be deleterious by at least one bioinformatics tool included in dbSNFP33a in the case of missense or stop variants (Liu et al., 2011; Liu et al., 2013; Liu et al., 2016); or (3b) predicted to be deleterious by MutationTaster for splice and frameshift variants (Steinhaus et al., 2021); 4) lie within DEGs that were previously identified using middle ear samples from the same six patients with exome data (Baschal et al., 2020); and 5) pass QC filters. In addition to the dbscSNV score, missense variants were also rechecked against equivalent hg19 databases then annotated using MutationTaster (Kent et al., 2002; Steinhaus et al., 2021). For the final set of candidate genes with rare deleterious variants, MAF was also checked in the GenomeAsia 100K database and variants were visualized from .bam files using the Integrative Genomics Viewer (IGV) v2.12.3 (Thorvaldsdóttir et al., 2013; GenomeAsia100K Consortium, 2019). For further validation, Sanger sequencing of the identified variants was performed using the DNA sample from one pediatric patient.
2.3 Network analysis
NetworkAnalyst software and the IMEx Interactome database were used to visualize protein-protein networks with the candidate genes as input (Xia et al., 2014; Xia et al., 2015; Zhou et al., 2019). For the resulting subnetworks, enrichment analysis was conducted using PANTHER BP (Mi et al., 2019). False discovery rate (FDR)-adjusted p < .05 was used as the threshold to denote significant pathways.
3 Results
3.1 Exome analysis
Among the six patients with cholesteatoma who provided both DNA and RNA samples, the average age was 42 years (range 9.6–77.3 years) and all patients were male (Baschal et al., 2020). Five patients identified as White and one patient as Hispanic. Clinical description of patients including otoscopy, imaging, and intra-operative findings can be found in Supplementary Table S1.
From the combined data of six patients, exome sequencing revealed a total of 113, 176 genetic variants within the coding regions of the human genome. Of these variants, 5,078 were rare with a MAF ≤.001 (Figure 1). Five-hundred thirty-nine of these rare variants had a scaled CADD score ≥3. Of these, 426 were single nucleotide variants (SNVs) predicted to be deleterious by at least one bioinformatics tool, and 41 of these SNVs lie within our previously identified DEGs for cholesteatoma (Baschal et al., 2020). On the other hand, there were 113 splice or frameshift variants, of which 84 were predicted to be deleterious, and 12 frameshift variants were found within DEGs. In total, 52 rare deleterious variants were found within DEGs and 12 passed QC filters. In summary, 12 rare deleterious variants, including one frameshift and 11 missense variants, were included in the final selection (Table 1).
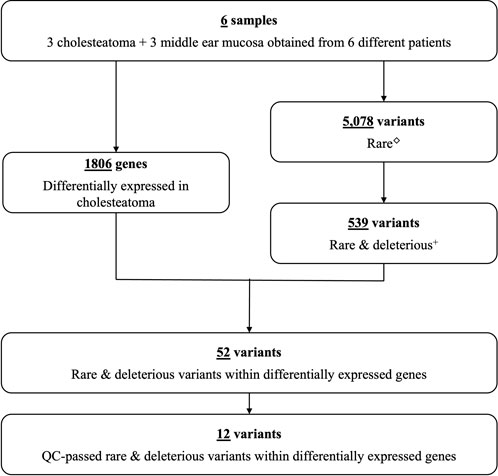
FIGURE 1. Study flow chart. Number of differentially expressed genes and genetic variants identified in six patients with cholesteatoma (Supplementary Table S1). ◇Rare variants with MAF ≤.001. +Predicted to lead to loss of gene function.
