 Gowri Nayar1Ignacio Terrizzano2Ed Seabolt2Akshay Agarwal2
Gowri Nayar1Ignacio Terrizzano2Ed Seabolt2Akshay Agarwal2 Christina Boucher3
Christina Boucher3 Jaime Ruiz3
Jaime Ruiz3 Ilya B. Slizovskiy4James H. Kaufman5
Ilya B. Slizovskiy4James H. Kaufman5 Noelle R. Noyes4*
Noelle R. Noyes4*- 1Department of Biomedical Informatics, Stanford University, Stanford, CA, United States
- 2IBM Research Almaden, San Jose, CA, United States
- 3Department of Computer and Information Science and Engineering, University of Florida, Gainesville, FL, United States
- 4Department of Veterinary Population Medicine, University of Minnesota, Minneapolis, MN, United States
- 5Altos Labs, Redwood City, CA, United States
Horizontal gene transfer mediated by conjugation is considered an important evolutionary mechanism of bacteria. It allows organisms to quickly evolve new phenotypic properties including antimicrobial resistance (AMR) and virulence. The frequency of conjugation-mediated cargo gene exchange has not yet been comprehensively studied within and between bacterial taxa. We developed a frequency-based network of genus-genus conjugation features and candidate cargo genes from whole-genome sequence data of over 180,000 bacterial genomes, representing 1,345 genera. Using our method, which we refer to as ggMOB, we revealed that over half of the bacterial genomes contained one or more known conjugation features that matched exactly to at least one other genome. Moreover, the proportion of genomes containing these conjugation features varied substantially by genus and conjugation feature. These results and the genus-level network structure can be viewed interactively in the ggMOB interface, which allows for user-defined filtering of conjugation features and candidate cargo genes. Using the network data, we observed that the ratio of AMR gene representation in conjugative versus non-conjugative genomes exceeded 5:1, confirming that conjugation is a critical force for AMR spread across genera. Finally, we demonstrated that clustering genomes by conjugation profile sometimes correlated well with classical phylogenetic structuring; but that in some cases the clustering was highly discordant, suggesting that the importance of the accessory genome in driving bacterial evolution may be highly variable across both time and taxonomy. These results can advance scientific understanding of bacterial evolution, and can be used as a starting point for probing genus-genus gene exchange within complex microbial communities that include unculturable bacteria. ggMOB is publicly available under the GNU licence at https://ruiz-hci-lab.github.io/ggMOB/
1 Introduction
Several mechanisms of horizontal gene transfer (HGT) allow bacteria to exchange genetic material. One of these mechanisms, termed conjugation, occurs when bacterial cells form direct physical contacts that allow for passage of genetic material from one bacterium to another. The machinery required to form these contacts and initiate genetic exchange is often contained within integrative and conjugative elements (ICE), plasmids, and other mobile genetic elements (MGEs) (Frost et al., 2005; Wozniak and Waldor, 2010; Roberts and Mullany, 2011; Wiedenbeck and Cohan, 2011; Perry and Wright, 2013; Johnson and Grossman, 2015; Singer et al., 2016). The conditions that induce excision and conjugation are not fully elucidated, but DNA damage and subsequent SOS response seem to be an important trigger (Waldor et al., 2004; Koraimann and Wagner, 2014). The cost of acquiring and maintaining the new genetic material also influences the success of transfer events (Uhlemann et al., 2021).
Genes exchanged between bacteria during conjugation include functional domains associated with conjugative machinery (e.g., excisionases, integrases, conjugative transport proteins) as well as intervening “accessory” genes that are not necessary for conjugation, often termed “cargo genes” (Johnson and Grossman, 2015). By pairing conjugative machinery with an array of diverse cargo genes, bacterial communities can significantly expand their genetic repertoire, including between bacteria of diverse taxonomy (Guglielmini et al., 2011; Bellanger et al., 2014; Neil and Allard, 2021). Functions commonly associated with conjugative cargo include antimicrobial resistance (AMR) and virulence (Roberts and Mullany, 2011; Perry and Wright, 2013; Johnson and Grossman, 2015; Cury et al., 2017), which can pose a risk to human and animal health if transferred into pathogens (Partridge et al., 2018). Therefore, understanding the microbial ecology of conjugative elements and cargo genes (i.e., their distribution and behavior across bacterial taxa) is important in assessing the risk posed by various bacterial communities (Gaston et al., 2021). For example, how often do different bacterial taxa carry conjugative machinery and AMR genes; what resistance phenotypes are commonly associated with the presence of conjugative machinery within the genome; how often do different commensal bacterial taxa carry out conjugation to exchange cargo genes with pathogens; and what conditions foster conjugative exchange of specific cargo genes between pathogens and non-pathogens? These questions are fundamental to understanding how bacterial communities respond to external stimuli, and how these responses increase the overall risk posed by microbial communities of varying composition (Martínez et al., 2015; Oh et al., 2018).
However, the process of conjugative HGT is highly stochastic and therefore, difficult to predict (Lopatkin and Collin, 2020). One reason for this stochasticity is variability in the conjugation competency of donor and recipient bacterial cells for a given conjugative MGE; as well as variability in the capacity of a given type of conjugative MGE to also transfer unrelated cargo genes. Recent meta-analyses of conjugation rates for specific bacterial species and/or MGEs have highlighted these complexities (Alderliesten et al., 2020; Sheppard et al., 2020). Historically, the scientific process for estimating conjugative likelihood has stemmed from highly controlled in vitro experiments between pairs of bacterial isolates and specific MGEs. Results from such studies have been crucial for uncovering the behavior of MGEs and their importance for functions such as AMR. However, reductive experiments typically do not generalize well to the complex microbial communities found in situ, including host and environmental microbiomes. Furthermore, these experiments are necessarily restricted in their ability to characterize the full bacterial host range of a given MGE, as they typically involve only several distinct bacterial taxa. One major challenge that remains is to generate a conjugation likelihood for every host-donor-MGE combination observed across all bacterial taxa and MGE.
Insight into this challenge can be gained through the plethora of whole genome sequence (WGS) data which is now publicly available. As an example, the analysis of HGT-associated genes from just 336 genomes across 16 phyla was sufficient to significantly improve bacterial phylogenies as compared to those obtained from conserved marker genes (Abby et al., 2012). An analysis of 1,000 genomes demonstrated that ICE machinery is ubiquitous across diverse prokaryotes, and likely one of the most common mechanisms of bacterial evolution (Guglielmini et al., 2011). Currently, public datasets contain orders of magnitude more WGS data, which can be used to improve our understanding of the mechanisms by which critically important genes and pathogens emerge and persist (Botelho et al., 2020). However, despite the importance of HGT in bacterial evolution and pathogenicity, there has not yet been a comprehensive, systematic survey of the frequency of conjugation and cargo genes within or between bacterial genera. The objective of this work was to describe intra- and inter-genus conjugation-cargo dynamics by leveraging the comprehensive set of WGS data and conjugation sequences currently available within the Reference Sequence (RefSeq) and Short Read Archive (SRA) databases at the National Center for Biotechnology Information (NCBI). In particular, we analyzed 186,887 WGS datasets to identify putative conjugation events and corresponding candidate cargo genes, as well as to characterize the frequency of AMR genes with respect to the frequency of their occurrence with conjugative proteins. We were able to identify over 95,000 genomes containing conjugative proteins, and more than 4 billion putative cargo genes between genomes. We summarize and disseminate this analysis through an open-source network that describes the genus-genus sharing of conjugation features and cargo genes, representing genomes from over 1,300 different genera. Our network, which we refer to as ggMOB, allows users to filter for both conjugation features and putative cargo genes. Using ggMOB we analyzed the ratio of AMR gene representation in conjugative versus non-conjugative genomes and found it to be greater than 5:1, confirming that conjugation is a critical force for AMR spread across genera. Finally, we demonstrated that clustering genomes by conjugation profile sometimes correlated well with taxonomic structuring, but in some cases was highly discordant, suggesting that the importance of the accessory genome in driving bacterial evolution may be highly variable across genera. These results demonstrates that ggMOB can be used to further probe potential genus-genus mobilization dynamics, and thus, provide insight into conjugative mobilization between unculturable bacteria and complex interactions involving multiple genera.
2 Results
2.1 Overview of ggMOB
The analyses conducted in this paper were derived from an existing resource (Seabolt et al., 2020), which was constructed by curating and annotating 186,887 genomes from NCBI (see MATERIALS AND METHODS). Using the sequence data and annotated features obtained from over 166,000 curated and high-quality WGS datasets, we identified genomes that contained conjugative features (Table 1), which we term conjugative genomes (see MATERIALS AND METHODS). By recording counts of shared features across these conjugative genomes, we constructed ggMOB (the “genus-genus mobilization” network), which contains information about features that are shared between conjugative genus-genus pairs.
