
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 05 January 2023
Sec. Evolutionary and Population Genetics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.1033437
This article is part of the Research TopicPopulation Genetics and Genomics of Plant Pathogenic Fungi and OomycetesView all 4 articles
 Bochra Amina Bahri1,2*
Bochra Amina Bahri1,2* Rajiv Krishna Parvathaneni1,2
Rajiv Krishna Parvathaneni1,2 Willis Turner Spratling1
Willis Turner Spratling1 Harshita Saxena2
Harshita Saxena2 Suraj Sapkota1†
Suraj Sapkota1† Paul L. Raymer2,3
Paul L. Raymer2,3 Alfredo D. Martinez-Espinoza1
Alfredo D. Martinez-Espinoza1Dollar spot is one of the most damaging diseases in turfgrass, reducing its quality and playability. Two species, Clarireedia monteithiana and C. jacksonii (formerly Sclerotinia homoeocarpa) have been reported so far in the United States To study the Clarireedia genome, two isolates H2 and H3, sampled from seashore paspalum in Hawaii in 2019 were sequenced via Illumina paired-end sequencing by synthesis technology and PacBio SMRT sequencing. Both isolates were identified as C. aff. paspali, a novel species in the United States Using short and long reads, C. aff. paspali H3 contained 193 contigs with 48.6 Mbp and presented the most completed assembly and annotation among Clarireedia species. Out of the 13,428 protein models from AUGUSTUS, 349 cytoplasmic effectors and 13 apoplastic effectors were identified by EffectorP. To further decipher Clarireedia pathogenicity, C. aff. paspali genomes (H2 and H3), as well as available C. jacksonii (LWC-10 and HRI11), C. monteithiana (DRR09 and RB-19) genomes were screened for fifty-four pathogenesis determinants, previously identified in S. sclerotiorum. Seventeen orthologs of pathogenicity genes have been identified in Clarireedia species involved in oxalic acid production (pac1, nox1), mitogen-activated protein kinase cascade (pka1, smk3, ste12), appressorium formation (caf1, pks13, ams2, rgb1, rhs1) and glycolytic pathway (gpd). Within these genes, 366 species-specific SNPs were recorded between Clarireedia species; twenty-eight were non-synonymous and non-conservative. The predicted protein structure of six of these genes showed superimposition of the models among Clarireedia spp. The genomic variations revealed here could potentially lead to differences in pathogenesis and other physiological functions among Clarireedia species.
Turfgrass is a valuable commodity worldwide and in the United States It is used in home lawns, golf courses, sport fields and recreational lands and shown to improve groundwater recharge, soil erosion and soil carbon sequestration (Held and Potter 2012). The turfgrass industry contributes more than 822,848 jobs and has a total economic impact of $57.94 billion annually in the United States (Haydu et al., 2006). However, dollar spot, the most common and damaging disease in turfgrass, represents a significant risk for the expansion and the economic sustainability of this commodity. It has a worldwide distribution and can infect most cultivated turfgrass species. The disease is typically controlled by repeated fungicide applications. Because of high demand in turfgrass aesthetic quality and playability, mitigating dollar spot disease represents a major expense for the turfgrass industry (Smiley et al., 2005) and fungicide applications, resulting in environmental safety issues including the emergence of fungicide-resistant dollar spot strains (Putman et al., 2015).
The disease was at first attributed to the ascomycete Sclerotinia homoeocarpa F.T. Bennett (Bennett, 1937). However, taxonomic classification of the causal agent remained unresolved until 2018. Based on three DNA markers (rDNA internal transcribed spaces ITS region, calmodulin CaM and DNA replication licensing factor Mcm7), Salgado-Salazar and colleagues (Salgado-Salazar et al., 2018) placed the causal agent in a new genus, Clarireedia, as a member of the family Rutstroemiaceae rather than the Sclerotiniaceae family. Their study described four species for the genus Clarireedia, C. homoeocarpa, C. bennettii, C. jacksonii, and C. monteithiana. C. jacksonii and C. monteithiana were the two species reported in the United States and the most prevalent worldwide species, infecting cool- and warm-season turfgrasses, respectively. C. homoeocarpa and C. bennettii were isolated on warm-season turfgrasses and were restricted to the United Kingdom (Salgado-Salazar et al., 2018). Recently, a new species, C. paspali, was reported on seashore paspalum (Paspalum vaginatum) in different provinces of China (Hu et al., 2019). This species is characterized by the presence of an intron at the 3′-end of the small subunit ribosomal ribonucleic acid region, which genetically separated C. paspali from C. jacksonii, and C. monteithiana. However, within C. paspali, a group named C. aff. paspali, which did not present this intronic region, was genetically differentiated (Hu et al., 2019). Typically, the molecular identification of Clarireedia pathogens and their differentiation at the species level requires conventional Sanger sequencing using universal primers. In their study, Salgado-Salazar et al. (2018) identified twenty-eight, twenty, and eight species-specific SNPs differentiating C. monteithiana, C. jacksonii, C. homoeocarpa, and C. bennettii in the ITS, CaM and Mcm7 sequences, respectively (Salgado-Salazar et al., 2018). However, recently Groben et al. (2020) were able to quickly diagnose Clarireedia from field samples using a quantitative real-time PCR assay. In addition, a co-dominant cleaved amplified polymorphic sequence assay differentiating C. monteithiana from C. jacksonii was recently developed (Stackhouse et al., 2021). Beside variations at the molecular level (Powell and Vargas 2001; DeVries et al., 2008), diversity in vegetative compatibility groups (VCGs) and mating-type locus were also reported in Clarireedia spp. (Powell and Vargas 2001; Viji et al., 2004). Overall, the clonal reproduction of the pathogens supported the low level of genetic diversity observed (Powell and Vargas 2001; Viji et al., 2004; DeVries et al., 2008). However, some Clarireedia studies provided evidence that heterokaryosis and random mating increased genetic variability in nature (Hsiang and Mahuku 1999; Kessler et al., 2018; Salgado-Salazar et al., 2018) and stimulated research interest in exploring the genomic diversity, population dynamics, and structure.
So far, few research studies have investigated the pathogenicity in Clarireedia and knowledge in turfgrass-Clarireedia interactions are currently lacking. Although, C. jacksonii and C. monteithiana have been predominantly isolated in United States fields from cool-and warm-season turfgrasses, respectively (Salgado-Salazar et al., 2018), artificial cross inoculations showed no host specificity for dollar spot infection (Aynardi et al., 2019; Sapkota et al., 2020). In fact, under artificial inoculations, no variability in host range and only differences in virulence level were observed between Clarireedia isolates (Chakraborty et al., 2006; Steketee et al., 2017). Several virulence factors in plant pathogenic fungi were shown to contribute to a successful infection. Oxalic acid was detected in pure cultures of Clarireedia spp. (Venu et al., 2009) as well as in infected turfgrass tissues (Orshinsky et al., 2012a; Rioux et al., 2020) and was suggested to be an important pathogenicity factor as it is in S. sclerotiorum, the closest well-characterized fungal pathogens (Bolton et al., 2006). This compound was identified to play a role in symptom development and host colonization (Rioux et al., 2020). Additionally, glycosyl hydrolase enzymes and serine proteases, previously reported as pathogenicity factors in several fungal pathogens with a wide host range (Rowe and Kliebenstein 2007; Li et al., 2010), were identified in Clarireedia spp. transcripts and up-regulated in dollar spot infected creeping bentgrass (Orshinsky et al., 2012a). Further studies are still needed to decipher the full pathogenicity pathways of Clarireedia species at the molecular and genomic levels.
Over the past two decades, the development of next-generation sequencing (NGS) technologies and the contemporary advances in computational biology and bioinformatics, have boosted species diagnosis and scientific discoveries. Several studies in plant pathogens have highlighted the use of NGS technologies in providing high throughput species/strain level characterization and investigating the changes in population dynamics (Hubbard et al., 2015; Malapi-Wight et al., 2016; Chalupowicz et al., 2019). By coupling Illumina with PacBio approaches, combining short-read and long-read sequencing datasets, scientists were able to reach a higher level of whole genome sequencing and genome assembly for several plant pathogen species (De Miccolis Angelini et al., 2019; Porto et al., 2019). Typically, a well-curated and assembled reference genome for mapping NGS reads is crucially needed to facilitate pathogen identification, resolve taxonomy of dollar spot species, and perform phylogenetic and comparative genomic analysis across strains and pathogen species. Whole genome assembly is also important to understand genomic structure of pathogen species and to infer important phenotypic characteristics (such as virulence and fungicide resistance) and predict pathogen dynamics over time. To date, draft genomes for ten Clarireedia isolates have been generated: HRI11, HRS10, LWC-10, MB-01, SH44, SE16F4, CPB17, and LT30 were classified as C. jacksonii; while DRR09 and RB-19 were identified as C. monteithiana (Green et al., 2016; Salgado-Salazar et al., 2018; Crouch et al., 2021; Sapkota et al., 2021). Eight out of the ten Clarireedia genomes assembled were only sequenced using Illumina technology. The maximum genome sizes for C. jacksonii and C. monteithiana were reported for HRI11 with 43.35 Mbp sequenced with Illumina and PacBio (Green et al., 2016) and DRR09 with 48.70 Mb sequenced with Illumina (Crouch et al., 2021), respectively. Overall, the genomes currently available for Clarireedia spp. are fragmented (with >231 scaffolds), and most genomes lack information on genome features, and are not annotated. In addition, there are no comprehensive studies on the genomes and the genomic diversity in Clarireedia spp. Annotation of the genomes and identification of the genetic variation between the different Clarireedia species responsible for dollar spot are essential for understanding the epidemiology, pathogenicity, evolution, and host specialization of the pathogens and for improving turfgrass disease management strategies.
The overall goal of this study is to genetically and molecularly characterize C. aff. paspali, a new species of the Clarireedia genus causing dollar spot in turfgrass. This study aims to gain insight into shared genomic features among Clarireedia species and between the Clarireedia species and the closely related fungal species of the Sclerotiniaceae and Rutstroemiaceae families. The objectives were to 1) present the first draft genome of C. aff. paspali, 2) compare the genome features of C. aff. paspali with other Clarireedia species, 3) perform a phylogenetic analysis to understand the relationship among Clarireedia species and between Clarireedia and its close fungal relatives, and 4) compare at the genomic and protein levels the genome of C. aff. paspali with other Clarireedia species for a range of possible determinants involved in pathogen morphogenesis and pathogenesis. In this study, S. sclerotiorum and R. sydowiana were used as representatives of the Sclerotiniaceae family and the Rutstroemiaceae family, respectively.
Plugs of seashore paspalum displaying dollar spot symptoms on leaves were sampled by a sod producer in Hawaii in September of 2019 (lat. 21.42827; long. -158.02432). The symptoms were characterized by white to straw-colored lesions, tip dieback, and irregular sunken patches in the turf stand (Sapkota et al., 2020). Two isolates, H2 and H3, were isolated and purified from the cores following the protocol described by Sapkota et al. (2020). Briefly, seashore paspalum leaves with dollar spot symptoms were surface sterilized with 10% bleach and 80% ethanol for 2 min each, rinsed with sterile autoclaved water three times, cut into 1-to-2 cm pieces, plated on potato dextrose agar (PDA) plates, and incubated at room temperature. White fluffy mycelium growth was visible 24-hr after incubation. The actively growing mycelium was transferred to another PDA plate and this step was repeated three times to obtain pure cultures. For each isolate, a seven-day old mycelial culture was scrubbed from the PDA medium and DNA extraction was performed using cetyl trimethylammonium bromide (CTAB) method (Doyle and Doyle 1987). The internal transcribed spacer (ITS) region of ribosomal DNA, calmodulin (CaM) gene, elongation factor 1 (EF) gene, and b-tubulin (TUB) gene of the two isolates were amplified using ITS5-ITS4 (White et al., 1990), CMD5-CMD6 (Hong et al., 2005), EF1F-EF1R (Carbone and Kohn 1999), and Bt2a- Bt2b (Glass and Donaldson 1995) primer sets, respectively. Two isolates, C. jacksonii (DS-CB, also known as DS3) and C. monteithiana (DS-SP, also known as DS8) sampled in Griffin, GA in 2019 from bentgrass and seashore paspalum, respectively, were also added as references (Sapkota et al., 2020). Amplicons were analyzed by 2% agarose gel electrophoresis, Sanger sequenced, and blasted against the NCBI database. Sequence alignment with ClustalW and comparison of ITS and EF regions from H2 and H3 isolates with Clarireedia reference isolates sampled from seashore paspalum from Hu et al. (2019) (reference isolates BH3, BH1, HK1, and SZ1 of C. jacksonii, C. monteithiana, C. aff. paspali, and C. paspali, respectively), was also performed under Mega X (Kumar et al., 2018). Species-specific single nucleotide polymorphisms (SNPs), identified by Salgado-Salazar et al. (2018), were also investigated in the aligned ITS sequences.