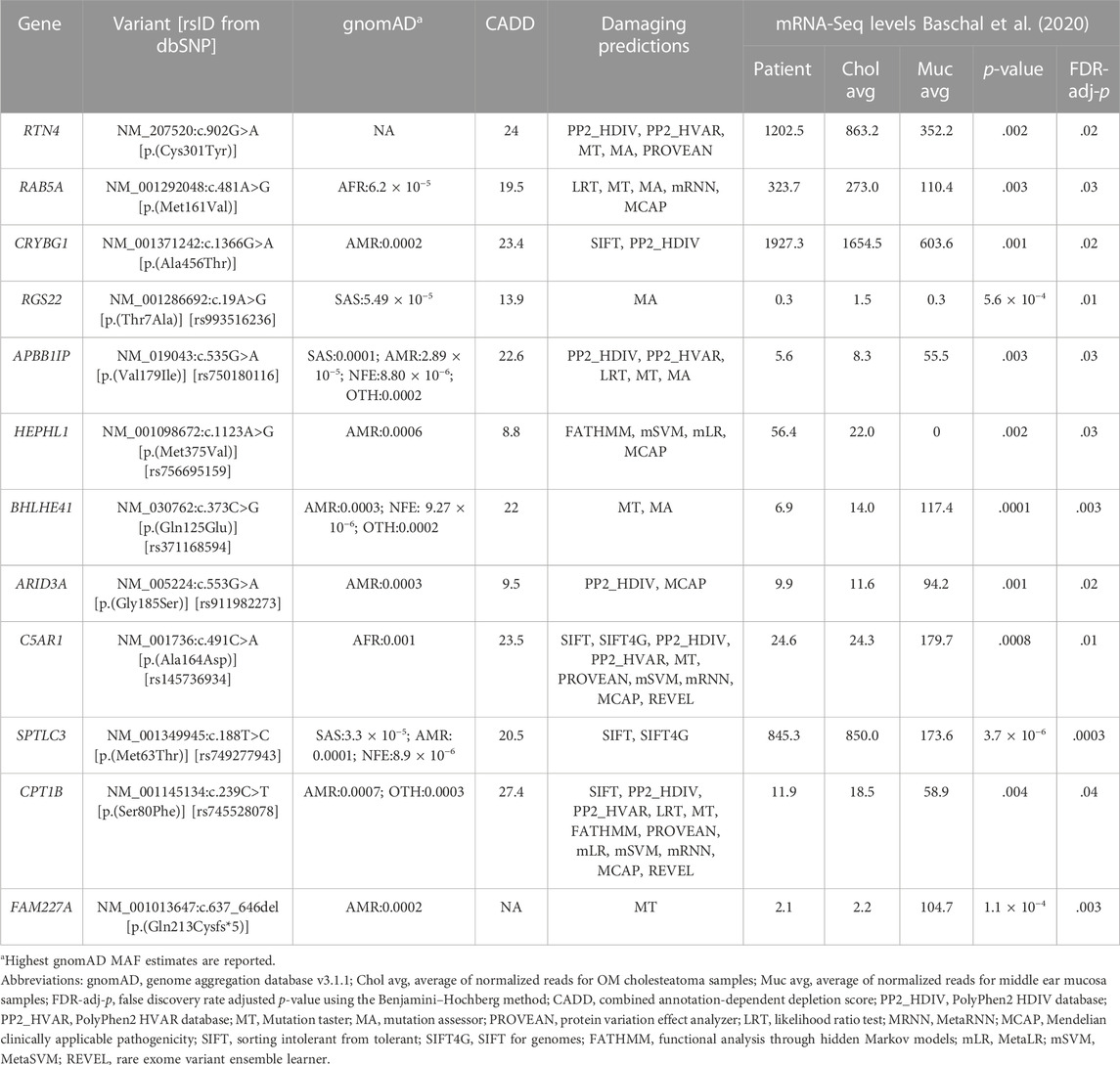
TABLE 1. Twelve candidate genes identified in a pediatric patient with cholesteatoma.
The 12 selected variants were identified as heterozygous in one pediatric patient and were verified by Sanger sequencing; however, no rare variants were identified in the other five adult patients (Table 1). These 12 variants were found in the following genes: APBB1IP, ARID3A, BHLHE41, C5AR1, CPT1B, CRYBG1, FAM227A, HEPHL1, RAB5A, RGS22, RTN4, and SPTLC3. All 12 genes are novel and have not been previously reported for DNA variants that confer human susceptibility to otitis media or cholesteatoma.
3.2 Network analysis
Network analysis is used to build a network of genes that interact to serve a set of biological functions. In this model, seeds are the genes from the list of input genes, in this case, our 12 candidate genes, which interact within the biological pathways identified in the network. Nodes represent genes that work collectively to serve a biological process and edges represent interactions between the nodes. Our analysis yielded a subnetwork consisting of 157 edges and 156 nodes (Figure 2A). Of the 12 candidate genes, eight genes were included as seeds in the network, including APBB1IP, ARID3A, BHLHE41, C5AR1, CPT1B, CRYBG1, RAB5A, and RTN4. There were 23 cellular pathways identified within the subnetwork, 10 of which were significant (Figure 2B). A PANTHER BP pathway was deemed significant if the module analysis on each subnetwork led to an FDR-adjusted p-value <.05. The cellular and biological functions were classified using PANTHER BP Gene Ontology Terms. The significant pathways identified in our network analysis included endocytosis, intracellular protein transport, protein transport, vesicle-mediated transport, viral process, regulation of catalytic activity, regulation of cell cycle, negative regulation of apoptotic process, apoptotic process, and rhythmic process. When compared to the network analysis conducted using DEGs from tissue mRNA-sequence data as input, 17 cellular processes out of the 23 identified in our study overlapped (Supplementary Table S2).