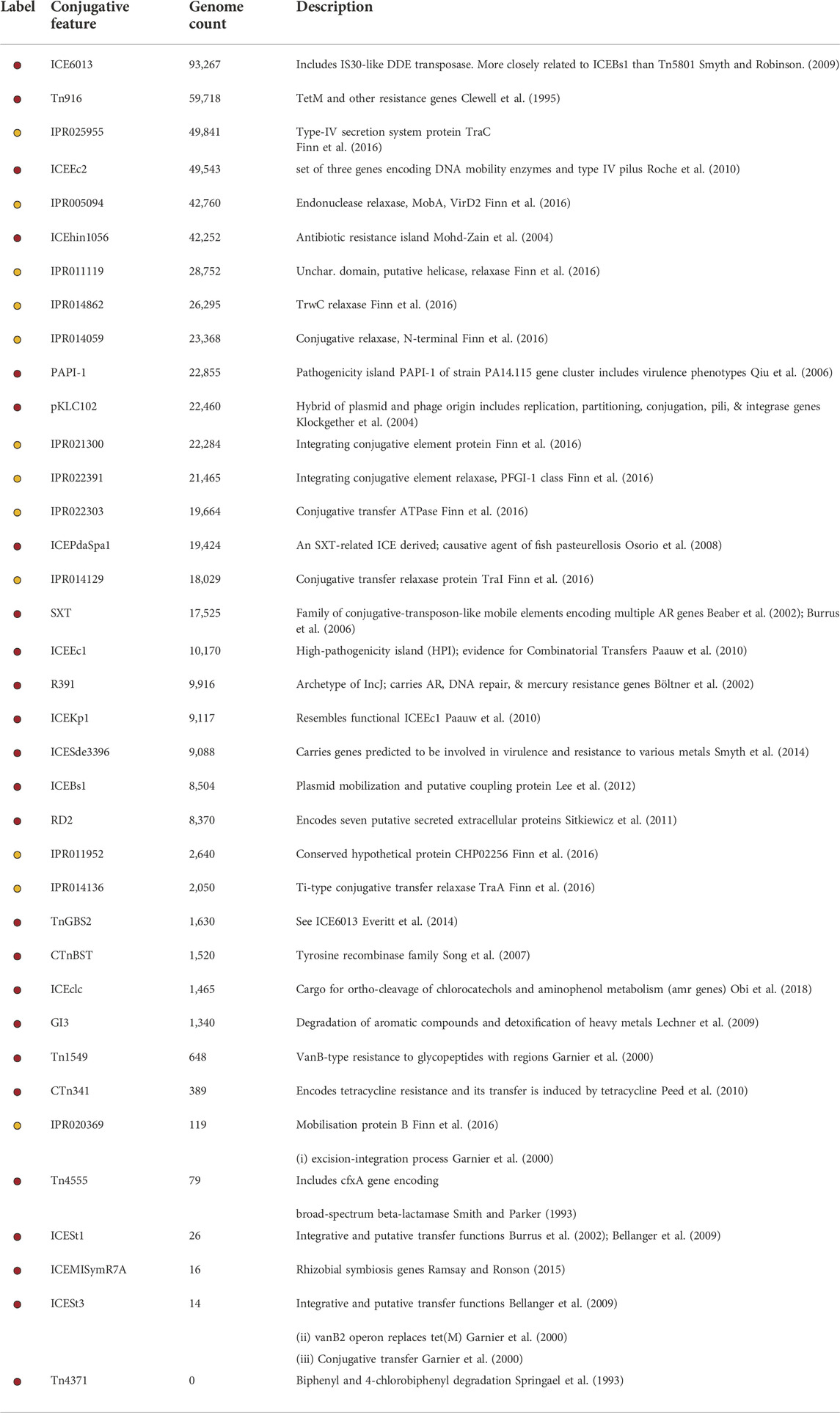
TABLE 1. Conjugative features included in this study. Color label indicates the color used to represent the conjugative feature in Figure 2. Yellow indicates a conjugative feature defined by a single IPR code and red indicates a family of codes. Genome count indicates the number of genomes that contain the conjugative feature.
2.2 Inter- and intra-genus conjugation profiles
Of the 106,443 genomes that contained at least one conjugative feature, 95,781 shared at least one conjugative feature with at least one other genome in the set, indicating a common evolutionary history. These 95,781 conjugative genomes represented close to 47% (631 of the 1,345) of the genera contained in the relational database (Seabolt et al., 2020). The lack of conjugative machinery in the other 714 genera may be a false negative finding (i.e., incomplete list of conjugative features, lack of representation in the utilized NCBI databases, or lack of inclusion in genome assemblies), or could indicate inherent differences in the conjugative ability of genera across the taxonomic tree. Similarly, one might reasonably expect the number of observed conjugative genomes to scale with the number of genomes available for each genus. However, genus representation in NCBI is not uniform across genera, leading to bias in available genomes per genus. To correct for this imbalance, we computed the conjugative genome proportion by normalizing the number of observed conjugative genomes to the total number of genomes per genus (Table 2). This analysis demonstrated that the genera with the largest fraction of conjugative genomes were not the genera with the most genomes in NCBI. For example, although Salmonella had by far the greatest number of high quality genomes (N = 39,574), it ranked fifth in terms of the proportion of genomes that contained a conjugative feature. The top 30 genera listed in Table 2 all had a conjugative genome frequency greater than 20%, with genomes from the genus Legionella containing conjugative proteins over 99% of the time. This high percentage may have been driven by sampling bias in the available NCBI WGS datasets (for example, the Legionella pneumophila WGS accessions appear to have been collected from a single site), or it may represent the propensity for conjugation-mediated processes to occur within individual genera.

TABLE 2. Proportion of genomes that contained conjugative feature(s), by genus. All genera with over 100 representative genomes are listed, in descending order by the proportion of conjugative genomes in each genus.
Next, we identified a total of 5,956 conjugation proteins across genus pairs, i.e., triples of the form (genus1, genus2, protein-name), associated with a total of 23,353,196,048 cargo protein sequences. These results are visualizable as connected nodes in the ggMOB network. This count is non-distinct by protein name as, for instance, Tyrosine recombinase XerD was found both between Salmonella-Salmonella genomes, as well as between Oligella-Proteus genomes and other genera pairs. Of these 5,956 triples, 1,680 contained genome pairs belonging to the same genus (i.e., intra-genus). Some genera were much more likely to contain genomes with conjugative features that matched to genomes from other genera, i.e., inter-genus. For example, we observed over 30 million instances of matching conjugation proteins in genomes from the Acinetobacter genus. Of these instances, 84% were to genomes from other genera (i.e., inter-genus), versus 16% from genomes within Acinetobacter (i.e., intra-genus). Members of the Acinetobacter genus are well-known for genome plasticity (Chan et al., 2015), which contributes to important phenotypes such as AMR and biofilm formation.
Genera that shared the highest number of conjugation proteins with Acinetobacter genomes included Escherichia, Shigella, Vibrio, Salmonella, Pseudomonas, Klebsiella, Enterobacter and Citrobacter. While this result was not unexpected given the list of known pathogens contained within these genera, our database of intra- and inter-genus exact-match conjugative features also revealed many unreported and unexpected associations. For example, genomes within the genus Nitrosomonas contained 38,034 instances of conjugative proteins, nearly 100% of which were shared with genomes from the Shigella genus. The specific conjugative feature involved in the vast majority of these exact matches was ICEEc2, a relatively recently discovered ICE MGE that was previously shown to transfer competently between Salmonella enterica serovar Typhimurium strain and into a Yersinia pseudotuberculosis strain. Our results suggest that ICEEc2 has a very broad host-donor range, including many genera that may not yet be described in the literature in reference to this particular ICE.
We found that the likelihood of identifying conjugative features in pairs of genomes within versus across genera was highly variable. Of the 36 conjugative features analyzed, 13 were only identified in pairs of genomes belonging to the same genus, i.e., intra-genus (Table 3). However, six conjugative features were more likely to be observed across genera than within genera, i.e.,
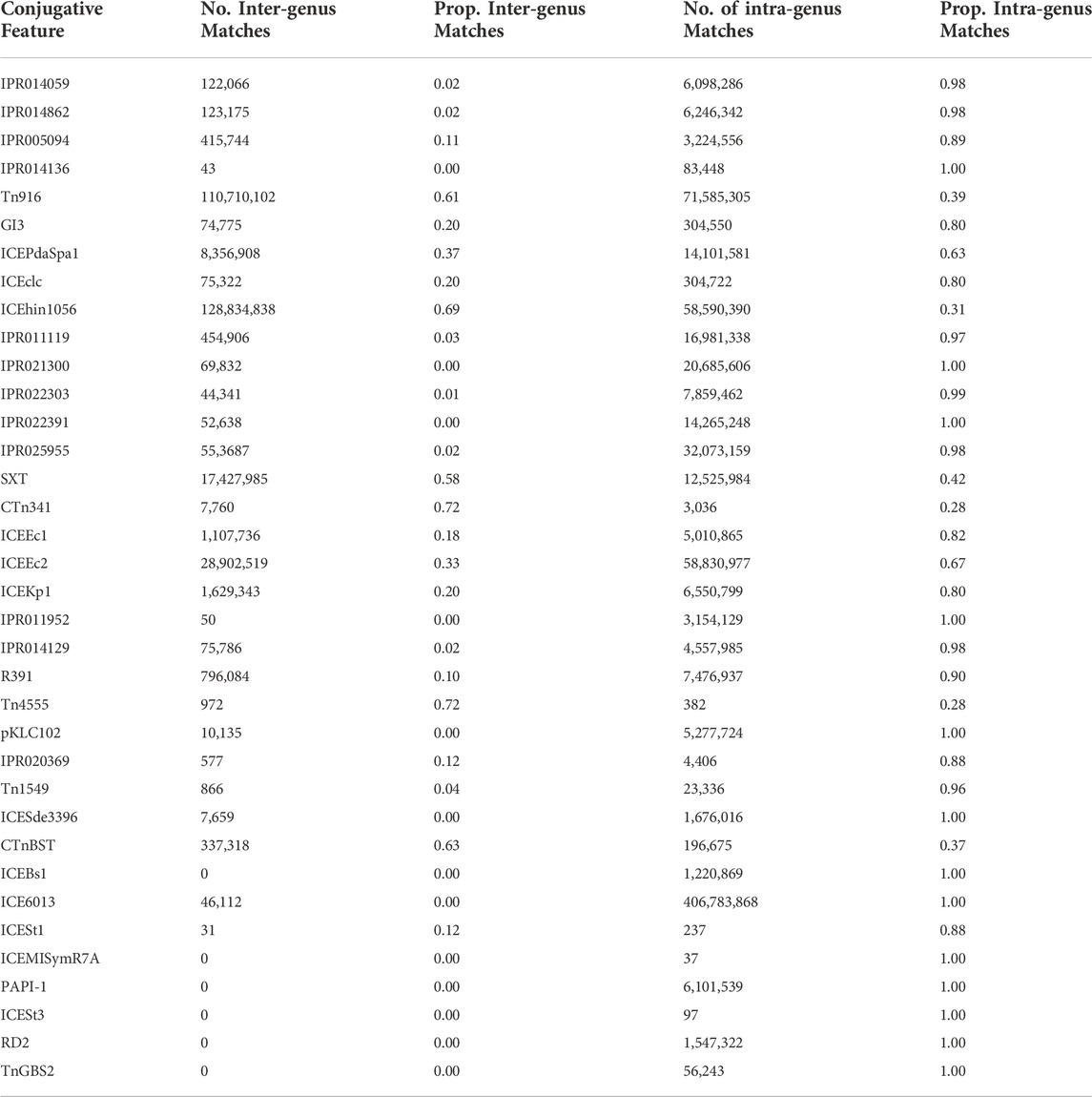
TABLE 3. Count and proportion of inter- and intra-genus detection of conjugative features.