Creeping bentgrass (Agrostis stolonifera) cv. A-1/A-4, zoysiagrass (Zoysia matrella) cv. Zorro, bermudagrass (Cynodon dactylon) cv. Princess, and seashore paspalum (Paspalum vaginatum) cv. SeaStar turfgrasses were used for the pathogenicity test of H2 and H3 isolates. Plants were grown in Kord nursery pots (3” × 3”) filled with Metro-MIX 852 RSI professional growing mix (Sun Gro Horticulture). Treatments consisted of H2-inoculated pots, H3-inoculated pots, and uninoculated control pots for each turfgrass species. Each treatment consisted of three replications for a total of thirty-six pots. H2 and H3 inoculum was prepared using sterile, water-soaked mixtures of wheat, barley, and oat seeds. One-week old H2- or H3-colonized Potato Dextrose Agar (Difco) plugs were independently added to grain mixes in 500 ml Erlenmeyer flasks. Mycelia of H2 and H3 were allowed to infect and proliferate through grain mixes for 2 weeks. Five infected grain seeds were then added to all corresponding treatment pots, and five uninfected grain seeds were added to each control pot. After inoculation, pots were arranged randomly in nursery trays and trays were covered with Mondi propagation domes that were sprayed with sterile water to increase and maintain humidity. Trays were then placed in a growth chamber (Conviron, Pembina, ND, United States) and covered with black plastic bags that had been sprayed with sterile water. The bags were removed after 2 days in the growth chamber, and disease symptoms were evaluated in the following days. The growth chamber used for the pathogenicity test was programmed to operate at 23°C and 90% relative humidity with a 12h/12 h photoperiod (day/night). Disease was visually scored using a severity scale (% turf area blighted) with an index of 0–10 (0 = asymptomatic; 1 = 1%–10% symptomatic tissue; 5 = 41%–50% symptomatic tissue; 10 = 91%–100% symptomatic tissue) (Viji et al., 2004; Aynardi et al., 2019). DS-SP C. monteithiana isolate sampled in 2019 in seashore paspalum in Georgia United States (Sapkota et al., 2020) was also added as reference in the pathogenicity test.
Seven-day old mycelial cultures of H2 and H3 isolates were scrapped from the PDA medium and sent on dry ice to BGI Americas Corporation (https://www.bgi.com/us/) for DNA extraction, library construction and DNA sequencing with NGS technologies. High-quality genomic DNA (gDNA) was extracted from both isolates using CTAB protocol (Doyle and Doyle 1987). DNA quantification and purity were evaluated using Qubit™ Fluorometric quantitation (Thermo Fisher Scientific, Inc., Waltham, MA, United States) and NanoDrop reading, and 0.6% TAE agarose gel was run to check the DNA quality in order to meet the required standards for sequencing (sample concentration≥50 ng/ul and sample purity A260/280 at 1.8–2.0).
For one of the two isolates (H3), a 20 K library was constructed for PacBio SMRT sequencing. A large amount of high-quality DNA (20 µg) was used for the long-read library preparation, per the manufacturer protocols (PacBio, Menlo Park, CA, United States). SMRTbell Express Template Prep kit (PacBio, Menlo Park, CA, United States) was used for library template preparation. Briefly, g-TUBE (Covaris, Inc., United States) and AMPure PB beads (Beckman Coulter Inc. United States) were used to shear the gDNA into ∼20-kb fragments and for fragment size selection, respectively. To remove the single-stranded ends and repair DNA damage, the sheared gDNA was treated with ExoVII enzyme. Then, single-molecule real time (SMRT) hairpin adapters were ligated to the ends of the double-strand fragments polished with T4 DNA Polymerase. Subsequently, the adapter-ligated SMRTbell template was digested with ExoIII and VII enzymes for the removal of failed ligation products. The library was again purified with AMPure PB beads, quantified using Qubit™ Fluorometric quantitation and quality checked using a 2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA, United States). Sequencing primer was then annealed to the SMRTbell template, followed by binding of sequence polymerase to the annealed template. Finally, one SMRT cell was run on the PacBio PacBio Sequel System to sequence the sample.
In addition, whole genome shotgun (WGS) sequencing approach was performed on both H2 and H3 isolates. 2 µg of gDNA of each isolate were fragmented and size selected as described above. Paired-end short read sequencing libraries (Illumina HiSeq 2500 System, 2 × 100 bp) were prepared with Illumina HiSeq Rapid v2 SBS Kits. The short-read libraries were sequenced on an Illumina HiSeq 2500 System, per standard Illumina protocols (Illumina, Inc., San Diego, CA, United States). The sequencing adapters in the PacBio CLR reads and the 2 × 100 Illumina HiSeq reads were removed by the sequencing company.
The genome size estimation was performed using the k-mer analysis of the 2 × 100 Illumina HiSeq reads from the H3 isolate using the k-mer of sizes 17, 19, and 21 under the Jellyfish software (Marçais and Kingsford 2011). The frequency of the k-mers and the depth were plotted for the k-mer frequency distribution. An error threshold of four was used to eliminate the noise peak likely due to sequencing errors. Genome size was estimated by dividing the total number of k-mers by the peak position. The genome coverage for Illumina was calculated by dividing the total number of reads by the genome size estimate with the different k-mer sizes. The genome coverage was also estimated from the assembled genome of C. aff. paspali H3.
The genome assembly of the C. aff. paspali was performed using PacBio reads of isolate H3 under the fork of the canu version 2.2 (Koren et al., 2017). Default parameters were used with an estimated genome size of 42 MB (Conservative genome estimate taken from the k-mer analysis of Jellyfish). The assemblies were polished using the Illumina reads (∼35X read coverage) of isolate H3 under Pilon using the settings “--fix all--mindepth 0.5.” The resulting contigs were BLAST to the NCBI “nr” database to contaminants. An e-value cutoff of 1e-10 was used. Contigs (74 from 194 total contigs) which had matches to the “nr” database at the stringent cut-off were manually inspected for the presence of non-plant contaminations. One contig (tig00001304_pilon; size = 2200 bp) showed homology to a herpes virus genome (e-value = 0) over its entire length and was removed from downstream analyses. The H2 genome assembly was performed using SPAdes version 3.15 (Bankevich et al., 2012). The genome coverage was estimated for the final genomes of the H2 and H3 isolates. The completeness of the genome of C. aff. paspali (H3 isolate) was assessed by the Benchmarking universal Single Copy Orthologs (BUSCO) (Simão et al., 2015) compared to a reference dataset of fungi (“fungi_odb10”) and eukaryote (“eukaryote_odb10”). Published genomes of C. jacksonii (LWC-10 isolate, Crouch et al., 2021), C. monteithiana (DRR09 isolate; Crouch et al., 2021) and S. sclerotiorum (1980 UF-70 isolate; GenBank Acc. No. GCA_001857865.1) were added for the comparative analysis. In addition, RepeatMasker v4.0.9 (http://www.repeatmasker.org/) was used to assess the repetitive content in the genome using the settings “-parallel 40 -html -gff -e rmblast -e ncbi.”
The soft masked genome generated from the RepeatMasker was used to for the gene prediction. The ab initio gene prediction was performed under AUGUSTUS v3.4 using the species Sclerotinia sclerotiorum genome (1980 UF-70 isolate; GenBank acc. no GCA_001857865.1) and using the hints file generated from the BLAST analysis of the EST sequences from C. monteithiana (DRR09; Crouch et al., 2021) and C. jacksonii (MB01; Crouch et al., 2021). The aligned ESTs were filtered for a minimum coverage of 80% (filterPSL.pl--best--minCover = 80) and the hint file was generated. Furthermore, the functional annotation prediction of the C. aff. paspali (H3) was performed using the INTERPRO scan (Blum et al., 2021) under default settings. We used the setting “--goterms--pathways--iprlookup” in INTERPRO scan that assigned the GO term to the genes. The top 10 occurrences of the GO terms were identified from this INTERPRO list for cellular component, biological process and molecular function.
The similarity in the nucleotides at the whole genome level among the Clarireedia species was performed to understand how they are related to each other. Two isolates for each species were used: C. jacksonii, C. monteithiana and C. aff. paspali were represented by LWC-10 (Crouch et al., 2021) and HRI11(Green et al., 2016), DRR09 and RB-19 (Crouch et al., 2021), H3 and H2 isolates, respectively. The Clarireedia genomes were also compared with S. sclerotiorum (1980 UF-70 isolate; GenBank acc. no GCA_001857865.1) and R. sydowiana (CBS115975 isolate; GenBank acc. no JWJB00000000). For this analysis, the program FASTAni (Jain et al., 2018) was used with default settings. Furthermore, the different Clarireedia species and S. sclerotiorum were compared on the orthologous genes. Identification of orthologous genes in C. aff. paspali (H3) was performed using the protein models from AUGUSTUS under Orthofinder (Emms and Kelly 2019) with default settings with published gene models of C. jacksonii (LWC-10), C. monteithiana (DRR09) and S. sclerotiorum (1980 UF-70 isolate). An orthogroup can comprise one or more gene models from at least one species. The overlap between ortho groups across species was plotted using the Upset plot (https://asntech.shinyapps.io/intervene/). Because no gene models were available for R. sydowiana, the comparison of orthogroups was not possible.
The molecular biology of C. aff. paspali morphogenesis and pathogenesis was investigated at the genomic and protein levels. We used the 13,428 protein models identified through AUGUSTUS and ran it through the EffectorP version 3 (Sperschneider and Dodds, 2022) using the default settings. The program identified weather the gene model is either a cytoplasmic effector, apoplastic effector, both or a non-effector. We used a cut-off of p-value > 0.9 to designate the cytoplasmic and apoplastic effectors. A BLASTP analysis to the protein database of NCBI was performed on these effectors and the top 10 hits were noted.
The genome of C. aff. paspali was compared with C. monteithiana and C. jacksonii as well as S. sclerotiorum (1980 UF-70 isolate), as a representative of the Sclerotiniaceae family, for the presence/absence of a range of fifty-four possible determinants involved in pathogen morphogenesis and pathogenesis (Supplementary Table S1). These determinants were previously characterized in S. sclerotiorum (Xia et al., 2019), the closest fully sequenced and well characterized relative species to Clarireedia. LWC-10 and HRI11 (Green et al., 2016; Crouch et al., 2021), DRR09 and RB-19 (Crouch et al., 2021), H3 and H2 isolates, representative of C. jacksonii, C. monteithiana, and C. aff. paspali were used in this analysis. For each gene present in Clarireedia, the average percentage of similarity between Clarireedia spp. and S. sclerotiorum at the DNA and protein levels were also recorded using NCBI blast. Species-specific SNP variations as well as non-synonymous and non-conservative SNPs between the Clarireedia species within these pathogenicity genes was also investigated for the genes that were present in all three species C. aff. paspali, C. monteithiana, and C. jacksonii. Species-specific SNPs are SNPs present in both isolates of a particular Clarireedia species and absent in isolates of the other two species. Non-conservative SNPs are non-synonymous substitutions that change the property of the corresponding amino acids. This comparative analysis was also performed on five conserved genomic sequences of the ITS, TUB, CaM, EF and Mcm7 regions. BLAST analysis of the curated pathogenicity and conserved genes was performed using the default settings to extract the sequences from the different assembled genomes. The S. sclerotiorum cDNA sequences were used as templates to assign the start and the end of the gene. We also included in this comparative analysis the sequences from the plant pathogen R. sydowiana (CBS115975 isolate) as a representative of the Rutstroemiaceae family, and the sequences from the plant pathogen Z. tritici (CBS115943 isolate; https://fungi.ensembl.org/Zymoseptoria_tritici/Info/Index) causal agent of septoria leaf blotch as an outgroup.
The nucleotide sequences of the 13 virulence genes (present across the Clarireedia species) from S. sclerotiorum were translated using MEGA X (Kumar et al., 2018) software, and the peptide sequences obtained were used to predict the three-dimensional structure of the selected virulence genes using a web-based protein structure prediction tool, Protein Homology/analogy Recognition Engine (Phyre2) version 2.0 (Kelley et al., 2015). The predicted structures having coverage >80% were selected, followed by prediction of structures of the respective virulence genes from C. jacksonii, C. monteithiana, and C. aff. paspali. The molecular graphics, structural alignment, and comparison of the proteins from S. sclerotiorum and the three Clarireedia spp. was performed using the MatchMaker extension of UCSF Chimera 1.15 (Pettersen et al., 2004). Along with constructing the molecular graphics of the predicted models, movie animation of the aligned models was created to distinguish the structural differences using UCSF Chimera 1.15. In addition, the quality of the structures predicted was validated using two model validation online tools, the Structure Analysis and Verification Server (SAVES) v6.0 and Qualitative Model Energy ANalysis (QMEAN) (Benkert et al., 2011). The different programs in SAVES v5.0 such as PROCHECK (Laskowski et al., 1996), VERIFY-3D (Bowie et al., 1991; Lüthy et al., 1992) and ERRAT (Colovos and Yeates 1993), were used to provide the stereochemical attributes of the structure in the form of Ramachandran plot, atomic-resolution coordinates of the 3D models, and comprehensive quality factor of the non-bonded atoms in the structure, respectively. The quality score of the model was also evaluated as Z-scores using QMEAN tool.