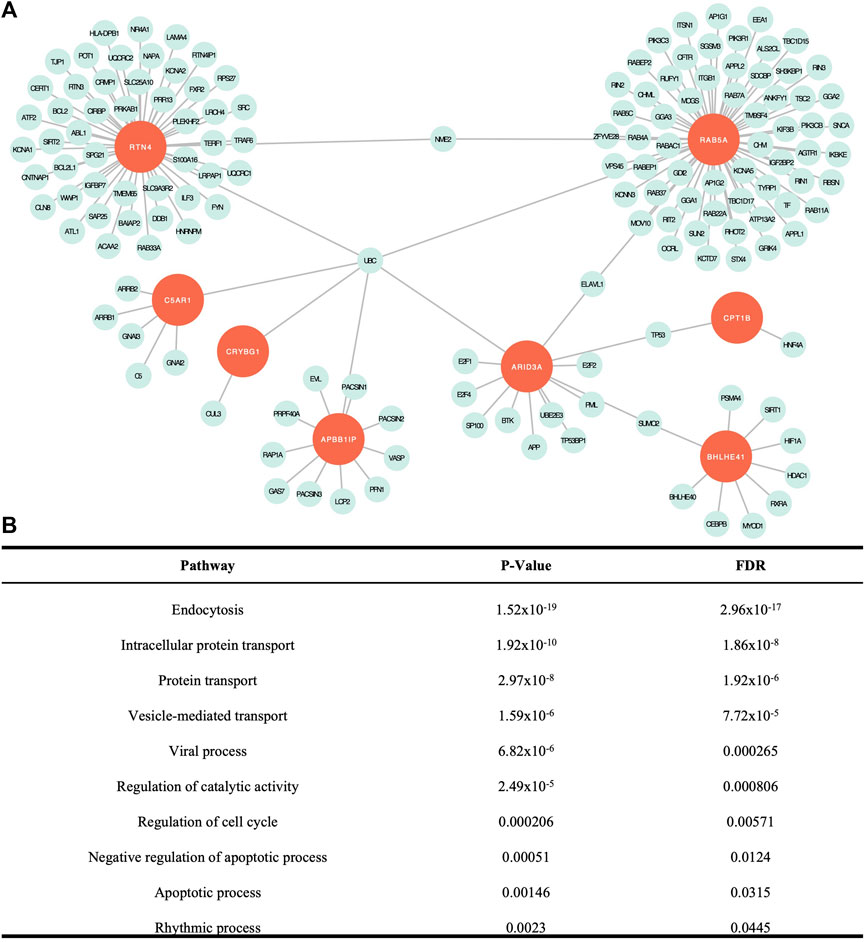
FIGURE 2. Network analysis findings. (A) Visual representation of the network created with the eight seed genes and associated proteins. (B) Significant cellular pathways determined by false discovery rate (FDR) adjusted p < .05. All except intracellular protein transport overlapped with significant pathways identified from DEG data (Baschal et al., 2020).
4 Discussion
In this study, we report 12 novel candidate genes for cholesteatoma that were identified from the exome sequence data while using DEGs from a prior mRNA-sequencing study of the same six unrelated patients (Baschal et al., 2020). All the 12 candidate genes were identified in the youngest of the cohort, who presented with cholesteatoma at 9 years old (Figure 3). The patient’s cholesteatoma is consistent with the acquired form that started with an episode of acute otitis media long before the diagnosis of right-sided conductive hearing loss at age 8 years. His otoscopic examination revealed a white mass behind a retracted left tympanic membrane and moderate conductive hearing loss in the same ear. Although the extent of his disease was limited to the middle ear and less severe than the other individuals in the study, the relatively early presentation of cholesteatoma in the context of an indistinct history of acute otitis media is notable. While acquired cholesteatoma is usually observed in patients with chronic otitis media, there have been previous reports of cholesteatoma diagnosed following an episode of acute otitis media, that is potentially infected by atypical bacteria (Abes et al., 2017). However, in our patient, it is more likely that there was inadequate documentation of chronic otitis media.