2.3 Cargo gene profiles
To characterize the set of genes that are most likely to have been associated with conjugative HGT events, we identified all proteins that were contained in at least two conjugative genomes with 100% sequence identity. Out of 51,362,178 total unique protein sequences in the source database, 28,042 were identified as conjugation-associated proteins (i.e., conjugation machinery), and 11,276,651 were identified as candidate cargo proteins. The full set of cargo proteins mapped to 20,550 distinct names (excluding “putative protein(s)” or “hypothetical protein(s)”), with a wide range of frequencies within and between genera. Annotation of the conjugative genomes demonstrated that the vast majority of conjugative and cargo proteins were adjacent within the genome (Figure 2). Moreover, within each genome, the conjugative features were more likely to be proximate to putative cargo protein versus non-cargo proteins, suggesting again a common evolutionary history.
2.4 Genotypic AMR and association with conjugation features
A subset of the observed cargo protein names were associated with a set of confirmed AMR protein names. We identified this subset by selecting only those names that Prokka assigned to sequences mapping to a name defined in MEGARes v1.0 (Lakin et al., 2016), a comprehensive AMR database. The entity relations in our database ensured a 1:1 mapping between gene and protein names and their respective sequences. Of the 3,824 distinct sequences contained in MEGARes, Prokka identified 3,674 as valid sequences coding for protein. These 3,674 distinct proteins were assigned 286 distinct names, excluding “putative protein” and “hypothetical protein”. These unnamed proteins comprised just 1% of the MEGARes protein set. While this highly curated set is certainly not a comprehensive list of all proteins contributing to AMR, it is an initial set to estimate the fraction of AMR proteins within the larger set of conjugation and cargo proteins.
To further investigate the microbial genomics of AMR in relation to HGT events, we used plasmid sequences from NCBI to identify the set of AMR-specific cargo proteins found in a plasmid sequence. We then compared the distribution of AMR proteins in plasmids versus conjugative genomes (Figure 3). The distribution of AMR genes differed between plasmids and conjugative genomes, with a high probability that AMR genes were identified in conjugative (versus non-conjugative) genomes, and a much lower probability of being identified in plasmids (Figure 3). While plasmids are often critical to the microbial ecology of AMR, this analysis suggests that other conjugative processes may drive the vast majority of AMR gene exchanges between bacteria. This dynamic has been reported for specific AMR gene groups, including carbapenem AMR genes (Botelho et al., 2020), and also supports previous analyses of conjugative machinery across bacterial genera (Guglielmini et al., 2011).
To analyze the distribution of AMR proteins across genomes, we calculated the probability that each AMR gene was identified in conjugative versus non-conjugative genomes (Table 4). From the complete list of 286 AMR protein names, 220 were found more often in conjugative genomes; three were found with equal probability in conjugative and non-conjugative genomes; and 63 were found more often in genomes that did not contain any conjugation proteins. Given that the proportion of conjugative genomes in our database was 51%, these results suggest that AMR genes are disproportionately represented within conjugative genomes.
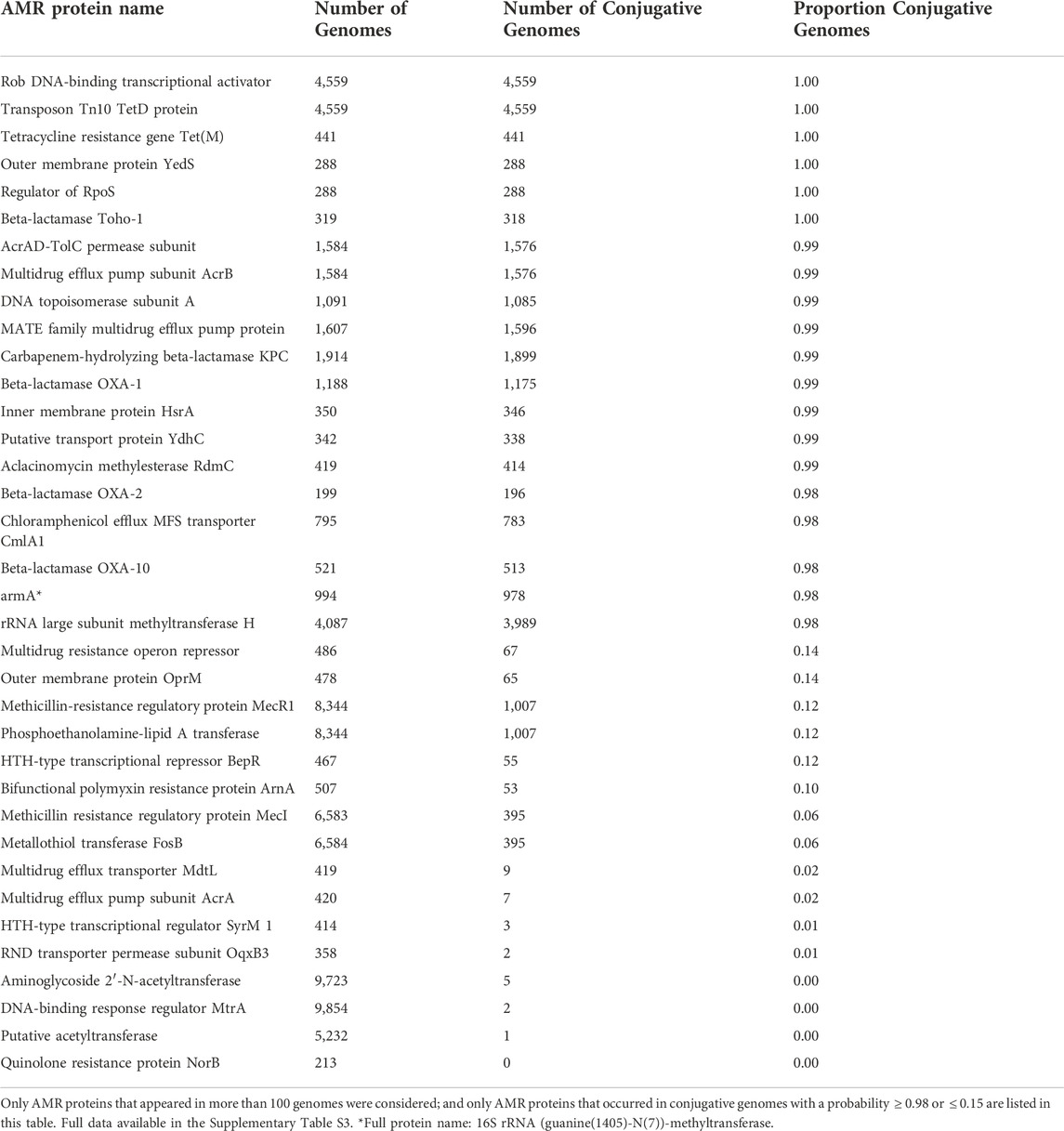
TABLE 4. Probability of observing AMR proteins in genomes that also contained conjugative features.
The above analysis does not distinguish between different types of conjugation features, and also treats AMR proteins as independent features. However, the data in Figure 2 demonstrate that many of the conjugation features defined in Table 1 often co-occur in the same genome, as do some of the AMR proteins. To gain insight into these correlations, and to identify groups of AMR proteins associated with different conjugation families, we performed a genomic co-occurrence analysis across all conjugation features for the 138 AMR proteins found in conjugative genomes with a frequency of at least 5 times compared to identification in non-conjugative genomes (i.e., these AMR proteins were highly represented in conjugative genomes, Figure 4). The results of this analysis demonstrated that some conjugation features frequently co-occurred within genomes with both other conjugation features as well as multiple AMR proteins. For example, IPR005094 co-occurred with the highest diversity of AMR protein names (N = 139, see Supplementary Table S2). Many co-occurrence patterns reflected known biological associations. For example, the Tn916 conjugation feature co-occurred most frequently with tetracycline ribosomal protection protein TetM, a genomic association discovered over 3 decades ago (Su et al., 1992). While TetM seemed to co-occur with a few select conjugation features (such as Tn916), other AMR protein names co-occurred with many conjugation features. For example, many of the AMR names associated with extended-spectrum beta-lactam and carbapenem resistance (e.g., beta-lactamases Toho-1, OXA-1, OXa-2, OXA-10, SHV-2, and KPC) co-occured with the majority of evaluated conjugation features, which may provide partial explanation for the observed rapid expansion of these important AMR genes within Enterobacteriaceae (Logan and Weinstein, 2017). Similarly, the recently widely-publicized mcr-1 protein co-occurred with multiple conjugation features, which both strengthens and expands upon recent findings that this AMR gene has been mobilized on numerous plasmid types (Wang et al., 2018). Co-occurrence data such as those provided in Figure 4 may represent a new and sustainable (i.e., easily updated) source of information regarding the potential for new and emerging AMR genes to expand within and across bacterial populations via HGT. This information, in turn, could help to prioritize and focus public health and human clinical decision-making regarding AMR.
Conversely, 29 AMR proteins occurred at least 5x more frequently in non-conjugative genomes compared to conjugative genomes (Supplementary Figure S1). Of note is the observation that no beta-lactam AMR protein names occur in this list of 29 AMR names, which contrasts starkly to the preponderance of beta-lactam-associated AMR names in Figure 4, again suggesting that beta-lactam resistance is tightly coupled with conjugative machinery, and that conjugation-mediated exchange is the primary evolutionary driver of beta-lactam resistance. By comparison, several mechanisms of multi-drug resistance (MDR) are contained within the list of 29 AMR proteins observed more frequently in non-conjugative versus conjugative genomes, i.e., AcrB, AcrE, OqxB7, mdtA, mdtE, mdtH, and MexB. These mechanisms of MDR tend to be multi-function, i.e., the proteins confer multiple functional benefits to bacteria, in addition to AMR. Together, the results of Figure 4 and Supplementary Figure S1 suggest that proteins with more specific AMR functions tend to be disproportionately represented amongst conjugative genomes, while more generalist proteins tend to be disproportionately represented within non-conjugative genomes. One hypothesis for this observation is that the fitness cost-benefit dynamics differ for generalist versus specialist genes, such that specialized genes are more likely to transiently yet rapidly spread within bacterial populations via the so-called ‘accessory genome’ (which includes conjugation-mediated exchange), whereas generalist genes are more likely to be maintained permanently within bacterial genomes, and thus are less likely to be identified as conjugation-associated cargo.
2.5 Phenotypic AMR and association with conjugative genomes
Given our hypothesis that conjugation-mediated spread of specialized AMR genes may be promoted by more specific evolutionary pressures such as antimicrobial drug exposures, we hypothesized that this signature of selective pressure may also manifest in the phenotypic properties of conjugative versus non-conjugative genomes. To evaluate this, we queried the NCBI BioSample assay metadata in our relational database, to identify isolates that had been subjected to phenotypic antibiotic susceptibility testing (AST) to known antibiotic compounds. For the 186,887 highest quality genomes, the NCBI assay metadata contained 15,286 phenotypically-confirmed resistant genome-compound AST results, representing 13,076 tests for conjugative genomes and 2,210 tests for non-conjugative genomes. Altogether, 1,242 genomes were used in these tests, of which 1,023 were conjugative genomes and 219 were non-conjugative genomes. For each antibiotic compound listed in the AST results, we computed the number of phenotypically resistant isolates with conjugative-vs. non-conjugative genomes.