The extracted sequences of six pathogenicity genes (shk1, ste12, smk3, pph1, gpd, nox2 that were present in all three Clarireedia species as well as in R. sydowiana) and the five conserved genes (ITS, TUB, CaM, EF and Mcm7) were concatenated into one contiguous sequence for each isolate, aligned with MUSCLE (Edgar 2004) and used for phylogenetic analysis. A neighbour joining (NJ) tree based on the maximum composite likelihood method was performed under Mega X (Kumar et al., 2018) with a 500 replicates bootstrap test. In addition, divergence-time estimates within Clarireedia species and between Clarireedia and S. sclerotiorum were also calculated using an average relative divergence time of 437 million years ago (Mya) between S. sclerotiorum and Z. tritici as standard (www.timetree.org). Furthermore, a molecular clock test was conducted in MEGA X (Kumar et al., 2018) and was performed by comparing the Maximum Likelihood value for the NJ topology with and without the molecular clock constraints under Tamura-Nei model (Tajima 1993). The analysis was performed on a total of 22,983 positions of the nine nucleotide sequences combining the virulence and the conserved gene regions. The nine nucleotide sequences were from the Clarireedia isolates LWC-10 and HRI11 (Green et al., 2016; Crouch et al., 2021), DRR09 and RB-19 (Crouch et al., 2021), H3 and H2, as well as from S. sclerotiorum (1980 UF-70 isolate), Rutstroemia sydowiana (CBS115975 isolate), Zymoseptoria tritici (CBS115943 isolate). In addition, the Tajima’s relative rate test (Tamura and Nei 1993) was performed under MEGA X (Kumar et al., 2018) between C. aff. paspali (H3), C. jacksonii (LWC-10) and C. monteithiana (DRR09), with Z. tritici (CBS115943) sequence as an outgroup.
Pure cultures of H2 and H3 presented an aspect with a white fluffy mycelium on PDA media, typical for dollar spot (Figures 1A,B). From the pathogenicity tests, aerial mycelial growth from both H2 and H3 inoculations, as well as for the DS-SP C. monteithiana isolate, was observed in all four turfgrass species 2 days after inoculation. Dollar spot foliar symptoms (lesions, blight, tip dieback) caused by H2, H3 and DS-SP infection were prominent in all turfgrass species 2 weeks after inoculation (Figures 1D–F). Symptoms were most severe on seashore paspalum. No differences in aggressiveness were observed between the 2 C. aff. paspali isolates and DS-SP C. monteithiana isolate when inoculated on the four different turfgrass species. Control plants showed no symptoms. The pathogen was re-isolated from the artificially inoculated plants. Results of the pathogenicity tests indicate that both H2 and H3 isolates were able to infect cool- and warm-season turfgrass species under favorable conditions. In addition, preliminary tests showed that H3 C. aff paspali isolate did not show any differences in aggressiveness compared to DS9 and DS7 C. monteithiana isolates (sampled at Griffin, GA in 2019 on berdumagrass and zoysiagrass, respectively) and DS-CB C. jasksonii isolate (sampled from Griffin, GA in 2019 on creeping bentgrass) when inoculated on seashore paspalum cv. SeaStar, bermudagrass cv. Princess, zoysiagrass cv. Zorro and creeping bentgrass cv. A-1/A-4 (data not shown). However, additional investigations need to be carried out at the population level to compare the aggressiveness of this new species C. aff. paspali to the previously described species in the United States.
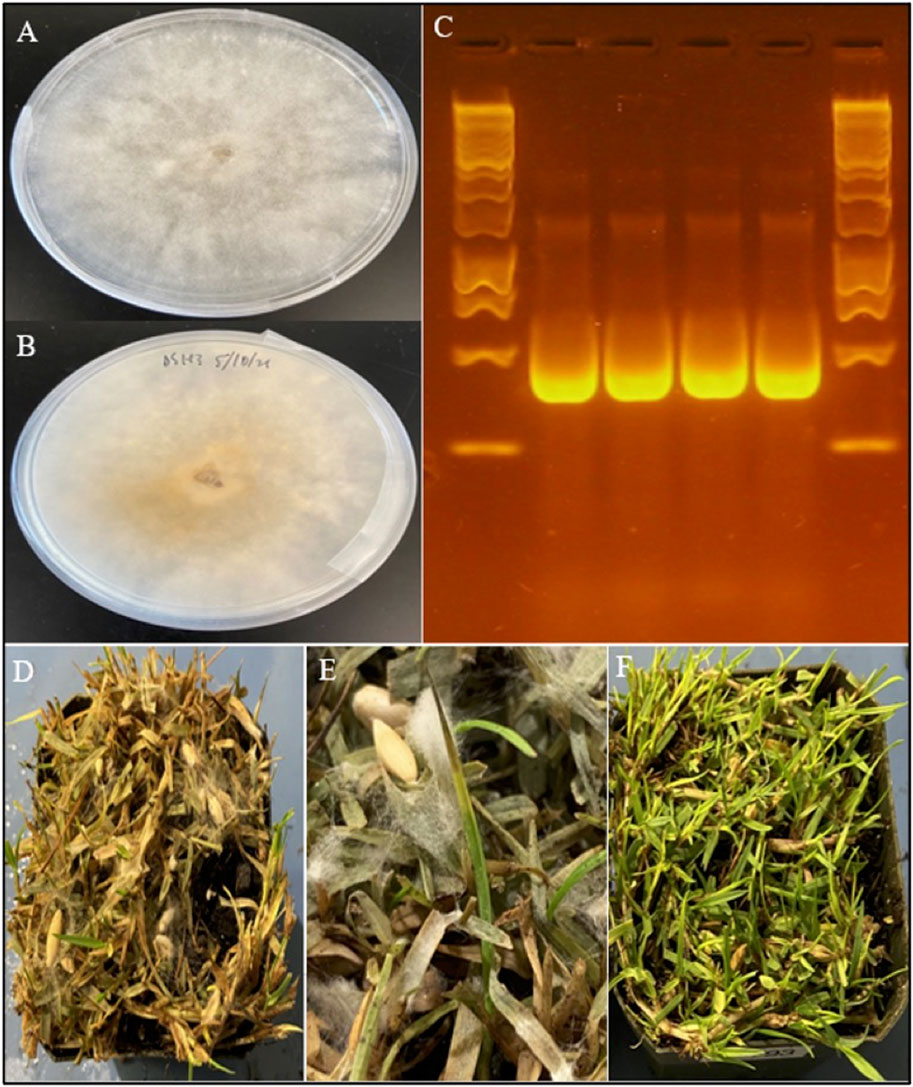
FIGURE 1. Morphology, molecular identification, and symptoms of C. aff. paspali. (A) Front and (B) back of a 5-day old C. aff. paspali culture (H3 isolate) on PDA media. (C) Molecular characterization of H2 (no. 1) and H3 isolates (no. 2) of C. aff. paspali along with two reference isolates DS-SP of C. monteithiana (no. 3) and DS-CB of C. jacksonii (no. 4) on the ITS region (from left to right) showing no differences in band sizes between Clarireedia species. A Thermo Scientific GeneRuler 1 kb DNA Ladder was used. (D) Pathogenicity tests showing signs and symptoms of C. aff. paspali infection and (E) foliar tip dieback and aerial mycelia growth. (F) Non-inoculated control.
BLAST results against the NCBI database of H2 and H3 Sanger sequences exhibited 98.07%–99.37% sequence homology to C. homeocarpa (GenBank acc. no. KF545301.1, MF969128.1, DQ448301.1, KF545258.1 for ITS, TUB, EF and CaM respectively). Based on sequence comparison with Clarireedia reference isolates from Hu et al. (2019), H2 and H3 isolates showed the highest homology (100–99.59%) with isolate HK1 of C. aff. paspali on the ITS and EF regions, and did not present the intron at the 3′-end of the SSU rDNA region, characteristic of C. paspali (Figure 1C). In addition, H2 and H3 did not carry any of the SNPs specific to C. jacksonii, C. monteithiana, C. homeocarpa or C. bennetti identified in the ITS region by Salgado-Salazar et al. (2018). However, H2 and H3 isolates presented 1 (“C” at position 131; Salgado-Salazar et al. (2018)) and 2 (“C” at position 64 and “C” at position 483; Salgado-Salazar et al. (2018)) species-specific SNPs for C. aff. paspali in the ITS and EF genes, respectively.
Draft genome sequence data, generated using an Illumina 550 bp PE-SRS library, generated 14,99,240,400 of total raw reads (H2 and H3 isolate) and 14,73,271,400 and 14,90,276,400 of total high-quality reads for the H2 and H3 isolates, respectively. The H3 isolate was sequenced with the long-read PacBio sequencing platform with a total of 163,709 sequenced reads for a total of 13.90 gigabases.
The main peak in the k-mer frequency distribution graph gives a matched k-mer value for the genome size estimation. Noise peaks were formed by very low coverage error k-mers (noise; below k = 4) from sequencing errors. The highest peak was observed at estimated depths of 35X, 34X, and 35X for the 17-, 19-, and 21-mers, respectively (Figure 2). The presence of a single dominant peak indicates that the genome is only slightly repetitive and haploid. According to the k-mer size estimations, the estimated genome size of C. aff. paspali H3 isolate ranged between 42–44 Mb with ∼316X of coverage (with 44 Mb genome size).
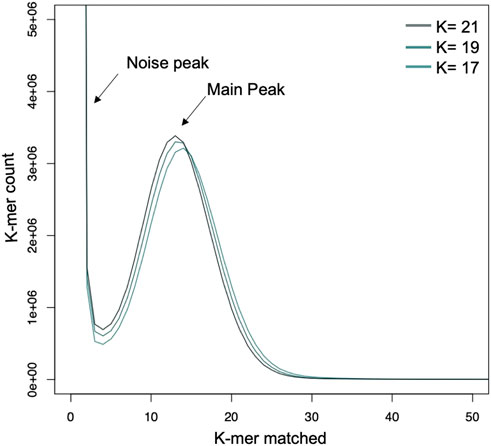
FIGURE 2. Genome estimation using k-mer analysis with 17-, 19- and 21- mers. The k-mers show a peak at a similar location which gives an estimate of 35 X genome coverage and a genome size of 48 MB for C. aff. paspali (H3 isolate).
The error corrected PacBio + HiSeq draft assembly of C. aff. paspali H3 isolate contained a total of 193 contigs with ∼48.6 Mbp and contig N50 length was 5 Mb (Table 1). The top 23 contigs represented >90% of the total genome (43.8 Mbp). The sizes of the two largest contigs for C. aff. paspali H3, tig00000001 and tig00002064, were 10.2 Mbp and 5.9 Mbp, respectively (Supplementary Table S2). The de novo assembly for C. aff. paspali H2 isolate produced 6,743 contigs with 39.26 Mbp, and ∼77 kb contig N50 with HiSeq sequencing method. Based on a 48.6 Mb estimate, we generated more than ∼286X coverage (size of genome/total # of bases) of the H3 C. aff. paspali genome using one SMRT cell. The H2 isolate was assembled using Illumina reads with ∼37.53 X coverage of the genome.
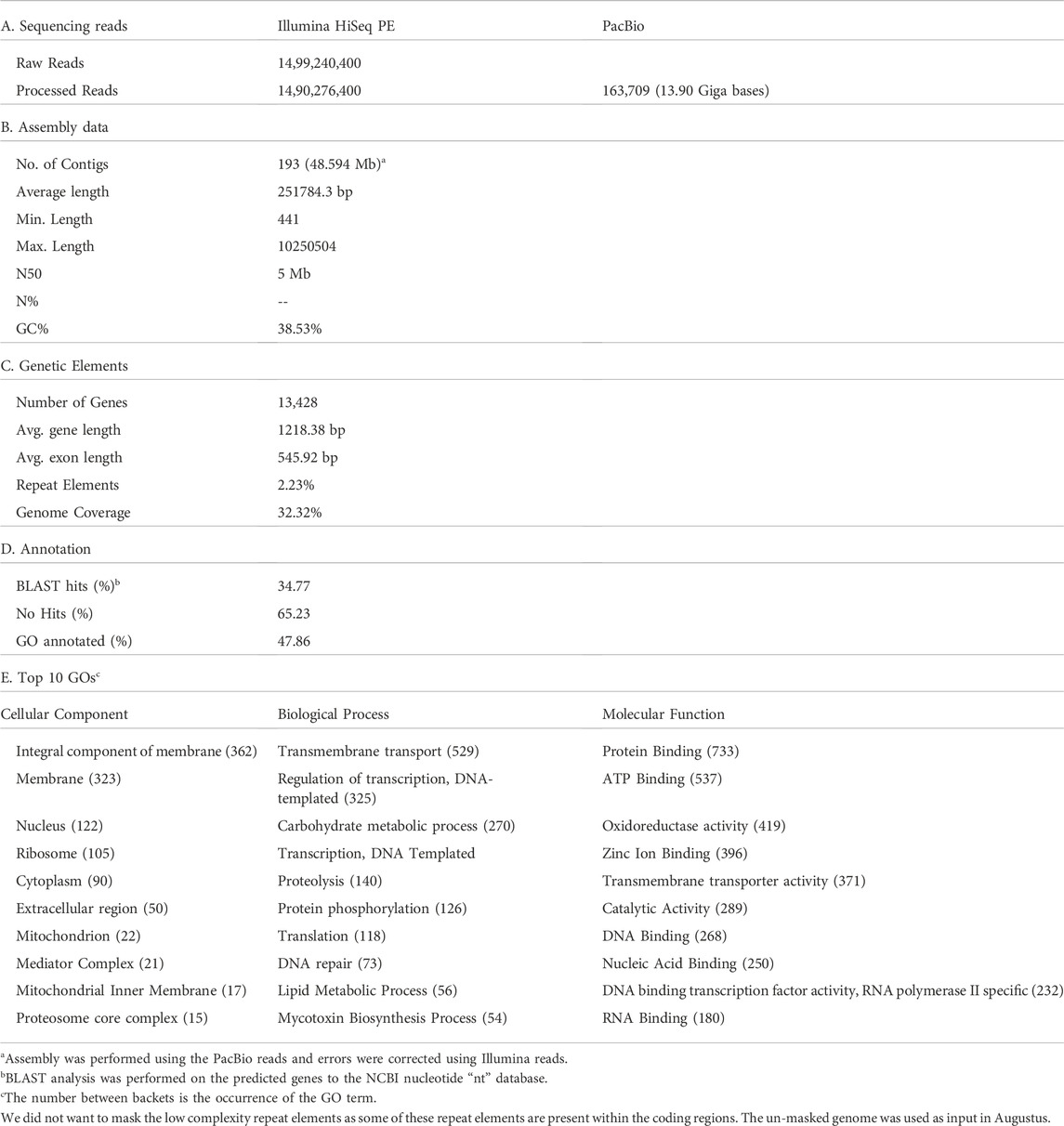
TABLE 1. Sequencing summary statistics of C. aff. paspali genome (H3 isolate).