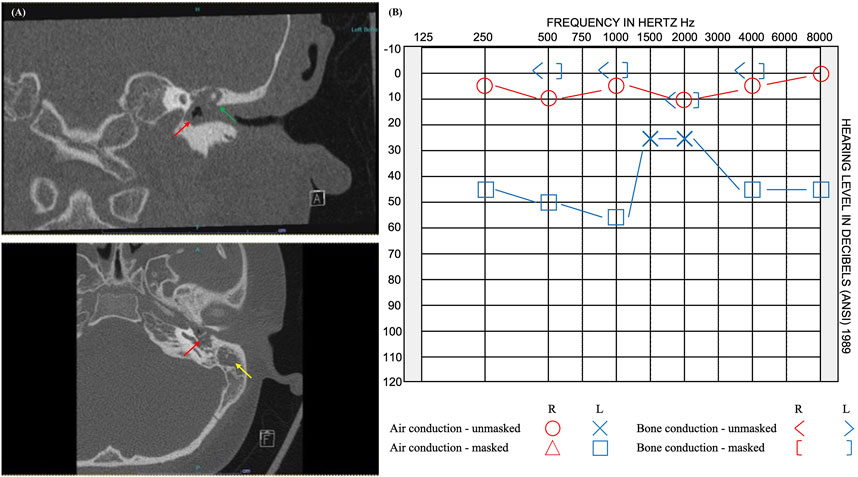
FIGURE 3. Imaging and audiogram results of pediatric patient with rare variants in 12 candidate genes. (A) CT imaging reveals opacification of the left middle ear cavity (red arrow), blunting of the scutum (green arrow), and complete opacification of the left mastoid (yellow arrow). (B) Audiogram results showed normal hearing at 250–8,000 Hz on the right and moderate rising to mild conductive hearing loss at 250–8,000 Hz on the left.
The expression profiles of the 12 candidate genes were assessed in the online databases, Genotype-Tissue Expression (GTEx) and Human Protein Atlas (Lonsdale et al., 2013; Uhlén et al., 2015). Due to the absence of middle ear tissues in these databases, esophageal mucosa, lungs, cervix, uterus, vagina, minor salivary glands, and skin were assessed as surrogates for middle ear mucosa (Supplementary Table S2). All 12 genes showed expression at the RNA or protein level in the tissues of interest (Supplementary Table S2). None of the genes had prior implications in OM or other middle ear pathologies to the best of our knowledge. However, in our own middle ear expression dataset from the same patients, all 12 genes were DEGs for cholesteatoma (Table 1): RTN4, RAB5A, CRYBG1, RGS22, HEPHL1, and SPTLC3 were upregulated, while APBB1IP, BHLHE41, ARID3A, C5AR1, CPT1B, and FAM227A were downregulated. The pediatric patient with variants in all 12 genes had similar trends in expression, except for RGS22, which had reduced expression with levels similar to the mucosal samples than to the other cholesteatoma samples (Table 1).
From the 12 candidate genes, eight were identified as seeds in our network analysis: APBB1IP, ARID3A, BHLHE41, C5AR1, CPT1B, CRYBG1, RAB5A, and RTN4. These seed genes all play a role in the innate immune response to infection or disease (Supplementary Table S3). The innate immune system has been historically implicated in the pathogenesis of otitis media (Ram and Chinen, 2011). A recent single-cell transcriptomic study of a normal murine middle ear illustrated the expression of innate immunity-related genes in all 17 mucosal cell types (Ryan et al., 2020).
Major cellular pathways in our network analysis relate to endoplasmic reticulum (ER) function, including intracellular protein transport, protein transport, and vesicle-mediated transport. When otitis media is induced, a rise in ER stress function is observed with an associated increase in proinflammatory gene expression (Zhao et al., 2020). ER dysfunction is known to cause various inflammatory diseases (Cao et al., 2013; Zhang et al., 2017). The additional basic cellular processes highlighted in our study include poor wound healing and cell renewal in cholesteatoma. While viral process was a significant pathway, our previous study on the microbiota of cholesteatoma did not detect any of the common respiratory viruses (Frank et al., 2022). Taken together, the 12 candidate genes may be part of key signaling pathways during the mucosal response to a middle ear infection.