The results of this analysis revealed that phenotypic resistance occurred in conjugative genomes with probability >80%, regardless of compound being tested (Table 5 and Supplementary Table S3). As with the disproportionate representation of AMR proteins within conjugative genomes, the phenotypic AMR data suggest that microbial AMR dynamics are driven largely by conjugation-mediated processes. However, it is also important to note that NCBI phenotypic assay data is likely biased due to the motivations for clinicians and researchers to submit isolates for phenotypic testing. Therefore, to test for SRA sampling bias with respect to these compounds, we also measured the phenotypically-resistant fraction expected for randomly selected genomes, based on the number of genomes tested per compound in Table 5 and the number of conjugative and non-conjugative genomes across the entire database. This null hypothesis was tested by running 100 bootstrapped trials for each compound (Table 3). The observed average probability that phenotypic AMR was expressed by an isolate with a conjugative genome was 0.85 ± 0.05 independent of antibiotic compound, i.e., weighted by total genomes tested per compound (Table 5). In a random process, the probability would be expected to be near 51% given the fraction of all genomes with conjugation features. These results further support the importance of conjugation in the microbial ecology and epidemiology of both genotypic and phenotypic AMR.

TABLE 5. Associations between phenotypic AMR and representation of conjugative versus non-conjugative genomes.
2.6 Genome clustering by conjugative feature profile
The specific proteins transferred between bacteria are known to vary by conjugation feature, for example as demonstrated by the co-occurrence patterns of conjugation features and AMR proteins within genomes (Figure 4 and Supplementary Figure S1). To demonstrate the structuring of bacterial populations by conjugation-cargo co-occurrence patterns, we generated the same co-occurrence matrix for all cargo proteins and all conjugative features within genomes that contained a high frequency of cargo proteins (Figure 5). We then calculated the genome-genome Euclidean distance for all genomes in the co-occurrence matrix, using a vector of normalized conjugation features (see Methods, Supplementary Figure S2). The results demonstrate clusters of genomes with similar conjugative features. While these clusters often reflect classical bacterial taxonomic structure, there are also instances of discordance between the clustering based on conjugation profile and traditional grouping based on taxonomy. These results reflect the evolutionary dynamics of bacterial populations, and suggest that the relative importance of vertical versus horizontal gene transfer events is highly variable. Such variability in the importance of HGT events has been previously reported, including differences in plasmid versus ICE-mediated exchange and interactions with bacterial host range (Cury et al., 2018). This finding has far-reaching and complex implications for applications that rely on a measure of phylogenetic relatedness, i.e., outbreak detection and source attribution. In some cases the use of the core genome may be sufficient to accurately reflect bacterial phylogenetic relatedness; while in others, the information in the core genome may obfuscate the true relationships between bacterial isolates. As WGS data become more widely used, these complexities must be considered, and in some cases, incorporated into phylogenetic analysis workflows.
The relational database underlying this work and the ggMOB tool is a necessary (yet not sufficient) component of improved interpretation and use of WGS data. We note that, in principle, one can use the co-occurrence matrix and vectorization procedure on genomic properties other than conjugative features. This provides a flexible and customizable approach for defining a different set of genomes of interest within the mobilome, which can then be used to (re)classify organisms not by name, but by genome-genome distance in a space of mobilization features. This capability could represent a powerful tool for improving our understanding of bacterial evolution while also informing the next generation of applied WGS computational and statistical pipelines.
3 Discussion
The public availability of large scale genomic data makes it possible to apply cloud computing technology and big data techniques to study important phenomena in molecular and microbiology. Curating these data in a relational database with biologically structured entity relations (i.e., linking genomes, genes, proteins, domains, and metadata) provides a powerful method with which to ask biological questions about the data. We leveraged this approach in the current study of cargo and conjugation, which is an essential mechanism by which bacteria acquire new phenotypes, transmit molecular functions, and adapt to stress. Furthermore, these events are critical for understanding bacterial evolution and phylogeny (Guglielmini et al., 2011; Abby et al., 2012; Bellanger et al., 2014). Our work not only sheds light on conjugation-mediated cargo transfers between and within genera, but also demonstrates the ability of mining and analyzing large datasets in improving our understanding of bacterial evolutionary dynamics. The network of putative genus-genus conjugation features and candidate cargo genes can be dynamically visualized using the ggMOB tool, which supports hypothesis generation and testing related to intra- and inter-genus conjugation dynamics.
In our analysis we identified sets of proteins with the strongest evidence as conjugation and cargo proteins. This was accomplished by selecting only those proteins that exhibited both 100% sequence identity and co-occurrence in pairs of genomes containing identical conjugation-associated sequences. With this strict selection process, the putative cargo proteins exhibited a high degree of spatial correlation within assembled contigs (i.e., they were highly adjacent to each other, as well as to the conjugation protein itself). Other proteins in these genomes may also have been transferred (or are transferable) by bacterial conjugation, but they did not meet our strict selection criteria. Considering only strictly-selected candidate cargo proteins, we were able to profile the frequency of conjugation-mediated protein exchange within and between genera.
Our results suggest that conjugation-mediated exchange is not uncommon, affirming prior studies (Guglielmini et al., 2011; Bellanger et al., 2014). Conjugation-related proteins were observable in 51% of bacterial genomes and in 631 of 1,345 genera (approximately 47%). Frequency of intra- and inter-genus conjugation-mediated exchange varied significantly depending on the taxa involved, suggesting that taxonomy greatly influences genetic exchange of, e.g., AMR or pathogenicity proteins (Delavat et al., 2017). By quantifying this across a large database of high-quality WGS data, we measured the “exchange likelihood” between different genera. These likelihoods can be visualized dynamically in the ggMOB tool, which reveals distinct clusters of genera that share conjugative features with exact sequence match. This suggests that the likelihood of protein transfer varies substantially by genus pair, and that the bacterial composition within a given environment is an important consideration when attempting to evaluate mobilization potential within a microbial community (Lopatkin and Collin, 2020; Neil and Allard, 2021).
While we have conducted this analysis for a specific set of conjugation features (Table 1), the analytic approach can be applied to any MGE(s) and cargo protein(s) of interest. As such, our overall approach represents a method for obtaining a long-range evolutionary view of transfer likelihood between diverse bacterial taxa, including pathogens and commensal bacteria (Guglielmini et al., 2011). These baseline exchange likelihoods are critical parameters for risk analysis at the microbial community level, including for applications such as personalized microbiome medicine, and microbiome-centric surveillance.
Bacterial taxon is not the only significant driver of exchange likelihood; we also observed that putative, successful transfer events were more likely to involve cargo proteins that infer fitness advantage to the involved bacterial populations, such as AMR. While any gene can, in principle, be transmitted as a cargo gene in conjugative exchange, only a subset of transferred proteins will increase the fitness of the receiving organism. The likelihood of observing successful transfer depends on a large number of factors including the environment, the existing proteins in the recipient chromosome, the cargo proteins themselves, and the survival probability of the organism (Cohen et al., 2010).
Conjugation-mediated protein transfer that improves fitness may increase survival probability. Therefore, chromosomal arrangements that group fitness-conferring cargo proteins near the conjugation machinery will be observed more frequently than those arrangements that involve neutral or disadvantageous proteins. Conversely, very common proteins that aid in stress response may be less likely to be transferred as cargo, since the relative fitness advantage is diminished for proteins that are already likely to be present within a bacterium (i.e., proteins that confer redundant function). The particular stressor—as well as the specific advantageous proteins of interest—depend on phenotype of interest. This view is exemplified by the data in Table 4 which shows that rare AMR proteins are more likely to be found as cargo in genomes that also contain conjugative proteins, as compared to genomes that do not. Conversely, common AMR proteins are less likely to be found in genomes that contain conjugative machinery. One might hypothesize that, with chromosomal rearrangement, nature effects a real world experiment to dynamically optimize cargo protein collections—thereby spreading rare (but useful) proteins and gene combinations over time.
The particular cargo proteins shared between chromosomes varied by conjugation feature, as demonstrated by the AMR proteins analyzed in Figures 1, 4. Considering all conjugation features used in this study, our results suggest that conjugation dynamics are important in structuring genomic content, and thus driving phylogenetic evolution. Based on Figure 5, it seems that sometimes these evolutionary conjugation dynamics can sometimes overpower other taxonomic drivers, such that genus-level genomes do not always cluster together. To demonstrate this conjugation-driven phylogeny, we used the data in Figure 5 to generate Figure 2, which represents the distance between all pairs of genomes based on Euclidean distance between their representations as normalized conjugation feature vectors. The resulting hierarchical clustering shows that the dominant conjugation features are represented in genomes across different genera and, conversely, that individual genera include genomes with differing conjugation profiles. This abrogation of genus-level taxonomy due to conjugation-related genomic content is an inevitable consequence of the inter-genus transfers visualized in ggMOB. Given the reality of conjugative exchange, there is no reason to expect that taxonomic classification by organism name will always predict the composition of conjugation-associated cargo proteins. However, by selecting genomes based on a particular phenotype of interest, it is possible to classify organisms and genome-genome distances based on a feature space defined by conjugation (or other mobilization) proteins, as in Figure 2. Given the ubiquity and diversity of conjugation and other types of HGT (Guglielmini et al., 2011), these types of genome clustering techniques may provide crucial information about bacterial evolution that is not contained within traditional phylogenies. In this regard, the ability to filter the ggMOB data based on conjugation features of interest may particularly useful.