We submitted ∼48.6 Mbp of the C. aff. paspali genome to RepeatMasker and observed that only 2.23% of the genome was classified as repetitive, indicating a low complexity genome. The majority (1.82%) of the repeat elements were the simple repeats (microsatellites, simple sequence repeats, or short tandem repeats) and a smaller percentage were low-complexity repeats (0.28%). Other repeat elements included LINEs (0.04%), DNA repeats (0.01%), total interspread repeats (0.05%) and small RNA (0.09%). No homology to DNA transposons and retrotransposons was noted in the RepeatMasker analyses. According to our RepeatMasker analysis, C. jacksonii, C. monteithiana, S. sclerotiorum, and R. sydowiana, presented similarly low repetitive genomes with 2.13%, 2.02%, 2.47%, and 1.89% of repeats, respectively. To assess the completion of the genome, we performed a BUSCO analysis. The genome of the C. aff. paspali H3 isolate comprised 99.3% of the complete BUSCOs (97.6% single copy, 1.7% duplicated copy) using the fungal database. In addition, 0% of the gene models were fragmented BUSCOs and 0.7% were missing (Figure 3). In comparison to other Clarireedia species, C. aff. paspali showed the highest number of Complete BUSCOs and similar statistics were also observed with the EUKARYOTIC BUSCO dataset (Figure 3).
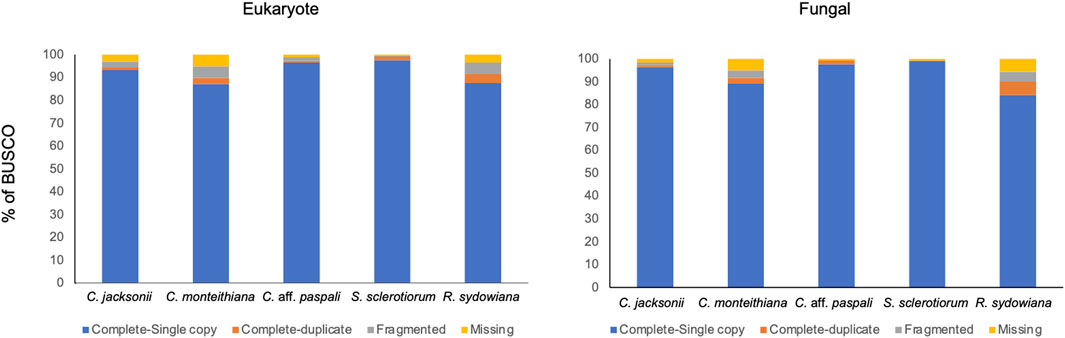
FIGURE 3. Benchmarking Universal Single Copy Orthologs (BUSCO) analysis of gene models using the eukaryote obd10 (n = 255; species = 70) and the fungal obd10 (n = 758; species = 549) BUSCOs. Gene models generated in this paper from the Augustus pipeline were used for the C. aff. paspali (H3 isolate). Published gene models were used for C. jacksonii (LWC-10), C. monteithiana (DRR09), and R. sydowiana (CBS115975), and S. sclerotiorum (1980 UF-70).
Using the trained de novo prediction from AUGUSTUS, we predicted 13,428 gene models (18.406 MB), with an average gene length of 1,360.7 bp, which constitutes 37.88% the C. aff. paspali genome (Table 1). We also identified that 34.77% (4,669 genes out of 13,428) of the discovered annotated gene models contain BLAST hits to the NCBI “nt” database. From the Interpro scan analysis, we assigned GO categories to 6,247 genes (47.86% of the total genes). Table 1 provides the top 10 GO terms in each of the categories of GO. The top GO term in cellular component, biological process and molecular function are Integral component of membrane (n = 362), Transmembrane transport (n = 529) and Protein Binding (n = 733) respectively.
A total of 11,340 ortho-clusters were identified in the analysis of the three Clarireedia species and S. sclerotiorum (Figure 4). The ortholog analysis revealed 10,580 orthologous clusters containing at least two Clarireedia species, which constitute 93.3% of the total orthologous clusters. In addition, 322 unique Clarireedia species-specific orthologous groups and 1,862 orthologous groups common to all the studied Clarireedia species, were identified. Interestingly, there were more orthogroups common between C. aff. paspali and C. monteithiana (n = 391) than C. aff. paspali and C. jacksonii (n = 160). Interestingly, at a pairwise level, C. jacksonii and C. monteithiana (n = 432) had the highest orthogroups in common. Within Clarireedia, C. monteithiana had the highest number of species-specific orthologous groups (270 orthologous groups), followed by C. aff. paspali (42) and C. jacksonii (10) (Figure 4).
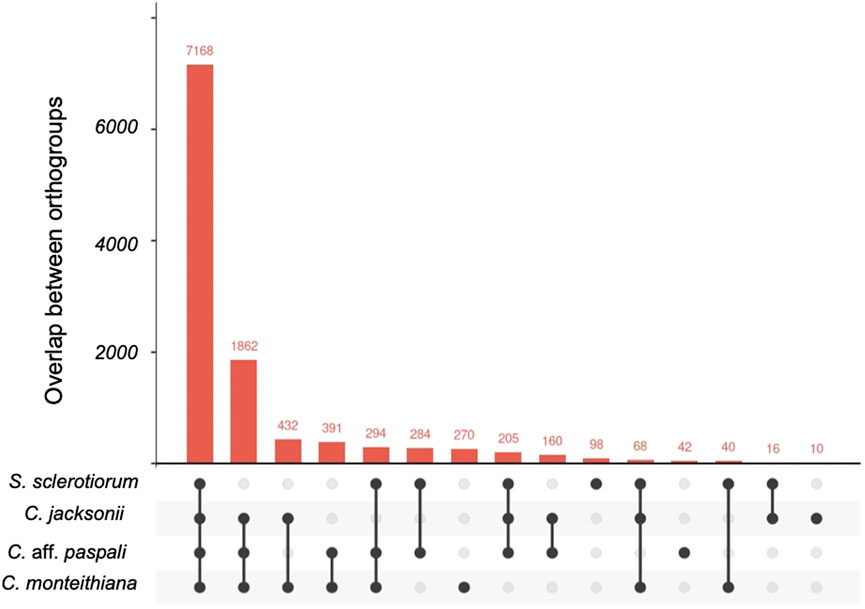
FIGURE 4. Upset plot of orthologous gene clusters between C. aff. paspali and other related species. One to several genes can be present within each ortho gene cluster. S. sclerotiorum, C. jacksonii, C. monteithiana, and C. aff. paspali are represented by the genome of isolates 1980 UF-70, LWC-10, DRR09, and H3, respectively.
From the FastANI analysis of genome similarity, we observed, as expected, more similarity within Clarireedia species (>98%) than between Clarireedia species (≤96%) (Figure 5A). 99.51% of sequence similarity within C. aff. paspali (between H2 and H3 isolates) was observed. Within species analysis showed the lowest genome similarity between the two isolates of C. monteihiana DRR09 and RB-19. On average, C. aff. paspali (H2 and H3) showed more genome similarity with C. monteihiana (96.82%) isolates (DRR09 and RB-19) than with C. jacksonii (96.51%) isolates (LWC-10 and HRI11). The genome similarity values of S. sclerotiorum and R. sydowiana with the Clarireedia species could not be identified as the similarity dropped below 80% and the program could not assign a value.
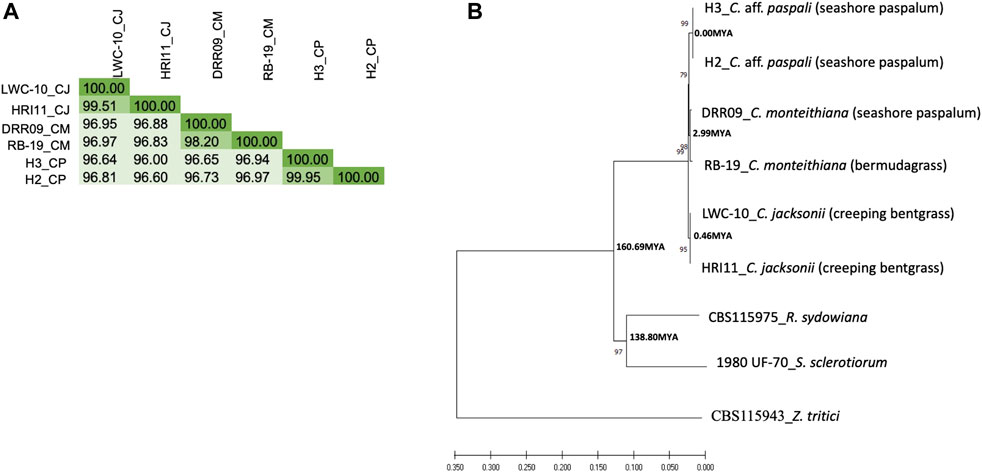
FIGURE 5. Genetic relationship of C. aff. paspali with related species. (A) DNA similarity matrix between the Clarireedia species. C. jacksonii (CJ), C. monteithiana (CM) and C. aff. paspali (CP) were represented by LWC-10 and HRI11, DRR09 and RB-19, H3 and H2 isolates, respectively. (B) Neighbor-joining tree of Clarireedia spp. and related species conducted from combined virulence and conserved gene sequences. Numbers associated with branches are bootstrap percentage. Two isolates for each Clarireedia species were used. S. sclerotiorum (1980 UF-70) and R. sydowiana (CBS115975) were used as representatives of the Sclerotiniaceae family and the Rutstroemiaceae family, respectively. Z. tritici (CBS115943) was used as an outgroup. Numbers associated with nodes are estimated divergence times. When known, the host origin of the isolate was indicated between brackets.
Out of the 13,428 protein models, we identified 357 cytoplasmic effectors and 13 apoplastic effectors (with p-values > 0.9) which in combine constitute 2.68% of the total identified protein models. 349 cytoplasmic effectors and 13 apoplastic effectors had BLAST hits to the NCBI “nr” protein database (Supplementary Table S3). Sixteen and 136 cytoplasmic effectors blasted against Sclerotinia spp. and Rutstroemia spp., respectively. In addition, five apoplastic effectors blasted against Rutstroemia spp., while none blasted against Sclerotinia spp. Three hundred and forty-nine cytoplasmic effectors and 13 apoplastic effectors were identified in 33 contigs of the C. aff. paspali genome; two major contigs (tig00000001 and tig00002064) grouped 36% of the effectors. The average densities were four cytoplasmic effectors every 100 kb and nine apoplastic effectors every 10 Mb. Three contigs (tig00000377, tig00000363, and tig00000354) harbored surprisingly a higher effector density (three cytoplasmic effectors every 10 kb) (Supplementary Table S3).
We screened the C. aff. paspali genome and related species for the fifty-four biological determinants compiled by Xia et al. (2019) involved in S. sclerotiorum morphogenesis and pathogenesis. Only seventeen genes (∼31% of the biological determinants) were present in at least 1 Clarireedia species (Table 2; Supplementary Table S1). Out of these seventeen pathogenesis genes, four were predicted by EffectorP as cytoplasmic effectors (caf1, pka1, nacα, and gpd genes) with p-values of 0.7-0-8 (Supplementary Table S1). Seven genes (cna1, caf1, nox1, pac1, pka1, ams2, and nacα) were present in all the Clarireedia species but absent in the sister genus R. sydowiana; however, three genes (pth2, bi1, and hex1) are absent in all the Clarireedia species but present in R. sydowiana. Four genes showed presence/absence variation between the Clarireedia species. rgb1 and rhs1, genes involved in virulence, appressoria formation and sclerotial formation, were exclusively present in both isolates of C. monteithiana and C. aff. paspali, respectively. Adenylate cyclase (sac1), a gene involved in virulence, hyphal growth and sclerotial formation was absent in both isolates of C. aff. paspali. Another gene, pks13, a polyketide synthase, involved in appresoria formation, was absent in both C. aff. paspali isolates and in 1 C. monteithiana isolate (RB-19 isolate sampled from bermudagrass). SNP variations among the genes were assessed in the thirteen virulence genes present across the Clarireedia species. A total of 366 species-specific SNPs were recorded. C. jacksonii (151 SNPs) and C. monteithiana (78 SNPs) presented the highest and lowest number of species-specific SNPs, respectively. The gene ste12, coding for downstream transcription factor of MAPK pathway, contained the highest number of species-specific SNPs for C. aff. paspali (25 SNPs) and for C. jacksonii (26 SNPs). However, the highest number of species-specific SNPs (11 SNPs) for C. monteithiana was observed in ams2 gene (GATA transcription factor; 11 SNPs) and caf1 gene (compound appressorium formation related gene 1; 10 SNPs). Of the 366 species-specific SNPs, only 28 SNPs were non-synonymous and non-conservative (7.6%). Overall, the percentage of protein similarity of S. sclerotiorum with Clarireedia species ranged from 58.39% (CNA1 protein) to 96.52% (AMS2 protein) with an average of 86.07%. NOX1 and NOX2 showed no polymorphism between Clarireedia species at the protein level and a high percentage of protein identity with S. sclerotiorum (91.76%–93.58%).