There are several limitations to this study. Due to the rarity of cholesteatoma, we have a very limited sample size with a methodology involving rare variant detection from exome sequence data. Note, however, that there has not been any previous genome-wide association studies published on a cholesteatoma cohort to identify significant genes (Baschal et al., 2020). Validation of the variants identified in this study may be best achieved using functional analysis with mutations induced in the 12 identified variants. The current lack of a clear understanding of the potential tissue source or cell type that evolves into cholesteatomatous tissue limits the design of functional experiments in a suitable cell line. On the other hand, our review of the literature demonstrated mouse model, cell line, or disease entities with the same direction of effect as in cholesteatoma samples in 7 of the 12 candidate genes (e.g., gene knockout vs. downregulation in cholesteatoma) with immune cell defects affecting the innate immune defenses (Supplementary Table S3).
5 Conclusion
We identified 12 novel candidate genes for cholesteatoma supported by identification of both genetic variants from exome sequence data and DEGs from mRNA-seq using cholesteatoma and middle ear mucosal tissues. Out of the 12 rare coding variants identified in a single Hispanic-American child with cholesteatoma, 11 were found as heterozygous in various gnomAD populations (Table 1), indicating that these variants may be replicated in DNA sequencing studies of cholesteatoma patients from other world populations. Our findings might also suggest that a few of the DEGs identified in cholesteatoma have a genetic basis, rather than being a product of chronic infectious processes. The occurrence of multiple rare variants might also play a role in earlier onset of cholesteatoma formation in chronic otitis media. Identification of these genes and pathways not only enriches our understanding of cholesteatoma formation but improve the potential for developing new prevention and treatment strategies for otitis media.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/gap/, phs001941 v1.p1.
Ethics statement
The studies involving human participants were reviewed and approved by the Colorado Multiple Institutional Review Board. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
RS-C conceptualized the study. SG, SC, MS, and HJ recruited patients and collected samples. NL performed bioinformatic and network analyses. HG performed Sanger sequencing. NL and RS-C wrote the manuscript. All authors read and approved the manuscript.
Funding
This work was funded by the National Institute on Deafness and Other Communication Disorders (NIDCD) of the National Institutes of Health through grants R01 DC015004 (to RS-C) and R01 DC019642 (to RS-C and Ivana V. Yang). NL was supported by the NIDCD training grant T32 DC012280 (to HJ and Sue Kinnamon).
Acknowledgments
We thank the patients who participated in this study, without whom this study would not have been possible.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1033965/full#supplementary-material
References
Abes, G. T., Abes, F. L. L., and Cruz, T. L. G. (2017). Development of attic cholesteatoma in acute otitis media with tuberculosis. Acta Med. Philipp. 51. doi:10.47895/amp.v51i1.649
Azaiez, H., Booth, K. T., Ephraim, S. S., Crone, B., Black-Ziegelbein, E. A., Marini, R. J., et al. (2018). Genomic landscape and mutational signatures of deafness-associated genes. Am. J. Hum. Genet. 103, 484–497. doi:10.1016/j.ajhg.2018.08.006
Bacciu, A., Pasanisi, E., Vincenti, V., Giordano, D., Caruso, A., Lauda, L., et al. (2005). Surgical treatment of middle ear cholesteatoma in children with Down syndrome. Otol. Neurotol. 26, 1007–1010. doi:10.1097/01.mao.0000185042.46523.9b
Baschal, E. E., Larson, E. D., Roberts, B., Pathak, S., Frank, G., Handley, E., et al. (2020). Identification of novel genes and biological pathways that overlap in infectious and nonallergic diseases of the upper and lower airways using network analyses. Front. Genet. 10, 1352. doi:10.3389/fgene.2019.01352