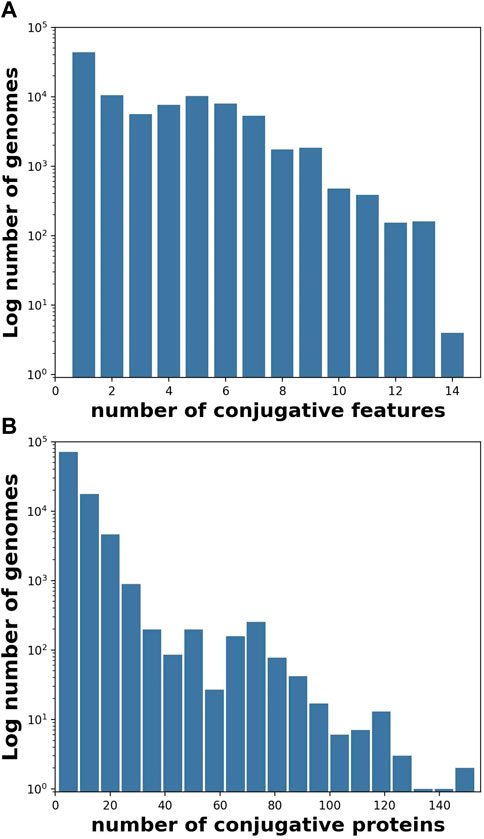
FIGURE 1. Individual genomes typically contained more than one conjugative feature (Table 1), and often contained more than one protein per feature. (A) Histogram (log frequency) of the number of conjugative features per genome, and (B) Histogram (log frequency) of the number of conjugative proteins per genome for all 106,433 genomes containing at least one conjugative feature.
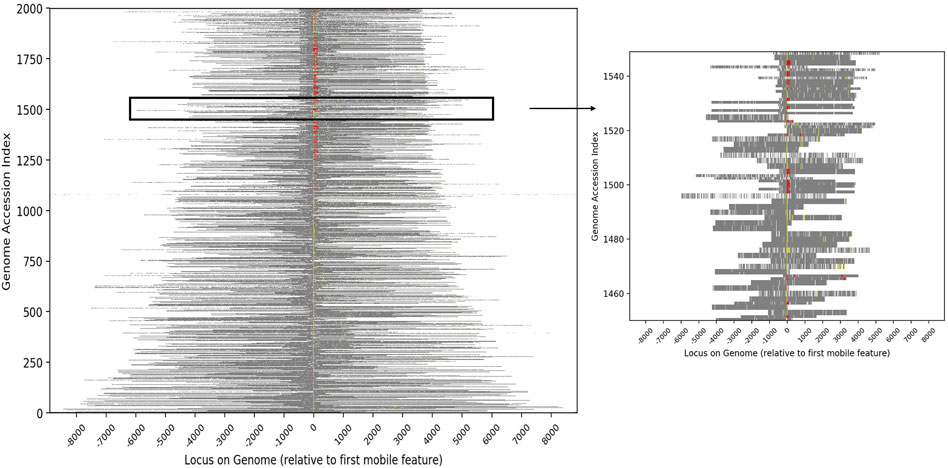
FIGURE 2. Heatmap showing the relative genomic positions of conjugative features and putative cargo proteins for the 2,000 genomes with the greatest number of cargo proteins. Conjugative features are represented as color pixels based on the colors shown in Table 1, with yellow representing proteins assigned specific IPR codes and red representing protein families from the literature. Cargo proteins are shown in grey and other chromosomal DNA in white. Each genome is bit shifted to the left until the first conjugative feature is centered in the figure. Most genomes contained more than one conjugative protein (all contain at least one). The inset highlights the genomes at indices 1450–1550 in order to expand a subset of the data.
4 Materials and methods
4.1 Creation of relational database
Here we briefly describe the process used to create the relational database that underlies ggMOB; further details can be found at (Seabolt et al., 2020). First, we downloaded whole genome sequences from NCBI’s Reference Sequence Database (RefSeq), which we then filtered to only obtain genomes that were identified as being of bacterial lineage, and as having an assembly level of “Complete Genome”. We added to this set of genomes non-assembled sequence data from NCBI’s Short Read Archive (SRA) by first downloading all datasets that had the following criteria: (1) the data consisted of WGS data generated from bacteria, as defined according to their taxonomic lineage; (2) the data were Illumina short-read sequence data from DNA; and (3) the sequence data were paired-end. Long-read and transcriptomic data were not considered. We note that we downloadeded all the SRA data in FASTQ format using the SRA toolkit (Sugawara and Shumway, 2010), and assembled them into contigs using SPAdes (Bankevich et al., 2012). We discarded any genome assemblies that contained more than 150 contigs (of size >500 bp) or had an N50 of less than 100 kbp. We note that only 48% of bacterial genomes met the aforementioned curation thresholds from the original corpus of SRA datasets. Next, we eliminated any assembled genomes in which a significant proportion of k-mers originated from multiple genomes across different genera. This removed another 13,044 genomes from consideration. This last step ensured all the genomes used for analysis were from a pure single bacterial isolate with valid genus-level classification, hence minimizing the probability of contamination. We obtained a total of 159,628 genome assemblies after filtering for all the above criteria, and a total of 186,887 genomes when including the genome assemblies from RefSeq.
Next, we annotated all the genomes using Prokka 1.12 (Prokka, 2014), resulting in the collation of the genome, gene, and protein data entities into CSV files. After annotation, we determined the protein domains using InterProScan 5.28–67.0 (Quevillon et al., 2005) with all available analyses provided by InterProScan (16). This resulted in 16 JSON files that were then parsed to create a set of CSV files. The annotation process yielded a total of 66,945,714 unique gene sequences; 51,362,178 unique protein sequences; and 138,327,556 unique protein domains along with related functional annotations.
Using the curated data, we created a relational database using IBM’s DB2 system, which contained the following five different entity types: genomes, genes, proteins, protein domains, and functional annotations. These entities were determined by the above data curation, assembly and annotation steps; each entity in the database was stored using the MD5 hash of the sequence itself to create a unique identifier. Thus, we can quickly query for an entity within the database using the unique identifiers as a key. We stored the relations between entities as tables in the relational database, e.g., the genes corresponding to a particular genome in a table. We note this saves storage because unique sequences are stored only once in their respective tables and the tables point to all the parent entities in which they are found. Thus, while database construction required 1468 CPUs, 6TB RAM, and 160 TB of hard drive space, the final relational database scales efficiently with the addition of new sequences (Seabolt et al., 2020).
4.2 Creation of ggMOB
After curation and annotation of the data, we identified all candidate conjugative and cargo proteins in order to create ggMOB. In particular, we used both the primary literature and the InterProScan coding system to generate a list of conjugative features for ggMOB. This led us to consider the following 12 InterProScan codes:
1. IPR005094 describes relaxases and mobilisation proteins, as exemplified by MobA/VirD2 (Pansegrau et al., 1993; Byrd and Matson, 1997; Quevillon et al., 2005).
2. IPR011119 represents a domain found in Proteobacteria annotated as helicase, conjugative relaxase or nickase (Street et al., 2003; Quevillon et al., 2005).
3. IPR014059 codes for a domain in the N-terminal region of a relaxase-helicase (TrwC) that acts in plasmid R388 conjugation. It has been associated with both DNA cleavage and strand transfer activities, and members of this family are frequently found in genomic proximity to conjugative proteins thought to indicate the presence of integrated plasmids when identified in bacterial chromosomes (Quevillon et al., 2005; Boer et al., 2006).
4. IPR014129 represents proteins in the relaxosome complex, exemplified by TraI, which mediates the single-strand nicking and ATP-dependent unwinding of the plasmid molecule via two separate domains in the protein (Matson and Ragonese, 2005; Quevillon et al., 2005).
5. IPR014862 represents a conserved domain found in the relaxosome complex, as exemplified by TrwC (Quevillon et al., 2005; Boer et al., 2006).
6. IPR021300 represents a conserved domain observed in ICE elements in the protein family PFL_4695, and originally identified in Pseudomonas fluorescens Pf-5 (Quevillon et al., 2005; Mavrodi et al., 2009).
7. IPR022303 describes a family of conjugative transfer ATPases representing predicted ATP-binding proteins associated with DNA conjugative transfer. They are found both in plasmids and bacterial chromosomal regions that appear to derive from integrative elements such as conjugative transposons.
8. IPR025955 describes a family of TraC-related proteins observed in Proteobacteria. TraC is a cytoplasmic, membrane protein encoded by the F transfer region of conjugative plasmids, and is required for the assembly of the F pilus structure, which creates and maintains contact between the donor and recipient cells during conjugation. The family includes predicted ATPases associated with DNA conjugative transfer (Schandel et al., 1992; Quevillon et al., 2005).
9. IPR022391 represents the N-terminal domain of proteins associated with conjugative relaxases in the PFGI-1 class, which includes TraI putative relaxases required for ICE function. While these relaxases are similar in function to TraI relaxases of the F plasmid, they have no sequence homology (Quevillon et al., 2005).
10. IPR011952 represents CD-NTase-associated protein 3, a group of proteins that function as part of CBASS (cyclic oligonucleotide-based antiphage signaling system), which provides immunity against bacteriophages (Millman et al., 2020).
11. IPR014136 encompasses TraA, a Ti-type conjugative transfer relaxase that likely nicks the OriT site and unwinds the coiled plasmid prior to conjugative transfer (similar to TraI(F) in this respect) (Harris et al., 1999).
12. IPR020369 represents mobilization protein B (MobB), which is thought to play a role in conjugative exchange by presenting MobA and its covalently-linked plasmid DNA to the conjugative pore for subsequent export (Meyer, 2011).
We supplemented the IPR features with additional conjugative features that provide essential functions in the conjugation process, including conjugative relaxases, nickases, helicases, and other mobilisation proteins (Schandel et al., 1992; Pansegrau et al., 1993; Byrd and Matson, 1997; Boer et al., 2006) (Table 1). These conjugative features contain substantial sequence diversity, (Frost et al., 2005; Wozniak and Waldor, 2010; Perry and Wright, 2013; Johnson and Grossman, 2015; Singer et al., 2016), but also represent conserved domains involved in the machinery required for conjugative transfer. Using standard SQL queries, we obtained a list of the unique identifiers that have one or more of the features described above, resulting in a list of 28,042 candidate conjugation proteins. From this list, we removed those that appeared exactly in only a single genome, which further reduced the list of potential conjugation proteins to 15,398 across 95,781 genomes. We refer to this resulting set of genomes as conjugative genomes because they contain putative conjugation features (Supplementary Table S4). We note that these genomes represent 51% of all genomes in the relational database.