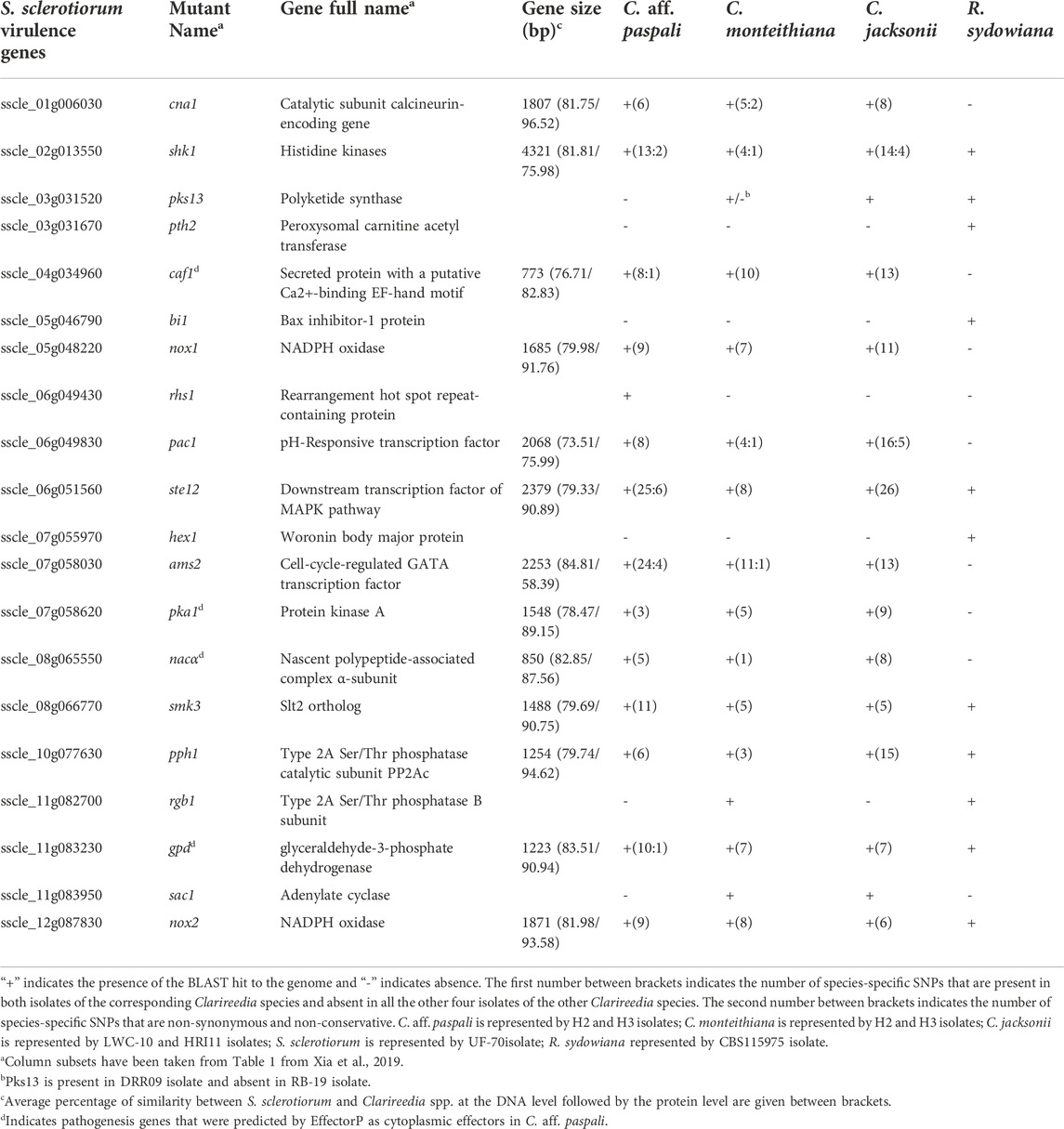
TABLE 2. Summary of S. sclerotiorum orthologs involved in pathogen morphogenesis and pathogenesis identified in C. aff. paspali and their genetic variation between Clarireedia species.
Protein modelling of the thirteen virulence genes of S. sclerotiorum was performed, and the models which were predicted with high coverage (more than 80%) were selected to assess the alignment of the respective proteins from Clarireedia spp. (Supplementary Table S4). Six models (GPD, NOX1, NOX2, PKA1, PPH1, and SMK3) from S. sclerotiorum was superimposed with the models from C. jacksonii, C. monteithiana, and C. aff. paspali, and different color codes were assigned to distinguish the shared and distinct regions among the proteins (Figure 6, Supplementary Figure S1; Supplementary Video S1). Overall, the comparative modeling of the six virulence genes between S. sclerotiorum and Clarireedia spp. suggested that the majority of the structural attributes were common to all isolates of Clarireedia spp. and S. sclerotiorum analyzed with only minor differences occurring at the structural level. The presence of similar sequences and identical structures is a strong indication of the regions being highly conserved amongst the two organisms, while the occurrence of distinct regions is suggestive of functional divergence among the proteins during molecular evolution (Centeno et al., 2005).

FIGURE 6. Three-dimensional protein models of six virulence genes [(A) gpd, (B) nox1, (C) nox2, (D) pka1, (E) pph1, and (F) smk3] from S. sclerotiorum aligned with the respective protein structures from C. jacksonii, C. monteithiana, and C. aff. paspali. The common structures between all species are in green. The species-specific structures of S. sclerotiorum that are distinct from all Clarireedia (C. jacksonii, C. monteithiana and C. aff. paspali) are highlighted in yellow. The single region in GPD (A) protein differentiating C. aff. paspli from C. jacksonii and C. monteithiana is color-coded in pink.
The quality assessment of the models predicted using ERRAT and VERIFY-3D programs yielded scores between 50.6667%–97.4299% and 59.27%–98.02%, respectively (Supplementary Table S4). Additionally, the PROCHECK server provided Ramachandran plot analysis results, wherein 70.8%–89.3% residues were found to be in the most favorable region, 10.4%–22.1% residues in allowed region and a few resides up to 1.7% were present in the disallowed region (Supplementary Table S4). The QMEAN Z-scores of the models ranged between −8.79 and −1.27. These model evaluation values suggest a relatively good quality of the predicted structures (Lüthy et al., 1992; Benkert et al., 2011; Messaoudi et al., 2013; Wlodawer 2017).
The phylogenetic analysis on the concatenated six pathogenicity and five conserved gene sequences (Supplementary Table S5) revealed that C. aff. paspali was more genetically related to C. monteihiana than to C. jacksonii. The NJ tree also pointed out that Clarireedia species were as divergent from R. sydowiana (CBS115975 isolate), the representative of the Rutstroemiaceae family, as from S. sclerotiorum (1980 UF-70 isolate), the representative of the Sclerotiniaceae family (Figure 5B). Using the divergence time of 437 Mya between S. sclerotiorum and Zymoseptoria tritici as reference (www.treetime.org), we estimated that isolates within Clarireedia species diverged from one another approximately 0.00, 2.99, and 0.46 Mya for C. aff. paspali, C. montheihiana, and C. jacksonii, respectively. In addition, Clarireedia species diverged from S. sclerotiorum approximately 160 Mya (Figure 5B). The molecular clock test showed that the null hypothesis of equal evolutionary rate throughout the NJ tree was rejected at a 5% significance level (p = 0.000E+000). The evolutionary rate between C. aff. paspali, C. monteithiana (p = 0.36917) and C. jacksonii (p = 0.09691) sequences were statistically equal. However, the evolutionary rates of R. sydowiana (p = 0.00509) and S. sclerotiorum (p = 0.00059) were significantly higher than in C. aff. paspali.
In this study, whole genome sequencing was performed for two isolates of C. aff. paspali. H3 isolate was sequenced by coupling Illumina paired-end with PacBio approaches, while H2 isolate was sequenced with HiSeq sequencing method only. The estimated genome size of C. aff. paspali was 48.6 Mb. As this is the first draft genome for C. aff. paspali, we relied on the assembly statistics of other published Clarireedia genomes for comparison. Similar genome lengths (36–48 Mb) were observed for C. monteithiana and C. jacksonii (Crouch et al., 2021). The genome of C. aff. paspali H3 and H2 isolates contained a total of 193 contigs and 5 Mb contig N50 and 6,743 contigs and ∼77 kb contig N50, respectively. In comparison, the de novo assemblies previously published for C. jacksonii produced 15.63–709.08 kb scaffold N50, and 231–31,623 contigs with PacBio and/or HiSeq sequencing methods (Green et al., 2016; Crouch et al., 2021). Clarireedia monteithiana available genome assemblies produced 8.76–20.76 kb scaffold N50, and 15,133–3,610 contigs with Illumina HiSeq sequencing data (Crouch et al., 2021). In this study, the de novo assembly for C. aff. paspali H2 isolate produced a better assembly than any other published Clarireedia assembly sequenced with just the short reads. The bioinformatic tools used for downstream analysis have different performance and could play an important role in the quality of the final assembly (Bankevich et al., 2012). In addition, by combining Illumina HiSeq short-read and PacBio long-read sequences, the hybrid assembly approach used in this study for C. aff. paspali H3 isolate was considered effective in reducing the number of contigs and increasing the contig N50 length of the Clarireedia genome. Short reads provided the accuracy in the bp read and long reads, which were more error prone, gave the required read length for a better assembly. The K-mer and RepeatMasker analysis revealed that the C. aff. paspali genome has low complexity (only 2.09% of repetitive regions), similarly to other Clarireedia species. In addition, AUGUSTUS predicted 12,894 gene models, representing 32.32% the C. aff. paspali genome. This number is similar to the range of the gene models present in other Clarireedia species (range between 10,821–11,089 genes (Crouch et al., 2021). Furthermore, the completeness of assembly and annotation were evaluated by the BUSCO analysis, and an annotation completeness of 81.9% was reached. Although this study presents the most complete assembly currently available for Clarireedia, further studies are needed to improve the genome assemblies and provide chromosome-level reference genomes. In this draft genome, more than 91.5% of the assembly is composed in 25 contigs (>250 kb per contig). Deciphering genome continuity and assembling the large PacBio contigs into chromosomes require additional techniques such as Hi-C, which was successfully applied in genome scaffolding for a wide range of eukaryotic species (Kaplan and Dekker 2013; Wang et al., 2020). Scaffolding using Hi-C data could provide full chromosome-level assemblies for each Clarireedia species. However, long-reads are still needed for C. monteithiana. Long-reads PacBio data sequenced on Sequel SMRT-cell (as shown in this study) or nanopore data sequenced on the MinION platform (Oxford Nanopore Technologies) would allow updates to the current C. monteithiana genomes. Chromosome-level genomes would reveal aspects of genome evolution and would open-up a more detailed view of genome plasticity within Clarireedia species. Karyotype studies to visually validate the chromosome number are needed in all the Clarireedia species. Flow cytometry, recommended to pre-calibrate genome assembly pipelines, would allow a more accurate genome assembly (Kooij and Pellicer 2020). C. aff. paspali presented on average, the largest genome (43.9 Mb; from this study) compared to C. monteithiana (42.4 Mb; Sapkota et al., 2021) and C. jacksonii (37.5 Mb; Sapkota et al., 2021). Difference in the genome size among Clarireedia spp. could correspond to a difference in the chromosome number. Large structural rearrangements leading to aneuploidy were observed in several plant pathogenic fungi including the haploid hemibiotrophic plant fungus Z. tritici. The top 23 contigs identified in this study, representing >90% of the genome (43.8 Mbp), could constitute the chromosome-level (core and accessory chromosomes) representation of C. aff. paspali genome. With a similar genome size, Z. tritici (39.7 Mbp) is represented by a total of 21 chromosomes and genome plasticity was common at the population level within Z. tritici, where eight out the 21 chromosomes constituting the reference genome, were considered as accessory because of their presence/absence variation (Goodwin et al., 2011). Chromosome counting may also provide additional information on genetic compatibility and mating patterns allowing genetic material exchange between Clarireedia species. Although sexual reproduction has not been reported so far in nature in Clarireedia (DeVries et al., 2008), heterothallism (Liberti et al., 2012; Putman et al., 2015) and vegetative compatibility groups were observed (Powell and Vargas, 2001; Viji et al., 2004).