Bergamaschi, R., Bergonzoni, C., Mazzanti, L., Scarano, E., Mencarelli, F., Messina, F., et al. (2008). Hearing loss in turner syndrome: Results of a multicentric study. J. Endocrinol. Invest. 31, 779–783. doi:10.1007/BF03349257
Cao, S. S., Zimmermann, E. M., Chuang, B.-M., Song, B., Nwokoye, A., Wilkinson, J. E., et al. (2013). The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice. Gastroenterology 144, 989–1000. e6. doi:10.1053/j.gastro.2013.01.023
Castle, J. T. (2018). Cholesteatoma pearls: Practical points and update. Head. Neck Pathol. 12, 419–429. doi:10.1007/s12105-018-0915-5
Collins, R., Ta, N. H., Jennings, B. A., Prinsley, P., Philpott, C. M., Steel, N., et al. (2020). Cholesteatoma and family history: An international survey. Clin. Otolaryngol. 45, 500–505. doi:10.1111/coa.13544
Diego, Z., Filippo, D. L., Maurizio, N., and Vincenzo, V. (2018). Surgical management of middle ear cholesteatoma in children with turner syndrome: A multicenter experience. Acta Bio Medica Atenei Parm. 89, 382–388. doi:10.23750/abm.v89i3.5409
Frank, D. N., Magno, J. P. M., Velasco, K. J. S., Bootpetch, T. C., Salud, J. E. D., David, K. J. V., et al. (2022). Microbiota associated with cholesteatoma tissue in chronic suppurative otitis media. Front. Cell. Infect. Microbiol. 12, 746428. doi:10.3389/fcimb.2022.746428
GenomeAsia100K Consortium (2019). The GenomeAsia 100K Project enables genetic discoveries across Asia. Nature 576, 106–111. doi:10.1038/s41586-019-1793-z
Jennings, B., Prinsley, P., Philpott, C., Willis, G., and Bhutta, M. F. (2018). The genetics of cholesteatoma. A systematic review using narrative synthesis. Clin. Otolaryngol. 43, 55–67. doi:10.1111/coa.12900
Kemppainen, H. O., Puh, H. J., Laippala, P. J., Sipila, M. M., Manninen, M. P., and Karma, P. H. (1999). Epidemiology and aetiology of middle ear cholesteatoma. Acta Otolaryngol. (Stockh.) 119, 568–572. doi:10.1080/00016489950180801
Kent, W. J., Sugnet, C. W., Furey, T. S., Roskin, K. M., Pringle, T. H., Zahler, A. M., et al. (2002). The human genome browser at UCSC. Genome Res. 12, 996–1006. doi:10.1101/gr.229102
Kircher, M., Witten, D. M., Jain, P., O’Roak, B. J., Cooper, G. M., and Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315. doi:10.1038/ng.2892
Kuo, C.-L., Shiao, A.-S., Yung, M., Sakagami, M., Sudhoff, H., Wang, C.-H., et al. (2015). Updates and knowledge gaps in cholesteatoma research. Biomed. Res. Int. 2015, e854024. doi:10.1155/2015/854024
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60, 706 humans. Nature 536, 285–291. doi:10.1038/nature19057
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinforma. Oxf. Engl. 25, 1754–1760. doi:10.1093/bioinformatics/btp324
Lim, D. B. N., Gault, E. J., Kubba, H., Morrissey, M. S. C., Wynne, D. M., and Donaldson, M. D. C. (2014). Cholesteatoma has a high prevalence in Turner syndrome, highlighting the need for earlier diagnosis and the potential benefits of otoscopy training for paediatricians. Acta Paediatr. Oslo Nor. 1992 (103), e282–e287. doi:10.1111/apa.12622
Lipkin, A. F., Coker, N. J., and Jenkins, H. A. (1986). Hereditary congenital cholesteatoma. A variant of branchio-oto dysplasia. Arch. Otolaryngol. Head. Neck Surg. 112, 1097–1100. doi:10.1001/archotol.1986.03780100085014
Liu, X., Jian, X., and Boerwinkle, E. (2013). dbNSFP v2.0: A database of human non-synonymous SNVs and their functional predictions and annotations. Hum. Mutat. 34, E2393–E2402. doi:10.1002/humu.22376
Liu, X., Jian, X., and Boerwinkle, E. (2011). dbNSFP: A lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 32, 894–899. doi:10.1002/humu.21517
Liu, X., Wu, C., Li, C., and Boerwinkle, E. (2016). dbNSFP v3.0: A one-stop database of functional predictions and annotations for human non-synonymous and splice site SNVs. Hum. Mutat. 37, 235–241. doi:10.1002/humu.22932