Next, we performed SQL queries in order to identify proteins most likely to be cargo based on evidence of conjugative transfer. To minimize misclassification of proteins as cargo, we applied two rules: (1) the proteins had to be present in at least two genomes with the same conjugative protein; and (2) they could not be present in any non-conjugative genome. To accomplish the query, we first queried the database for all proteins in the 95,781 conjugative genomes, which produced a list of 387,682,038 distinct < conjugative genome accession number, protein > tuples. In many cases a unique sequence was observed in more than one genome, and therefore, in total there were 21,207,794 distinct protein sequences in the set of conjugative genomes. Next, we filtered this list in order to identify the set of proteins that appeared in two or more conjugative genomes. To further reduce false positive identification of transfer by conjugation (vs. being vertically transferred), we discarded any protein that appeared in any of the 99,052 non-conjugative genomes. With this strict selection process, we identified 11,276,651 distinct sequences that we refer to as cargo proteins, i.e., proteins with the greatest evidence of conjugative transfer. Lastly, we tabulated these results to produce a list of triples of the form < cargo protein, conjugative genome A, conjugative genome B> which describe genome A and genome B contained at least one identical conjugative protein sequence, yielding a total of 4,938,737,476 putative transfers. We used this file as input to a custom python script that identifies all intra- and inter-genera cargo protein transfers for each protein by comparing all pairs of genomes in order to identify the intersection of conjugative proteins of each pair of genomes, and the cargo proteins (if any) in this intersection. The output of this script was used to create the ggMOB network, which contains a node for each genus, and an edge between any pair of nodes in which the value of co-occurrence of <conjugative protein, cargo protein> is non-empty (see https://github.com/Ruiz-HCI-Lab/ggMOB for source files and code).
4.3 Additional analyses
4.3.1 Conjugation and cargo gene proximity
To characterize the genomic proximity of conjugative and cargo genes within the conjugative genome pairs, we used our compiled list of genera, genomes, conjugative and cargo proteins, along with Prokka’s accession index, which indicates the position of a gene or protein sequence within the assembled sequences. While this approach was limited by the fact that the order of assembled sequences is unknown, the Prokka index does indicate position of annotated sequences within each assembled sequence; this information was used in a visual display of genomic distance (Figure 2).
4.3.2 Characterizing AMR genes
We identified all AMR proteins by querying our relational database with all sequences in the MEGARes database (Lakin et al., 2016). To obtain consistent annotations, we annotated the sequences in MEGARes in the same format as was used to annotate the set of all proteins in the database. We note that we were able to maintain high confidence that these annotations represent AMR proteins because the annotations were derived by exact sequence matching. Next, we extracted the set of cargo proteins with (self-consistent) names that matched any MEGARes AMR protein name. These data were then used to compute the frequency of observing each AMR gene in genomes that contained and did not contain conjugative features.
4.3.3 NCBI antibiotic susceptibility testing BioSample data
To analyze associations between phenotypic AMR and genomic conjugation features, we retrieved metadata for each NCBI accession that contained antimicrobial susceptibility testing that contained AST data, which include genomic accession number for each isolate, the antibiotic compound(s) against which it was tested, the AST type, and the phenotypic outcome (resistant, susceptible, or intermediate). We only considered those isolates with a resistant phenotypic outcome to be resistant. By linking the BioSample accession with the SRA accessions in our relational database, we were able to identify genomes for which corresponding AST data were available. These genomes were used in our analysis of phenotyic AMR and conjugative features.
4.3.4 Classification of plasmid and conjugation proteins
We note that many of the InterPro codes listed in Table 1 contain conjugative machinery found in both plasmids and ICEs. Since these two groups of MGEs exhibit unique microbial ecological and epidemiological dynamics regarding AMR, we attempted to further annotate conjugative proteins identified within our set of genomes. To accomplish this, we first queried NCBI for bacterial plasmids. All of these assembled bacterial plasmids were downloaded from NCBI and annotated via our Prokka and InterProScan pipelines. The annotated plasmid associated proteins were then placed in a database table. The MD5 hash was used as the primary key for entries in this table, as was true for every sequence entity in the FGP database. Determining whether a protein had been observed on a plasmid in any of the reference genomes was then accomplished by querying for the primary (i.e., FGP) key of each protein within the table of plasmid-associated proteins. If the primary key existed in both tables, we considered that protein to be a plasmid-associated protein. These data were used in the analyses shown in Figure 3.
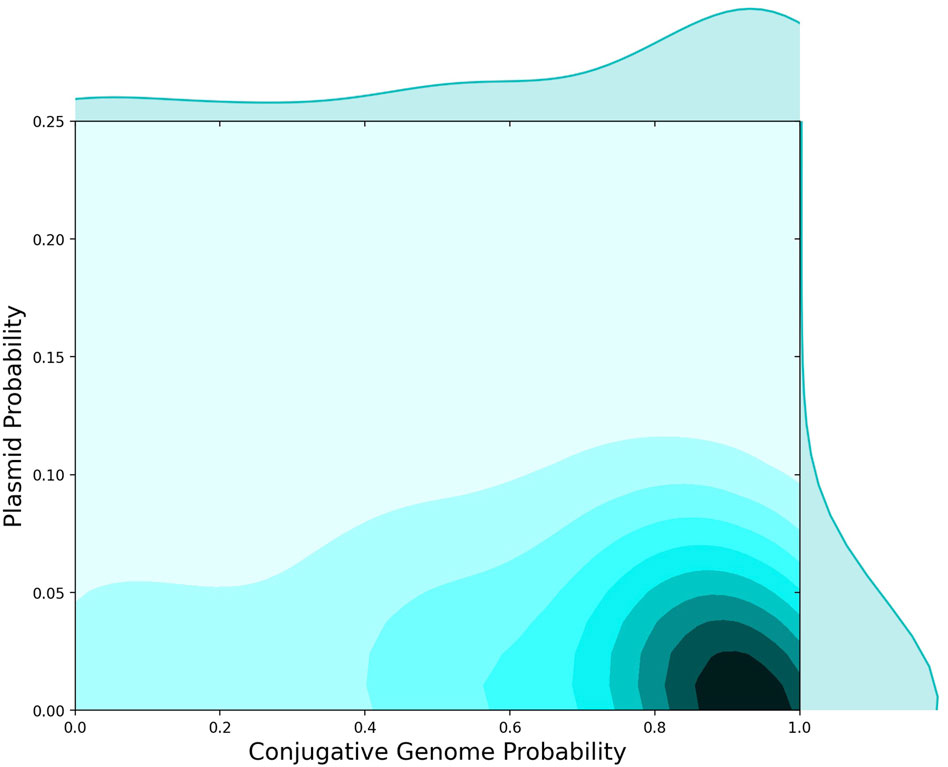
FIGURE 3. AMR Protein Detection in Plasmids versus Conjugative Genomes. 2D histogram showing the probability that AMR proteins were found in conjugative genomes (x-axis) versus the fraction of AMR proteins observed on plasmids (y-axis). Independent of presence on conjugative genomes, 5%–10% of AMR proteins were observed on plasmids, whereas the majority of AMR proteins were found in conjugative genomes.
4.3.5 Tabulation of data
Conjugative genomes were tabulated by genus as shown in Table 2. Observed conjugative features from Table 1 were tabulated by genome for the analysis shown in Figures 1, 5. Similarly, proteins assigned AMR-associated annotations were tabulated by number of conjugative and non-conjugative genomes, and the fraction of unique sequences observed in plasmids was tabulated as well (See Figure 4).

FIGURE 4. Co-occurrence of AMR proteins with conjugative features, for the 138 AMR proteins observed in conjugative genomes with a frequency greater than 5x the frequency of observation in non-conjugative genomes. Of these AMR proteins, 43 are only observed in conjugative genomes.
4.3.6 Hierarchical clustering and co-occurrence
Hierarchical clustering was used to characterize the co-occurrence of proteins or genomes by conjugation feature (Figures 1, 4, 5. The co-occurrence matrix was generated using the Seaborn clustermap algorithm, which performs single linkage clustering to generate heatmaps and dendrograms (Waskom, 2015). The vector of features used to generate the heatmap shown in Figure 2 was the vector of conjugation features for each genome, which in order to compute the (Euclidean) distance between each genome. Again, Seaborn clustermap was used for hierarchical clustering and visualization.
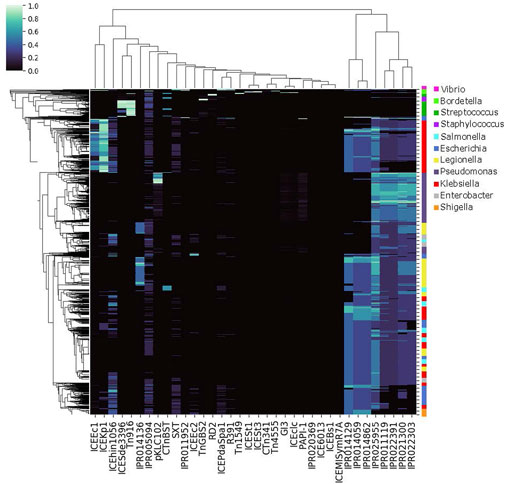
FIGURE 5. Genome-IPR Co-Occurence Map. Co-occurrence of conjugation features by genome, for the 6,000 genomes with the highest cargo protein fraction and the 500 genomes with the rarest conjugation features (see text). Co-occurrence of particular conjugation features does vary by taxonomic group.
Data availability statement
The original contributions presented in the study are included in the article Supplementary Material and at https://github.com/Ruiz-HCI-Lab/ggMOB. Further inquiries can be directed to the corresponding author.
Author contributions
GN, JK, IT, and IS performed and designed research, compiled supporting data, analyzed data, and wrote the paper. GN and AA developed big data visualizations, classified AMR genes and assay data, and contributed to the paper. ES built the database, pipeline, and contributed to the paper. CB provided input on research design, methods and visualizations, and contributed to the paper. NN proposed the study, provided input into the research design, and wrote the paper. JR built the online ggMOB dynamic network and contributed to the paper.
Funding
Financial support for this study was provided by the National Institute of Allergy and Infectious Diseases (NIAID) of the US. National Institutes of Health (NIH), Project No. 1R01AI141810-01 and the National Institute of Food and Agriculture of the United States Department of Agriculture (USDA NIFA), Project No. MINV62-058.
Acknowledgments
The authors would like to acknowledge contributions from M. Roth, H. Krishnareddy, K. Clarkson, and E. Kandogan.
Conflict of interest
ES and AA are currently employed by IBM, GN and JK were employed by IBM at the time of this work. JK is currently employed by Altos Labs.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1024577/full#supplementary-material
Supplementary Figure S1 | Co-occurrence of AMR proteins with conjugative features.Co-occurrence of AMR proteins with conjugative features for the 29 AMR proteins observed in non-conjugative genomes with a frequency at least 5x the frequency of observation in conjugative genomes..
Supplementary Figure S2 | Heatmap of Genome-Genome Distances Based on Conjugation Feature Profiles. Genome-genome Euclidean distance between vectors of conjugation features for the 6500 genomes used in 5. For visualization purposes, 1 in 75 labels were rendered on each axis. The figure shows that different sets of genomes cluster based on different co-occurring conjugation features..