In this study, symptoms observed in seashore paspalum on a sod farm in Hawaii presented typical signs of dollar spot. Prior to this finding, C. jacksonii and C. monteithiana were the two species identified as responsible for dollar spot in turfgrass in the United States (Salgado-Salazar et al., 2018). However, in this study, Sanger sequencing and BLAST results revealed the presence of C. aff. paspali. To date, this species has only been identified on seashore paspalum in China based on multi-locus analysis (ITS, EF, and Mcm7) (Hu et al., 2019). The major hypothesis supported by our study on the emergence of C. aff. paspali in the United States is its introduction from China rather than natural local evolution from pre-existing Clarireedia species. Because Clarireedia spp. do not produce spores, the pathogen dissemination through long-distances is most likely human-caused and attributed to movement of infected plant materials (Walsh et al., 1999; Horvath et al., 2007; Rioux et al., 2014). In addition, because C. aff. paspali H2 and H3 isolates showed similar genome sequences and HK1 sequences were 100% identical on ITS and EF, the introduction of C. aff. paspali to the United States from China must have been recent. Further dollar spot sampling in Hawaii and genomic comparison of C. aff. paspali isolates from United States and China would help us to explain C. aff. paspali emergence in the United States
At the genome level, Clarireedia species are closely related. C. aff. paspali isolates (H2 and H3), C. monteihiana isolates (DRR09 and RB-19) and C. jacksonii isolates (LWC-10 and HRI11) showed high genome similarity (>96%). In addition, similar evolutionary rates in all Clarireedia species were detected in the phylogenetic analysis on 22,983 bp of aligned sequence. A high level of synteny was observed between C. aff. paspali and the published genome of C. jacksonii HRI (data not shown). In addition, 25% of the orthologous clusters identified were shared by at least two Clarireedia species and about 16% of the orthologous clusters were common to all three Clarireedia species. However, phylogenetic analysis on the concatenated six pathogenicity and five conserved gene sequences, as well as the genome comparison between Clarireedia species showed clear differentiation between the three Clarireedia species. 322 orthologous groups were unique to one Clarireedia species. In comparison, C. aff. paspali showed the lowest fragmented and missing BUSCOs when using the fungal BUSCOs. The highest and lowest number of species-specific SNPs in the pathogenicity genes studied as well as the highest and lowest number of species-specific orthologous groups were observed for C. aff. paspali and C. monteithiana, respectively. In addition, three species-specific SNPs for C. aff. paspali have been identified in the ITS region and EF gene. Clarireedia species showed genomic variations at potential pathogenesis factors. In fact, four genes involved in morphogenesis and pathogenesis, Adenylate cyclase (sac1), a Type 2A Ser/Thr phosphatase B subunit (rgb1), a rearrangement hotspot repeat-containing protein (rhs1), and a polyketide synthase (pks13), showed presence/absence variation between Clarireedia species. In addition, our phylogenetic and genome comparison studies also revealed that C. aff. paspali was more closely related to C. monteithiana than to C. jacksonii. There were 2.4 times more orthogroups common between C. aff. paspali and C. monteithiana than between C. aff. paspali and C. jacksonii.
Furthermore, from within species analysis, C. aff. paspali and C. monteithiana showed the lowest and the highest diversity in terms of polymorphic positions, respectively. Phylogenetic analysis point-out the most recent divergence between C. aff. paspali isolates. However, DRR09 and RB-19 showed the least recent divergence (2.99 Myo). At the genome level, H2 and H3 isolates were almost genetically identical (99.51% of sequence similarity) while C. monteihiana isolates DRR09 and RB-19 showed the lowest genome similarity. DRR09 and RB-19 also differed by presence/absence of one gene (pks13) involved in appresoria formation. This pattern could be explained because C. aff. paspali isolates were sampled in the same year, on the same host and at the same location, while C. monteihiana isolates DRR09 and RB-19 were sampled in the Dominican Republic on seashore paspalum and in Mississippi, United States on bermudagrass, respectively.
Although Clarireedia were removed from the Sclerotiniaceae and reclassified as part of the Rutstroemiaceae family (Salgado-Salazar et al., 2018), Clarireedia species were found to be as genetically divergent from R. sydowiana as from S. sclerotiorum. In this study, S. sclerotiorum and R. sydowiana showed several genetic dissimilarities with Clarireedia. The evolutionary rate between C. aff. paspali and R. sydowiana or S. sclerotiorum were significantly different. The DNA similarity between S. sclerotiorum and the Clarireedia species was <80% on the genome level overall and 81.04% on the concatenated six pathogenicity and five conserved gene sequences studied. In addition, only seventeen of the fifty-four pathogenicity genes of S. sclerotiorum were identified in at least one Clarireedia species. However, the six pathogenicity genes compared at the protein level showed superimposition of the predicted models between S. sclerotiorum and Clarireedia spp., as well as among Clarireedia species. Overall, the results supported the differentiation in the virulence patterns and therefore host-range between Clarireedia, apparent hemibiotrophic turfgrass pathogen with its closely related necrotrophic dicto pathogen S. sclerotiorum. Clarireedia species and R. sydowiana differed by presence/absence of ten genes involved in pathogenesis. Three genes (pth2, bi1 and hex1) absent in all the Clarireedia species but present in R. sydowiana may have been selectively lost in the Clarireedia lineage. Seven genes (cna1, caf1, nox1, pac1, pka1 and nacα, and ams2) present in all the Clarireedia species but absent in R. sydowiana have been selected in the Clarireedia lineage and may play an important function in dollar spot development. In addition, more than 56% of the cytoplasmic effectors identified by EffectorP in Clarireedia spp. blasted neither against Rutstroemia spp. nor Sclerotinia spp. However, eight time more Clarireedia cytoplasmic effectors blasted against Rutstroemia spp. than Sclerotinia spp., suggesting a closer similarity of Clarireedia to Rutstroemia spp. than to Sclerotinia spp. in their pathogenic process.
Within the seventeen potential pathogenicity genes identified in Clarireedia species in this study, several similar proteins have already been confirmed in a sequencing-by-synthesis library of creeping bentgrass inoculated with dollar spot (Orshinsky et al., 2012a). In fact, based on RNA-seq analysis, two protein kinase (SHT_6900; SHT_8743:2.87), a glycerol-3-phosphate dehydrogenase (SHT_2740), an hybrid nrps pks (SHT_9252), a polyketide synthase (SHT_2432), an histidine kinase (SHT_9426), a GATA type transcription factor (SHT_2779) and two protein kinase regulator (SHT_7172 (ste50); SHT_4675) were either significantly down or up-regulated (>2 log fold change) during creeping bentgrass infection with dollar spot. In addition, some of these transcripts were enriched in NADPH binding domain, sac1 homology domain and polyketide synthase domain (Orshinsky et al., 2012b).
S. sclerotiorum, a necrotrophic fungal pathogen which can infect more than 400 plant hosts, has been demonstrated to produce oxalic acid and utilize it for successful pathogenesis (Bolton et al., 2006). Typically, for pathogens within the Sclerotinia genus, the host is infected by hyphal colonization of host tissues through stomata or wounded sections of leaves. Hyphae then exude oxalic acid into the intracellular space which lowers the pH and provides an optimal environment for pectolytic enzymes, such as polygalacturonase, to degrade cellular wall components (Heller and Witt-Geiges 2013). Initially, oxalic acid inhibits the oxidative burst within the cell and creates a reducing environment (Williams et al., 2011; Kabbage et al., 2013). After the infection is established, oxalic acid induces the production of reactive oxygen species (ROS), such as hydrogen peroxide and superoxide ions. Both these ROS’s are messengers that initiate cell death (Williams et al., 2011). The possibility that oxalic acid is also involved in pathogenicity of Clarireedia spp. was first raised following its detection in the pathogen grown in pure culture (Venu et al., 2009). Later, other studies showed the increase in oxalic acid content in dollar spot infected turfgrass (Orshinsky et al., 2012a; Rioux et al., 2020). Although oxalic acid was identified as an important pathogenicity factor in species of Sclerotinia, in this study, only two (pac1, nox1) out of the six genes involved in oxalic acid production (odc2, oah, sod1, pth2) in S. sclerotiorum have orthologs in Clarireedia species. Recently, Townsend and colleagues (Townsend et al., 2020) suggested that C. jacksonii could potentially present a different pathogenesis than S. sclerotiorum. In addition, beside nox1, another gene (nox2) belonging to the NOX family of NADPH oxidases has been identified in Clarireedia species. At the genomic level, nox1 and nox2 carried 27 and 23 species-specific SNPs respectively; however NOX1 and NOX2 proteins were not polymorphic between Clarireedia species and presented a high identity with S. sclerotiorum proteins, supporting its important role in the virulence and the wide host range patterns of Clarireedia spp. NOX family enzymes are critical sources of cellular superoxide anions and ROS via electron transfer. These enzymes contribute to a wide range of physiological functions including pathogenicity in several necrotrophic fungi with a wide host range from the Ascomycota division, such as Magnaporthe oryzae (Egan et al., 2007), Botrytis cinerea (Segmüller et al., 2008), Verticillium dahliae (Zhou et al., 2017), Claviceps purpurea (Giesbert et al., 2008), Alternaria alternata (Yang and Chung, 2012) and Sclerotinia sclerotiorum (Kim et al., 2011). NOX family enzymes have also been shown to be involved in sclerotia formation and development of fungal multicellular structures in S. sclerotiorum (Kim et al., 2011). These two nox genes could have distinct cellular functions in the pathogenicity of Clarireedia spp. For example, nox1 was shown to regulate fungal proliferation within plant tissues while nox2 is necessary for the penetration of host cells through the cuticle in M. grisea, V. dahliae, and B. cinerea (Segmüller et al., 2008; Egan et al., 2007; Giesbert et al., 2008; Ryder et al., 2013; Zhou et al., 2017). Typically during infection, mycelium of Clarireedia spp. penetrates the plant directly via the cuticle by forming an appressorium or indirectly through cut leaf tips and natural openings (stomata), grows to invade plant tissues and at a later stage forms fungal stroma in dead tissue to survive from one season to the next (Rioux et al., 2014). In addition, nox1 was shown to regulate differentiation of sexual structures (cleistothecia, perithecia development and ascospore production) in several fungi (Lara-Ortiz et al., 2003; Lalucque and Silar, 2003; Cano-Dominguez et al., 2008; Wang et al., 2014). Although, in the case of Clarireedia spp., no sexual reproduction has been observed in nature so far (DeVries et al., 2008), mating type, genetic diversity and linkage disequilibrium studies pointed-out the potential for occurrence of the sexual cycle in Clarireedia (Hsiang and Mahuku 1999; Liberti et al., 2012; Putman et al., 2015).
Furthermore, three other pathogenesis genes (pka1, smk3 and ste12) involved in mitogen-activated protein kinase (MAPK) cascade have been identified in Clarireedia spp. MAPK are involved in fungal pathogen responses to host and environmental stimuli and several are conserved in plant pathogenic ascomycetes (Jiang et al., 2018). pka, protein kinases, such as pka1 identified here, participate in surface recognition in the early stages of the MAPK cascade during plant infection. smk3 is an ortholog of stl2 in yeast. stl2 is a conserved gene in all plant pathogenic filamentous ascomycetes regulating cell wall integrity and pathogenesis (Turrà et al., 2014). In Sclerotinia sclerotiorum, smk3 was shown to be involved in sclerotia formation and aerial hyphal growth (Bashi et al., 2016). Its ortholog (mps1) in M. grisea is essential for early stages of appressorium development (Li et al., 2012). Thus, because Clarireedia spp. is an appressorium-forming pathogen, smk3 could play an important role in its pathogenesis. In addition, ste12, a downstream transcription factor of MAPK pathway of yeast fus3 and kss1, is also essential for plant infection in pathogenic fungi. With the deletion of ste12, M. oryzae was unable to infect and develop appressorium for plant penetration (Li et al., 2012). Other genes identified here and potentially involved in appressorium formation were pks13, caf1, ams2, rgb1, and rhs1. caf1 presented several species-specific SNPs for each Clarireedia species but only 1 SNP, identified in C. aff. paspali, was non-synonymous and non-conservative. Similarly, four and one non-synonymous and non-conservative SNPs were identified in ams2 for C. aff. paspali and C. monteithiana, respectively. pks13 was absent in C. aff. paspali but present in C. jacksonii. rgb1 and rhs1 were exclusively present in both isolates of C. monteithiana and C. aff. paspali, respectively. Additional studies are needed to evaluate the contribution of these genes and their SNP to the variation in gene function and virulence between Clarireedia spp.
In addition, gpd, a glyceraldehyde-3-phosphate dehydrogenase (gpd), was also identified in all three Clarireedia species. GAPDH is a key enzyme in the glycolytic pathway that plays a significant role in pathogenesis of filamentous fungi (Barbosa et al., 2006; Egea et al., 2007). gpd transforms the glyceraldehyde 3-phosphate to glycerate-1, 3-biphosphate, generating the production of NADPH. gpd is generally present as a single copy (Liao et al., 2008), is highly conserved in filamentous fungi and is typically used for species identification (Ghuffar et al., 2018; Huang et al., 2018; Torres-Calzada et al., 2018). In this study, C. aff. paspali, C. monteithiana and C. jacksonii presented ten, seven and seven species-specific SNPs within the gdp gene that could be used for species identification, respectively.
In summary, we report the first draft genome of C. aff. paspali. This genome presents the most complete annotation among Clarireedia species so far and should be considered as the reference for future genomic and proteomic studies. This is also the first report of C. aff. paspali in the United States Several genes potentially involved in Clarireedia morphogenesis and pathogenesis were highlighted from this genomic analysis. Phylogenetic analysis revealed genetic differentiation among Clarireedia species. Because of the limited genomic resources published for members of the Rutstroemiaceae family and for Clarireedia species more specifically, our study provides valuable genomic information for future research discoveries on plant pathogens.