Lonsdale, J., Thomas, J., Salvatore, M., Phillips, R., Lo, E., Shad, S., et al. (2013). The genotype-tissue expression (GTEx) project. Nat. Genet. 45, 580–585. doi:10.1038/ng.2653
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi:10.1101/gr.107524.110
Mi, H., Muruganujan, A., Ebert, D., Huang, X., and Thomas, P. D. (2019). PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426. doi:10.1093/nar/gky1038
Prinsley, P. (2009). Familial cholesteatoma in East Anglia, UK. J. Laryngol. Otol. 123, 294–297. doi:10.1017/S0022215108002673
Prinsley, P., Jennings, B. A., Bhutta, M., Swan, D., Willis, G., and Philpott, C. (2019). The genetics of cholesteatoma study. Loss-of-function variants in an affected family. Clin. Otolaryngol. 44, 826–830. doi:10.1111/coa.13365
Ram, G., and Chinen, J. (2011). Infections and immunodeficiency in Down syndrome: Immunodeficiency in Down syndrome. Clin. Exp. Immunol. 164, 9–16. doi:10.1111/j.1365-2249.2011.04335.x
Ryan, A. F., Nasamran, C. A., Pak, K., Draf, C., Fisch, K. M., Webster, N., et al. (2020). Single-cell transcriptomes reveal a complex cellular landscape in the middle ear and differential capacities for acute response to infection. Front. Genet. 11, 358. doi:10.3389/fgene.2020.00358
Scott, E. M., Halees, A., Itan, Y., Spencer, E. G., He, Y., Azab, M. A., et al. (2016). Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat. Genet. 48, 1071–1076. doi:10.1038/ng.3592
Steinhaus, R., Proft, S., Schuelke, M., Cooper, D. N., Schwarz, J. M., and Seelow, D. (2021). MutationTaster2021. Nucleic Acids Res. 49, W446–W451. doi:10.1093/nar/gkab266
Thorvaldsdóttir, H., Robinson, J. T., and Mesirov, J. P. (2013). Integrative Genomics viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192. doi:10.1093/bib/bbs017
Uhlén, M., Fagerberg, L., Hallström, B. M., Lindskog, C., Oksvold, P., Mardinoglu, A., et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. doi:10.1126/science.1260419
Vaglio, A., Milunsky, A., Huang, X.-L., Quadrelli, A., Mechoso, B., Maher, T. A., et al. (2008). A 21 years follow-up of a girl patient with a pseudodicentric bisatellited chromosome 22 associated with partial trisomy 22pter-->22q12.1: Clinical, cytogenetic and molecular observations. Eur. J. Med. Genet. 51, 332–342. doi:10.1016/j.ejmg.2008.01.001
Wang, K., Li, M., and Hakonarson, H. (2010). Annovar: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164. doi:10.1093/nar/gkq603
Xia, J., Benner, M. J., and Hancock, R. E. W. (2014). NetworkAnalyst–integrative approaches for protein-protein interaction network analysis and visual exploration. Nucleic Acids Res. 42, W167–W174. doi:10.1093/nar/gku443
Xia, J., Gill, E. E., and Hancock, R. E. W. (2015). NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 10, 823–844. doi:10.1038/nprot.2015.052
Zhang, C., Syed, T. W., Liu, R., and Yu, J. (2017). Role of endoplasmic reticulum stress, autophagy, and inflammation in cardiovascular disease. Front. Cardiovasc. Med. 4, 29. doi:10.3389/fcvm.2017.00029
Zhao, H., Wang, Y., Li, B., Zheng, T., Liu, X., Hu, B. H., et al. (2020). Role of endoplasmic reticulum stress in otitis media. Front. Genet. 11, 495. doi:10.3389/fgene.2020.00495
Keywords: cholesteatoma, otitis media, exome sequencing, network analysis, chronic otitis media
Citation: Lee NK, Cass SP, Gubbels SP, Gomez HZ, Scholes MA, Jenkins HA and Santos-Cortez RLP (2023) Novel candidate genes for cholesteatoma in chronic otitis media. Front. Genet. 13:1033965. doi: 10.3389/fgene.2022.1033965
Received: 01 September 2022; Accepted: 26 December 2022;
Published: 09 January 2023.
Edited by:
Desheng Liang, Central South University, ChinaReviewed by:
Barbara Jennings, University of East Anglia, United KingdomHenrique Pauna, Hospital IPO, Brazil
Copyright © 2023 Lee, Cass, Gubbels, Gomez, Scholes, Jenkins and Santos-Cortez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Regie Lyn P. Santos-Cortez, cmVnaWUuc2FudG9zLWNvcnRlekBjdWFuc2NodXR6LmVkdQ==
†Present address: Melissa A. Scholes, Department of Otolaryngology, University of Mississippi Medical Center, Jackson, MS, United States