References
Abby, S. S., Tannier, E., Gouy, M., and Daubin, V. (2012). Lateral gene transfer as a support for the tree of life. Proc. Natl. Acad. Sci. U. S. A. 109 (13), 4962–4967. doi:10.1073/pnas.1116871109
Alderliesten, J. B., Duxbury, S. J. N., Zwart, M. P., de Visser, J. A. G. M., Stegeman, A., and Fischer, E. A. J. (2020). Effect of donor-recipient relatedness on the plasmid conjugation frequency: A meta-analysis. BMC Microbiol. 20, 135. doi:10.1186/s12866-020-01825-4
Bacic, M., Parker, A. C., Stagg, J., Whitley, H. P., Wells, W. G., Jacob, L. A., et al. (2005). Genetic and structural analysis of the Bacteroides conjugative transposon CTn341. J. Bacteriol. 187 (8), 2858–2869. doi:10.1128/JB.187.8.2858-2869.2005
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19 (5), 455–477. doi:10.1089/cmb.2012.0021
Beaber, J. W., Hochhut, B., and Waldor, M. K. (2002). Genomic and functional analyses of SXT, an integrating antibiotic resistance gene transfer element derived from Vibrio cholerae. J. Bacteriol. 184 (15), 4259–4269. doi:10.1128/jb.184.15.4259-4269.2002
Bellanger, X., Payot, S., Leblond-Bourget, N., and Guédon, G. (2014). Conjugative and mobilizable genomic islands in bacteria: Evolution and diversity. FEMS Microbiol. Rev. 38 (4), 720–760. doi:10.1111/1574-6976.12058
Bellanger, X., Roberts, A. P., Morel, C., Choulet, F., Pavlovic, G., Mullany, P., et al. (2009). Conjugative transfer of the integrative conjugative elements ICESt1 and ICESt3 from Streptococcus thermophilus. J. Bacteriol. 191 (8), 2764–2775. doi:10.1128/JB.01412-08
Boer, R., Russi, S., Guasch, A., Lucas, M., Blanco, A. G., Pérez-Luque, R., et al. (2006). Unveiling the molecular mechanism of a conjugative relaxase: The structure of TrwC complexed with a 27-mer DNA comprising the recognition hairpin and the cleavage site. J. Mol. Biol. 358 (3), 857–869. doi:10.1016/j.jmb.2006.02.018
Böltner, D., MacMahon, C., Pembroke, J. T., Strike, P., and Osborn, A. M. (2002). R391: A conjugative integrating mosaic comprised of phage, plasmid, and transposon elements. J. Bacteriol. 184 (18), 5158–5169. doi:10.1128/jb.184.18.5158-5169.2002
Botelho, J., Mourao, J., Roberts, A. P., and Peixe, L. (2020). Comprehensive genome data analysis establishes a triple whammy of carbapenemases, ICEs and multiple clinically relevant bacteria. Microb. Genom. 6. doi:10.1099/mgen.0.000424
Burrus, V., Marrero, J., and Waldor, M. K. (2006). The current ICE age: Biology and evolution of SXT-related integrating conjugative elements. Plasmid 55 (3), 173–183. doi:10.1016/j.plasmid.2006.01.001
Burrus, V., Pavlovic, G., Decaris, B., and Guédon, G. (2002). The ICESt1 element of Streptococcus thermophilus belongs to a large family of integrative and conjugative elements that exchange modules and change their specificity of integration. Plasmid 48 (2), 77–97. doi:10.1016/s0147-619x(02)00102-6
Byrd, D. R., and Matson, S. W. (1997). Nicking by transesterification: The reaction catalysed by a relaxase. Mol. Microbiol. 25 (6), 1011–1022. doi:10.1046/j.1365-2958.1997.5241885.x
Chan, A. P., Sutton, G., DePew, J., Krishnakumar, R., Choi, Y., Huang, X. Z., et al. (2015). A novel method of consensus pan-chromosome assembly and large-scale comparative analysis reveal the highly flexible pan-genome of Acinetobacter baumannii. Genome Biol. 16, 143. doi:10.1186/s13059-015-0701-6
Clewell, D. B., Flannagan, S. E., and Jaworski, D. D. (1995). Unconstrained bacterial promiscuity: The tn916–tn1545 family of conjugative transposons. Trends Microbiol. 3 (6), 229–236. doi:10.1016/s0966-842x(00)88930-1
Cohen, O., Gophna, U., and Pupko, T. (2010). The complexity hypothesis revisited: Connectivity rather than function constitutes a barrier to horizontal gene transfer. Mol. Biol. Evol. 28 (4), 1481–1489. doi:10.1093/molbev/msq333
Cury, J., Oliveira, P. H., de la Cruz, F., and Rocha, E. P. C. (2018). Host range and genetic plasticity explain the coexistence of integrative and extrachromosomal mobile genetic elements. Mol. Biol. Evol. 35 (9), 2850–2239. doi:10.1093/molbev/msy182
Cury, J., Touchon, M., and Rocha, E. P. C. (2017). Integrative and conjugative elements and their hosts: Composition, distribution and organization. Nucleic Acids Res. 45 (15), 8943–8956. doi:10.1093/nar/gkx607
Delavat, F., Miyazaki, R., Carraro, N., Pradervand, N., and van der Meer, J. R. (2017). The hidden life of integrative and conjugative elements. FEMS Microbiol. Rev. 41 (4), 512–537. doi:10.1093/femsre/fux008
Everitt, R. G., Didelot, X., Batty, E. M., Miller, R. R., Knox, K., Young, B. C., et al. (2014). Mobile elements drive recombination hotspots in the core genome of Staphylococcus aureus. Nat. Commun. 5, 3956. doi:10.1038/ncomms4956
Finn, R. D., Attwood, T. K., Babbitt, P. C., Bateman, A., Bork, P., Bridge, A. J., et al. (2016). Interpro in 2017 – beyond protein family and domain annotations. Nucleic Acids Res. 45 (D1), D190-D199. doi:10.1093/nar/gkw1107
Frost, L. S., Leplae, R., Summers, A. O., and Toussaint, A. (2005). Mobile genetic elements: The agents of open source evolution. Nat. Rev. Microbiol. 3 (9), 722–732. doi:10.1038/nrmicro1235
Garnier, F., Taourit, S., Glaser, P., Courvalin, P., and Galimand, M. (2000). Characterization of transposon Tn1549, conferring VanB-type resistance in Enterococcus spp. Microbiology 146 (6), 1481–1489. doi:10.1099/00221287-146-6-1481
Gaston, J. M., Zhao, S., Poyet, M., Groussin, M., et al. (2021). An omics-based framework for assessing the health risk of antimicrobial resistance genes. Nat. Commun. 12, 4765. doi:10.1038/s41467-021-25096-3
Guglielmini, J., Quintais, L., Garcillán-Barcia, M. P., de La Cruz, F., and Rocha, E. P. C. (2011). The repertoire of ICE in prokaryotes underscores the unity, diversity, and ubiquity of conjugation. PLoS Genet. 7 (8), e1002222. doi:10.1371/journal.pgen.1002222
Harris, R. L., Sholl, K. A., Conrad, M. N., Dresser, M. E., and Silverman, P. M. (1999). Interaction between the F plasmid TraA (F-pilin) and TraQ proteins. Mol. Microbiol. 34 (4), 780–791. doi:10.1046/j.1365-2958.1999.01640.x
Johnson, C. M., and Grossman, A. D. (2015). Integrative and conjugative elements (ICEs): What they do and how they work. Annu. Rev. Genet. 49, 577–601. doi:10.1146/annurev-genet-112414-055018
Klockgether, J., Reva, O., Larbig, K., and Tümmler, B. (2004). Sequence analysis of the mobile genome island pKLC102 of Pseudomonas aeruginosa C. J. Bacteriol. 186 (2), 518–534. doi:10.1128/jb.186.2.518-534.2004
Koraimann, G., and Wagner, M. A. (2014). Social behavior and decision making in bacterial conjugation. Front. Cell.. Infect. Microbiol. 4, 54. doi:10.3389/fcimb.2014.00054
Lakin, S. M., Dean, C., Noyes, N. R., Dettenwanger, A., Ross, A. S., Doster, E., et al. (2016). MEGARes: An antimicrobial resistance database for high throughput sequencing. Nucleic Acids Res. 45 (D1), D574-D580–D580. doi:10.1093/nar/gkw1009
Lechner, M., Schmitt, K., Bauer, S., Hot, D., Hubans, C., Levillain, E., et al. (2009). Genomic island excisions in Bordetella petrii. BMC Microbiol. 9 (1), 141. doi:10.1186/1471-2180-9-141
Lee, C. A., Thomas, J., and Grossman, A. D. (2012). The Bacillus subtilis conjugative transposon ICEBs1 mobilizes plasmids lacking dedicated mobilization functions. J. Bacteriol. 194 (12), 3165–3172. doi:10.1128/JB.00301-12
Logan, L. K., and Weinstein, R. A. (2017). The epidemiology of carbapenem-resistant Enterobacteriaceae: The impact and evolution of a global menace. J. Infect. Dis. 215 (1), S28–S36. S28–S36. doi:10.1093/infdis/jiw282
Lopatkin, A. J., and Collin, J. J. (2020). Predictive biology: Modelling, understanding and harnessing microbial complexity. Nat. Rev. Microbiol. 18, 507–520. doi:10.1038/s41579-020-0372-5
Martínez, J. L., Coque, T. M., and Baquero, F. (2015). What is a resistance gene? Ranking risk in resistomes. Nat. Rev. Microbiol. 13 (2), 116–123. doi:10.1038/nrmicro3399
Matson, S. W., and Ragonese, H. (2005). The F-plasmid TraI protein contains three functional domains required for conjugative DNA strand transfer. J. Bacteriol. 187 (2), 697–706. doi:10.1128/JB.187.2.697-706.2005
Mavrodi, D. V., Loper, J. E., Paulsen, I. T., and Thomashow, L. S. (2009). Mobile genetic elements in the genome of the beneficial rhizobacterium Pseudomonas fluorescens Pf-5. BMC Microbiol. 9 (1), 8. doi:10.1186/1471-2180-9-8
Meyer, R. (2011). Functional organization of MobB, a small protein required for efficient conjugal transfer of plasmid R1162. J. Bacteriol. 193 (15), 3904–3911. doi:10.1128/JB.05084-11
Millman, A., Melamed, S., Amitai, G., and Sorek, R. (2020). Diversity and classification of cyclic-oligonucleotide-based anti-phage signalling systems. Nat. Microbiol. 5 (12), 1608–1615. doi:10.1038/s41564-020-0777-y