The two C. aff. paspali isolates, H2 and H3, were deposited in GenBank under the BioSample SAMN22087743 (H2) and SAMN22083495 (H3). In addition, the ITS, TUB, CaM and EF sequences of the two C. aff. paspali isolates were deposited in GenBank: MZ578438, MZ620645, MZ620646, and MZ620647 for H2 isolate, respectively; MZ578439, MZ620643, MZ620644, and MZ620642 for H3 isolate, respectively. Raw reads of this Whole Genome Shotgun sequencing project were deposited at GenBank as Sequence Read Archive (SRA) under the BioProject PRJNA769080. Free access to the raw reads and the assembled genomes and annotation is also available through https://bahrilab.caes.uga.edu (under Genomic data tab).
BB designed and conducted the study. PR sampled the isolates. RP performed the genome assembly. RP and BB performed genomic data analysis. SS and WS isolated the dollar spot sample and performed the molecular identification with Sanger sequencing and pathogenicity tests. HS performed the protein structure prediction and visualization. BB, RP, WS, HS, SS, PR and AM-E wrote and edited the manuscript.
This research was supported by the State of Georgia funding, by the USDA National Institute of Food and Agriculture Hatch project 1023739 and by the Georgia Department of Agriculture_2020 Specialty Crop Block Grant program (FP00020738).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1033437/full#supplementary-material
SUPPLEMENTARY VIDEO S1 | Movie animations of the predicted protein structures of six virulence genes from S. sclerotiorum with Clarireedia spp. showing the structural similarities and differences at the protein level.
Aynardi, B. A., Jiménez-Gasco, M. M., and Uddin, W. (2019). Effects of isolates of Clarireedia jacksonii and Clarireedia monteithiana on severity of dollar spot in turfgrasses by host type. Eur. J. Plant Pathol. 155, 817–829. doi:10.1007/s10658-019-01813-z
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi:10.1089/cmb.2012.0021
Barbosa, M. S., Báo, S. N., Andreotti, P. F., Faria, F. P. de, Felipe, M. S. S., Feitosa, L., et al. (2006). Glyceraldehyde-3-Phosphate dehydrogenase of paracoccidioides brasiliensis is a cell surface protein involved in fungal adhesion to extracellular matrix proteins and interaction with cells. Infect. Immun. 74 (1), 382–389. doi:10.1128/IAI.74.1.382-389.2006
Bashi, Z. D., Gyawali, S., Bekkaoui, D., Coutu, C., Lee, L., Poon, J., et al. (2016). The Sclerotinia sclerotiorum Slt2 mitogen-activated protein kinase ortholog, SMK3, is required for infection initiation but not lesion expansion. Can. J. Microbiol. 62, 836–850. doi:10.1139/cjm-2016-0091
Benkert, P., Biasini, M., and Schwede, T. (2011). Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27, 343–350. doi:10.1093/bioinformatics/btq662
Bennett, F. T. (1937). Dollar spot disease of turf and its causal organism Sclerotinia homoeocarpa n. sp. Ann. Appl. Biol. 24, 236–257. doi:10.1111/j.1744-7348.1937.tb05032.x
Blum, M., Chang, H.-Y., Chuguransky, S., Grego, T., Kandasaamy, S., Mitchell, A., et al. (2021). The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 49, D344–D354. doi:10.1093/nar/gkaa977
Bolton, M. D., Thomma, B. P. H. J., and Nelson, B. D. (2006). Sclerotinia sclerotiorum (lib.) de Bary: Biology and molecular traits of a cosmopolitan pathogen. Mol. Plant Pathol. 7, 1–16. doi:10.1111/j.1364-3703.2005.00316.x
Bowie, J. U., Lüthy, R., and Eisenberg, D. (1991). A method to identify protein sequences that fold into a known three-dimensional structure. Science 253, 164–170. doi:10.1126/science.1853201
Cano-Domínguez, N., Álvarez-Delfín, K., Hansberg, W., and Aguirre, J. (2008). NADPH oxidases NOX-1 and NOX-2 require the regulatory subunit NOR-1 to control cell differentiation and growth in neurospora crassa. Eukaryot. Cell 7 (8), 1352–1361. doi:10.1128/EC.00137-08
Carbone, I., and Kohn, L. M. (1999). A method for designing primer sets for speciation studies in filamentous ascomycetes. Mycologia 91, 553–556. doi:10.1080/00275514.1999.12061051
Centeno, N. B., Planas-Iglesias, J., and Oliva, B. (2005). Comparative modelling of protein structure and its impact on microbial cell factories. Microb. Cell Fact. 4, 20. doi:10.1186/1475-2859-4-20
Chakraborty, N., Chang, T., Casler, M. D., and Jung, G. (2006). Response of bentgrass cultivars to Sclerotinia homoeocarpa isolates representing 10 vegetative compatibility groups. Crop Sci. 46, 1237–1244. doi:10.2135/cropsci2005.04-0031
Chalupowicz, L., Dombrovsky, A., Gaba, V., Luria, N., Reuven, M., Beerman, A., et al. (2019). Diagnosis of plant diseases using the Nanopore sequencing platform. Plant Pathol. 68, 229–238. doi:10.1111/ppa.12957
Colovos, C., and Yeates, T. O. (1993). Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 2, 1511–1519. doi:10.1002/pro.5560020916
Crouch, J. A., Beirn, L. A., Boehm, M. J., Carbone, I., Clarke, B. B., Kerns, J. P., et al. (2021). Genome resources for seven fungal isolates that cause dollar spot disease in turfgrass, including Clarireedia jacksonii and C. monteithiana. Plant Dis. 105, 691–694. doi:10.1094/PDIS-06-20-1296-A
De Miccolis Angelini, R. M., Romanazzi, G., Pollastro, S., Rotolo, C., Faretra, F., and Landi, L. (2019). New high-quality draft genome of the Brown rot fungal pathogen Monilinia fructicola. Genome Biol. Evol. 11, 2850–2855. doi:10.1093/gbe/evz207
DeVries, R. E., Trigiano, R. N., Windham, M. T., Windham, A. S., Sorochan, J. C., Rinehart, T. A., et al. (2008). Genetic analysis of fungicide-resistant Sclerotinia homoeocarpa isolates from Tennessee and Northern Mississippi. Plant Dis. 92, 83–90. doi:10.1094/PDIS-92-1-0083
Doyle, J. J., and Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Edgar, R. C. (2004). Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi:10.1093/nar/gkh340
Egan, M. J., Wang, Z.-Y., Jones, M. A., Smirnoff, N., and Talbot, N. J. (2007). Generation of reactive oxygen species by fungal NADPH oxidases is required for rice blast disease. Proc. Natl. Acad. Sci. 104 (28), 11772–11777. doi:10.1073/pnas.0700574104
Egea, L., Aguilera, L., Giménez, R., Sorolla, M. A., Aguilar, J., Badía, J., et al. (2007). Role of secreted glyceraldehyde-3-phosphate dehydrogenase in the infection mechanism of enterohemorrhagic and enteropathogenic Escherichia coli: Interaction of the extracellular enzyme with human plasminogen and fibrinogen. Int. J. Biochem. Cell Biol. 39 (6), 1190–1203. doi:10.1016/j.biocel.2007.03.008
Emms, D. M., and Kelly, S. (2019). OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238. doi:10.1186/s13059-019-1832-y
Ghuffar, S., Irshad, G., Naz, F., Rosli, H. B., Hyder, S., Mehmood, N., et al. (2018). First report of two penicillium spp. causing postharvest fruit rot of grapes in Pakistan. Plant Dis. 102 (5), 1037. doi:10.1094/PDIS-10-17-1616-PDN
Giesbert, S., Schürg, T., Scheele, S., and Tudzynski, P. (2008). The NADPH oxidase Cpnox1 is required for full pathogenicity of the ergot fungus Claviceps purpurea. Mol. Plant Pathol. 9 (3), 317–327. doi:10.1111/j.1364-3703.2008.00466.x
Glass, N. L., and Donaldson, G. C. (1995). Development of primer sets designed for use with the PCR to amplify conserved genes from filamentous ascomycetes. Appl. Environ. Microbiol. 61, 1323–1330. doi:10.1128/aem.61.4.1323-1330.1995
Goodwin, S. B., M’barek, S. B., Dhillon, B., Wittenberg, A. H. J., Crane, C. F., Hane, J. K., et al. (2011). Finished genome of the fungal wheat pathogen Mycosphaerella graminicola reveals dispensome structure, chromosome plasticity, and stealth pathogenesis. PLoS Genet. 7, e1002070. doi:10.1371/journal.pgen.1002070
Green, R., Sang, H., Chang, T., Allan-Perkins, E., Petit, E., and Jung, G. (2016). Draft genome sequences of the turfgrass pathogen Sclerotinia homoeocarpa. Genome Announc. 4, e01715–15. doi:10.1128/genomeA.01715-15
Groben, G., Clarke, B. B., Murphy, J., Koch, P., Crouch, J. A., Lee, S., et al. (2020). Real-time PCR detection of clarireedia spp., the causal agents of dollar spot in turfgrasses. Plant Dis. 104 (12), 3118–3123. doi:10.1094/PDIS-04-20-0726-RE
Haydu, J. J., Hodges, A. W., and Hall, C. R. (2006). Economic impacts of the turfgrass and lawncare industry in the United States. Fla. Coop. Ext. Serv. FE 632. Gainesville: Univ. Fl. Available at: https://edis.ifas.ufl.edu/pdf/FE/FE63200.pdf.
Held, D. W., and Potter, D. A. (2012). Prospects for managing turfgrass pests with reduced chemical inputs. Annu. Rev. Entomol. 57, 329–354. doi:10.1146/annurev-ento-120710-100542
Heller, A., and Witt-Geiges, T. (2013). Oxalic acid has an additional, detoxifying function in Sclerotinia sclerotiorum pathogenesis. PLoS One 8, e72292. doi:10.1371/journal.pone.0072292
Hong, S.-B., Go, S.-J., Shin, H.-D., Frisvad, J. C., and Samson, R. A. (2005). Polyphasic taxonomy of Aspergillus fumigatus and related species. Mycologia 97, 1316–1329. doi:10.3852/mycologia.97.6.1316
Horvath, B. J., Kravchenko, A. N., Robertson, G. P., and Vargas, J. M. (2007). Geostatistical analysis of dollar spot epidemics occurring on a mixed sward of creeping bentgrass and annual bluegrass. Crop Sci. 47 (3), 1206–1216. doi:10.2135/cropsci2006.09.0565
Hsiang, T., and Mahuku, G. S. (1999). Genetic variation within and between southern Ontario populations of Sclerotinia homoeocarpa. Plant Pathol. 48, 83–94. doi:10.1046/j.1365-3059.1999.00306.x
Hu, J., Zhou, Y., Geng, J., Dai, Y., Ren, H., and Lamour, K. (2019). A new dollar spot disease of turfgrass caused by Clarireedia paspali. Mycol. Prog. 18, 1423–1435. doi:10.1007/s11557-019-01526-x
Huang, L., Zhu, Y.-N., Yang, J.-Y., Li, D.-W., Li, Y., Bian, L.-M., et al. (2018). Shoot blight on Chinese fir (Cunninghamia lanceolata) is caused by bipolaris oryzae. Plant Dis. 102 (3), 500–506. doi:10.1094/PDIS-07-17-1032-RE
Hubbard, A., Lewis, C. M., Yoshida, K., Ramirez-Gonzalez, R. H., de Vallavieille-Pope, C., Thomas, J., et al. (2015). Field pathogenomics reveals the emergence of a diverse wheat yellow rust population. Genome Biol. 16, 23. doi:10.1186/s13059-015-0590-8
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 5114. doi:10.1038/s41467-018-07641-9
Jiang, C., Zhang, X., Liu, H., and Xu, J.-R. (2018). Mitogen-activated protein kinase signaling in plant pathogenic fungi. PLoS Pathog. 14, e1006875. doi:10.1371/journal.ppat.1006875
Kabbage, M., Williams, B., and Dickman, M. B. (2013). Cell death control: The interplay of apoptosis and autophagy in the pathogenicity of Sclerotinia sclerotiorum. PLoS Pathog. 9, e1003287. doi:10.1371/journal.ppat.1003287
Kaplan, N., and Dekker, J. (2013). High-throughput genome scaffolding from in vivo DNA interaction frequency. Nat. Biotechnol. 31, 1143–1147. doi:10.1038/nbt.2768
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. E. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi:10.1038/nprot.2015.053
Kessler, D., Sang, H., Bousquet, A., Hulvey, J. P., Garcia, D., Rhee, S., et al. (2018). Nucleic adaptability of heterokaryons to fungicides in a multinucleate fungus, Sclerotinia homoeocarpa. Fungal Genet. Biol. 115, 64–77. doi:10.1016/j.fgb.2018.01.005
Kim, H., Chen, C., Kabbage, M., and Dickman, M. B. (2011). Identification and characterization of Sclerotinia sclerotiorum NADPH oxidases. Appl. Environ. Microbiol. 77 (21), 7721–7729. doi:10.1128/AEM.05472-11
Kooij, P. W., and Pellicer, J. (2020). Genome size versus genome assemblies: Are the genomes truly expanded in polyploid fungal symbionts? Genome Biol. Evol. 12, 2384–2390. doi:10.1093/gbe/evaa217
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi:10.1101/gr.215087.116
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). Mega X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi:10.1093/molbev/msy096
Lalucque, H., and Silar, P. (2003). NADPH oxidase: An enzyme for multicellularity? Trends Microbiol. 11 (1), 9–12. doi:10.1016/s0966-842x(02)00007-0
Lara-Ortíz, T., Riveros-Rosas, H., and Aguirre, J. (2003). Reactive oxygen species generated by microbial NADPH oxidase NoxA regulate sexual development in Aspergillus nidulans. Mol. Microbiol. 50 (4), 1241–1255. doi:10.1046/j.1365-2958.2003.03800.x
Laskowski, R. A., Rullmannn, J. A., MacArthur, M. W., Kaptein, R., and Thornton, J. M. (1996). AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486. doi:10.1007/BF00228148
Li, G., Zhou, X., and Xu, J.-R. (2012). Genetic control of infection-related development in Magnaporthe oryzae. Curr. Opin. Microbiol. 15, 678–684. doi:10.1016/j.mib.2012.09.004
Li, J., Yu, L., Yang, J., Dong, L., Tian, B., Yu, Z., et al. (2010). New insights into the evolution of subtilisin-like serine protease genes in Pezizomycotina. BMC Evol. Biol. 10, 68. doi:10.1186/1471-2148-10-68
Liao, X., Fang, W., Zhang, Y., Fan, Y., Wu, X., Zhou, Q., et al. (2008). Characterization of a highly active promoter, PBbgpd, in beauveria bassiana. Curr. Microbiol. 57 (2), 121–126. doi:10.1007/s00284-008-9163-3
Liberti, D., Rollins, J. A., and Harmon, P. F. (2012). Evidence for morphological, vegetative, genetic, and mating-type diversity in Sclerotinia homoeocarpa. Phytopathology 102, 506–518. doi:10.1094/PHYTO-06-11-0180
Lüthy, R., Bowie, J. U., and Eisenberg, D. (1992). Assessment of protein models with three-dimensional profiles. Nature 356, 83–85. doi:10.1038/356083a0
Malapi-Wight, M., Salgado-Salazar, C., Demers, J. E., Clement, D. L., Rane, K. K., and Crouch, J. A. (2016). Sarcococca Blight: Use of whole-genome sequencing for fungal plant disease Diagnosis. Plant Dis. 100, 1093–1100. doi:10.1094/PDIS-10-15-1159-RE
Marçais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi:10.1093/bioinformatics/btr011
Messaoudi, A., Belguith, H., and Ben Hamida, J. (2013). Homology modeling and virtual screening approaches to identify potent inhibitors of VEB-1 β-lactamase. Theor. Biol. Med. Model. 10, 22. doi:10.1186/1742-4682-10-22
Orshinsky, A. M., Boehm, M. J., and Boland, G. J. (2012a). Plant wounding and Ophiostoma mitovirus 3a (OMV3a) influence infection of creeping bentgrass by Sclerotinia homoeocarpa. Can. J. Plant Pathology 34, 493–506. doi:10.1080/07060661.2012.678886
Orshinsky, A. M., Hu, J., Opiyo, S. O., Reddyvari-Channarayappa, V., Mitchell, T. K., and Boehm, M. J. (2012b). RNA-Seq analysis of the Sclerotinia homoeocarpa-creeping bentgrass pathosystem. PLoS One 7, e41150. doi:10.1371/journal.pone.0041150
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi:10.1002/jcc.20084
Porto, B. N., Caixeta, E. T., Mathioni, S. M., Vidigal, P. M. P., Zambolim, L., Zambolim, E. M., et al. (2019). Genome sequencing and transcript analysis of Hemileia vastatrix reveal expression dynamics of candidate effectors dependent on host compatibility. PLoS One 14, e0215598. doi:10.1371/journal.pone.0215598
Powell, J. F., and Vargas, J. M. (2001). Vegetative compatibility and seasonal variation among isolates of Sclerotinia homoeocarpa. Plant Dis. 85, 377–381. doi:10.1094/PDIS.2001.85.4.377
Putman, A. I., Tredway, L. P., and Carbone, I. (2015). Characterization and distribution of mating-type genes of the turfgrass pathogen Sclerotinia homoeocarpa on a global scale. Fungal Genet. Biol. 81, 25–40. doi:10.1016/j.fgb.2015.05.012
Rioux, R. A., Shultz, J., Garcia, M., Willis, D. K., Casler, M., Bonos, S., et al. (2014). Sclerotinia homoeocarpa overwinters in turfgrass and is present in commercial seed. PLoS One 9, e110897. doi:10.1371/journal.pone.0110897
Rioux, R. A., Stephens, C. M., Koch, P. L., Kabbage, M., and Kerns, J. P. (2020). Identification of a tractable model system and oxalic acid-dependent symptom development of the dollar spot pathogen Clarireedia jacksonii. Plant Pathol. 70, 722–734. doi:10.1111/ppa.13319
Rowe, H. C., and Kliebenstein, D. J. (2007). Elevated genetic variation within virulence-associated Botrytis cinerea polygalacturonase loci. Mol. Plant. Microbe. Interact. 20, 1126–1137. doi:10.1094/MPMI-20-9-1126
Ryder, L. S., Dagdas, Y. F., Mentlak, T. A., Kershaw, M. J., Thornton, C. R., Schuster, M., et al. (2013). NADPH oxidases regulate septin-mediated cytoskeletal remodeling during plant infection by the rice blast fungus. Proc. Natl. Acad. Sci. U. S. A. 110 (8), 3179–3184. doi:10.1073/pnas.1217470110
Salgado-Salazar, C., Beirn, L. A., Ismaiel, A., Boehm, M. J., Carbone, I., Putman, A. I., et al. (2018). Clarireedia: A new fungal genus comprising four pathogenic species responsible for dollar spot disease of turfgrass. Fungal Biol. 122, 761–773. doi:10.1016/j.funbio.2018.04.004
Sapkota, S., Catching, K. E., Raymer, P. L., Martinez-Espinoza, A. D., and Bahri, B. (2021). New approaches to an old problem: Dollar spot of turfgrass. Phytopathology 112, 469–480. doi:10.1094/PHYTO-11-20-0505-RVW
Sapkota, S., Martinez-Espinoza, A. D., Ali, E., Vermeer, C. B., and Bahri, B. A. (2020). Taxonomical identification of Clarireedia species causing dollar spot disease of turfgrass in Georgia. Plant Dis. 104, 3063. doi:10.1094/PDIS-03-20-0603-PDN
Segmüller, N., Kokkelink, L., Giesbert, S., Odinius, D., van Kan, J., and Tudzynski, P. (2008). NADPH oxidases are involved in differentiation and pathogenicity in botrytis cinerea. Mol. Plant. Microbe. Interact. 21 (6), 808–819. doi:10.1094/MPMI-21-6-0808
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi:10.1093/bioinformatics/btv351
Smiley, R. W., Dernoeden, P. H., and Clarke, B. B. (2005). Compendium of turfgrass diseases. Third Edition. The American Phytopathological Society. doi:10.1094/9780890546154
Sperschneider, J., and Dodds, P. (2022). EffectorP 3.0: Prediction of apoplastic and cytoplasmic effectors in fungi and oomycetes. Mol. Plant. Microbe. Interact. 35, 146–156. doi:10.1094/MPMI-08-21-0201-R
Stackhouse, T., Waliullah, S., Martinez-Espinoza, A. D., Bahri, B., and Ali, M. E. (2021). Development of a co-dominant cleaved amplified polymorphic sequences assay for the rapid detection and differentiation of two pathogenic Clarireedia spp. associated with dollar spot in turfgrass. Agronomy 11, 1489. doi:10.3390/agronomy11081489
Steketee, C. J., Martinez-Espinoza, A. D., Harris-Shultz, K. R., Henry, G. M., and Raymer, P. L. (2017). Evaluation of seashore Paspalum germplasm for resistance to dollar spot. Int. Turfgrass Soc. Res. J. 13, 175–184. doi:10.2134/itsrj2016.05.0411
Tajima, F. (1993). Simple methods for testing the molecular evolutionary clock hypothesis. Genetics 135, 599–607. doi:10.1093/genetics/135.2.599
Tamura, K., and Nei, M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10, 512–526. doi:10.1093/oxfordjournals.molbev.a040023
Torres-Calzada, C., Tapia-Tussell, R., Quijano-Ramayo, A., Martin-Mex, R., Rojas-Herrera, R., Higuera-Ciapara, I., et al. (2011). A species-specific polymerase chain reaction assay for rapid and sensitive detection of Colletotrichum capsici. Mol. Biotechnol. 49 (1), 48–55. doi:10.1007/s12033-011-9377-7
Townsend, R. V., Rioux, R. A., Kabbage, M., Stephens, C., Kerns, J. P., and Koch, P. (2020). Oxalic acid production in Clarireedia jacksonii is dictated by pH, host tissue, and xylan. Front. Microbiol. 11, 1732. doi:10.3389/fmicb.2020.01732
Turrà, D., Segorbe, D., and Di Pietro, A. (2014). Protein kinases in plant-pathogenic fungi: Conserved regulators of infection. Annu. Rev. Phytopathol. 52, 267–288. doi:10.1146/annurev-phyto-102313-050143
Venu, R. C., Beaulieu, R. A., Graham, T. L., Medina, A. M., and Boehm, M. J. (2009). Dollar spot fungus Sclerotinia homoeocarpa produces oxalic acid. Pap. Plant Pathology 401. Available at: https://digitalcommons.unl.edu/plantpathpapers/401.
Viji, G., Uddin, W., O’Neill, N. R., Mischke, S., and Saunders, J. A. (2004). Genetic diversity of Sclerotinia homoeocarpa isolates from turfgrasses from various regions in North America. Plant Dis. 88, 1269–1276. doi:10.1094/PDIS.2004.88.11.1269
Walsh, B., Ikeda, S. S., and Boland, G. J. (1999). Biology and management of dollar spot (Sclerotinia homoeocarpa); an important disease of turfgrass. Hort Science 34, 13–21. doi:10.21273/HORTSCI.34.1.13
Wang, L., Mogg, C., Walkowiak, S., Joshi, M., and Subramaniam, R. (2014). Characterization of NADPH oxidase genes NoxA and NoxB in Fusarium graminearum. Can. J. Plant Pathology 36 (1), 12–21. doi:10.1080/07060661.2013.868370
Wang, P., Yi, S., Mu, X., Zhang, J., and Du, J. (2020). Chromosome-level genome assembly of Cerasus humilis using PacBio and Hi-C technologies. Front. Genet. 11, 956. doi:10.3389/fgene.2020.00956
White, T. J., Bruns, T. D., Lee, S. B., and Sninsky, J. (1990). “Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics,” in PCR protocols: A guide to methods and applications. Editors M. A. Innis, D. H. Gelfand, J. J. Sninsky, and T. J. White (New York: Academic Press), 315–322. doi:10.1016/B978-0-12-372180-8.50042-1
Williams, B., Kabbage, M., Kim, H.-J., Britt, R., and Dickman, M. B. (2011). Tipping the balance: Sclerotinia sclerotiorum secreted oxalic acid suppresses host defenses by manipulating the host redox environment. PLoS Pathog. 7, e1002107. doi:10.1371/journal.ppat.1002107
Wlodawer, A. (2017). Stereochemistry and validation of macromolecular structures. Methods Mol. Biol. 1607, 595–610. doi:10.1007/978-1-4939-7000-1_24
Xia, S., Xu, Y., Hoy, R., Zhang, J., Qin, L., and Li, X. (2019). The notorious soilborne pathogenic fungus Sclerotinia sclerotiorum: An update on genes studied with mutant analysis. Pathogens 9, 27. doi:10.3390/pathogens9010027
Yang, S. L., and Chung, K.-R. (2013). Similar and distinct roles of NADPH oxidase components in the tangerine pathotype of Alternaria alternata: The Alternaria alternata NADPH oxidase. Mol. Plant Pathol. 14 (6), 543–556. doi:10.1111/mpp.12026
Keywords: Clarireedia paspali, PacBio and Illumina technologies, gene prediction, protein structure prediction, genome annotation, phylogeny, pathogenicity, effectors
Citation: Bahri BA, Parvathaneni RK, Spratling WT, Saxena H, Sapkota S, Raymer PL and Martinez-Espinoza AD (2023) Whole genome sequencing of Clarireedia aff. paspali reveals potential pathogenesis factors in Clarireedia species, causal agents of dollar spot in turfgrass. Front. Genet. 13:1033437. doi: 10.3389/fgene.2022.1033437
Received: 31 August 2022; Accepted: 21 November 2022;
Published: 05 January 2023.
Edited by:
Chongjing Xia, Southwest University of Science and Technology, ChinaReviewed by:
James P. Kerns, North Carolina State University, United StatesCopyright © 2023 Bahri, Parvathaneni, Spratling, Saxena, Sapkota, Raymer and Martinez-Espinoza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bochra Amina Bahri, YmJhaHJpQHVnYS5lZHU=
†Present address: Suraj Sapkota, USDA-ARS, Crop Genetics and Breeding Research Unit, Tifton, GA, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.