Mohd-Zain, Z., Turner, S. L., Cerdeno-Tárraga, A. M., Lilley, A. K., Inzana, T. J., Duncan, A. J., et al. (2004). Transferable antibiotic resistance elements in Haemophilus influenzae share a common evolutionary origin with a diverse family of syntenic genomic islands. J. Bacteriol. 186 (23), 8114–8122. doi:10.1128/JB.186.23.8114-8122.2004
Obi, C. C., Vayla, S., De Gannes, V., Berres, M. E., Walker, J., Pavelec, D., et al. (2018). The integrative conjugative element clc (ICEclc) of Pseudomonas aeruginosa JB2. Front. Microbiol. 9, 1532. doi:10.3389/fmicb.2018.01532
Oh, M., Pruden, A., Chen, C., Heath, L. S., Xia, K., and Zhang, L. (2018). MetaCompare: A computational pipeline for prioritizing environmental resistome risk. FEMS Microbiol. Ecol. 94 (6), fiy079. doi:10.1093/femsec/fiy079
Osorio, C. R., Marrero, J., Wozniak, R. A. F., Lemos, M. L., Burrus, V., and Waldor, M. K. (2008). Genomic and functional analysis of ICEPdaSpa1, a fish-pathogen-derived SXT-related integrating conjugative element that can mobilize a virulence plasmid. J. Bacteriol. 190 (9), 3353–3361. doi:10.1128/JB.00109-08
Paauw, A., Leverstein-van Hall, M. A., Verhoef, J., and Fluit, A. C. (2010). Evolution in quantum leaps: Multiple combinatorial transfers of HPI and other genetic modules in Enterobacteriaceae. PLoS ONE 5 (1), e8662. doi:10.1371/journal.pone.0008662
Pansegrau, W., Schoumacher, F., Hohn, B., and Lanka, E. (1993). Site-specific cleavage and joining of single-stranded DNA by VirD2 protein of agrobacterium tumefaciens Ti plasmids: Analogy to bacterial conjugation. Proc. Natl. Acad. Sci. U. S. A. 90 (24), 11538–11542. doi:10.1073/pnas.90.24.11538
Parker, B. J., Wearsch, P. A., Veloo, A. C. M., and Rodriguez-Palacios, A. (2020). The genus Alistipes: Gut bacteria with emerging implications to inflammation, cancer, and mental health. Front. Immunol. 11, 906. doi:10.3389/fimmu.2020.00906
Partridge, S. R., Kwong, S. M., Firth, N., and Jensen, S. O. (2018). Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. 31. doi:10.1128/cmr.00088-17
Peed, L., Parker, A. C., and Smith, C. J. (2010). Genetic and functional analyses of the mob operon on conjugative transposon CTn341 from Bacteroides spp. J. Bacteriol. 192 (18), 4643–4650. doi:10.1128/JB.00317-10
Perry, J., and Wright, G. (2013). The antibiotic resistance “mobilome”: Searching for the link between environment and clinic. Front. Microbiol. 4, 138. doi:10.3389/fmicb.2013.00138
Prokka, T. S. (2014). Prokka: Rapid prokaryotic genome annotation. Bioinformatics 30 (14), 2068–2069. doi:10.1093/bioinformatics/btu153
Qiu, X., Gurkar, A. U., and Lory, S. (2006). Interstrain transfer of the large pathogenicity island (PAPI-1) of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 103 (52), 19830–19835. doi:10.1073/pnas.0606810104
Quevillon, E., SilVentoinen, V., Pillai, S., HarteN., , MulderN., , ApweileR, R., et al. (2005). InterProScan: Protein domains identifier. Nucleic Acids Res. 33 (2), W116–W120. –W120. doi:10.1093/nar/gki442
Ramsay, J. P., and Ronson, C. W. (2015). Genetic regulation of symbiosis island transfer in Mesorhizobium loti. New York, NY: John Wiley & Sons, 219.
Roberts, A. P., and Mullany, P. (2011). Tn916-like genetic elements: A diverse group of modular mobile elements conferring antibiotic resistance. FEMS Microbiol. Rev. 35 (5), 856–871. doi:10.1111/j.1574-6976.2011.00283.x
Roche, D., Flechard, M., Lallier, N., Reperant, M., Bree, A., Pascal, G., et al. (2010). ICEEc2, a new integrative and conjugative element belonging to the pKLC102/PAGI-2 family, identified in Escherichia coli strain BEN374. J. Bacteriol. 192 (19), 5026–5036. doi:10.1128/JB.00609-10
Neil, K., and Allard, N. (2021). Molecular mechanisms influencing bacterial conjugation in the intestinal microbiota. Front. Microbiol. 12, 673260. doi:10.3389/fmicb.2021.673260
Schandel, K. A., Muller, M. M., and Webster, R. E. (1992). Localization of TraC, a protein involved in assembly of the F conjugative pilus. J. Bacteriol. 174 (11), 3800–3806. doi:10.1128/jb.174.11.3800-3806.1992
Seabolt, E., Nayar, G., Krishnareddy, H., Agarwal, A., Beck, K. L., Terrizzano, I., et al. (2020). Functional genomics platform, A cloud-based platform for studying microbial life at scale. IEEE/ACM Trans. Comput. Biol. Bioinform. 1 (–1), 940–952. doi:10.1109/TCBB.2020.3021231
Sheppard, R. J., Beddis, A. E., and Barraclough, T. G. (2020). The role of hosts, plasmids and environment in determining plasmid transfer rates: A meta-analysis. Plasmid 108, 102489. doi:10.1016/j.plasmid.2020.102489
Singer, A. C., Shaw, H., Rhodes, V., and Hart, A. (2016). Review of antimicrobial resistance in the environment and its relevance to environmental regulators. Front. Microbiol. 7, 1728. doi:10.3389/fmicb.2016.01728
Sitkiewicz, I., Green, N. M., Guo, N., Mereghetti, L., and Musser, J. M. (2011). Lateral gene transfer of streptococcal ICE element RD2 (region of difference 2) encoding secreted proteins. BMC Microbiol. 11 (1), 65. doi:10.1186/1471-2180-11-65
Smith, C. J., and Parker, A. C. (1993). Identification of a circular intermediate in the transfer and transposition of Tn4555, a mobilizable transposon from Bacteroides spp. J. Bacteriol. 175 (9), 2682–2691. doi:10.1128/jb.175.9.2682-2691.1993
Smyth, D. J., Shera, J., Bauer, M. J., Cameron, A., McNeilly, C. L., Sriprakash, K. S., et al. (2014). Conjugative transfer of ICE Sde 3396 between three β-hemolytic streptococcal species. BMC Res. Notes 7 (1), 521. doi:10.1186/1756-0500-7-521
Smyth, D. S., and Robinson, D. A. (2009). Integrative and sequence characteristics of a novel genetic element, ICE6013, in Staphylococcus aureus. J. Bacteriol. 191 (19), 5964–5975. doi:10.1128/JB.00352-09
Song, B., Shoemaker, N. B., Gardner, J. F., and Salyers, A. A. (2007). Integration site selection by the Bacteroides conjugative transposon CTnBST. J. Bacteriol. 189 (18), 6594–6601. doi:10.1128/JB.00668-07
Springael, D., Kreps, S., and Mergeay, M. (1993). Identification of a catabolic transposon, Tn4371, carrying biphenyl and 4-chlorobiphenyl degradation genes in Alcaligenes eutrophus A5. J. Bacteriol. 175 (6), 1674–1681. doi:10.1128/jb.175.6.1674-1681.1993
Street, L. M., Harley, M. J., Stern, J. C., Larkin, C., Williams, S. L., Miller, D. L., et al. (2003). Subdomain organization and catalytic residues of the F factor TraI relaxase domain. Biochim. Biophys. Acta 1646 (1-2), 86–99. doi:10.1016/s1570-9639(02)00553-8
Su, Y., He, P., and Clewell, D. B. (1992). Characterization of the tet (M) determinant of Tn916: Evidence for regulation by transcription attenuation. Antimicrob. Agents Chemother. 36 (4), 769–778. doi:10.1128/aac.36.4.769
Sugawara, H., and Shumway, M. (2010). The sequence read archive. Nucleic Acids Res. 39 (1), D19–D21. doi:10.1093/nar/gkq1019
Uhlemann, A. C. L., Aj Prensky, H., and Gomez-Simmonds, A. (2021). Conjugation dynamics depend on both the plasmid acquisition cost and the fitness cost. Mol. Syst. Biol. 17, e9913. doi:10.15252/msb.20209913
Waldor, M. K., Beaber, J. W., and Hochhut, B. (2004). SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature 427, 72–74. doi:10.1038/nature02241
Wang, R., van Dorp, L., Shaw, L. P., Bradley, P., Wang, Q., Wang, X., et al. (2018). The global distribution and spread of the mobilized colistin resistance gene mcr-1. Nat. Commun. 9 (1), 1179–9. doi:10.1038/s41467-018-03205-z
Waskom, M., Botvinnik, O., Hobson, P., Warmenhoven, J., Cole, J. B., Halchenko, Y., et al. (2015). Seaborn: V0. 6.0 (June 2015). Zenodo. doi:10.5281/zenodo.19108
Wiedenbeck, J., and Cohan, F. M. (2011). Origins of bacterial diversity through horizontal genetic transfer and adaptation to new ecological niches. FEMS Microbiol. Rev. 35 (5), 957–976. doi:10.1111/j.1574-6976.2011.00292.x
Keywords: antimicobial resistance, genomics, metagenomics, big data, mobile genetic elements (MGE)
Citation: Nayar G, Terrizzano I, Seabolt E, Agarwal A, Boucher C, Ruiz J, Slizovskiy IB, Kaufman JH and Noyes NR (2022) ggMOB: Elucidation of genomic conjugative features and associated cargo genes across bacterial genera using genus-genus mobilization networks. Front. Genet. 13:1024577. doi: 10.3389/fgene.2022.1024577
Received: 21 August 2022; Accepted: 25 October 2022;
Published: 08 December 2022.
Edited by:
Marco Pellegrini, National Research Council (CNR), ItalyReviewed by:
Ahmed Moustafa, American University in Cairo, EgyptDaljeet Singh Dhanjal, Lovely Professional University, India
Copyright © 2022 Nayar, Terrizzano, Seabolt, Agarwal, Boucher, Ruiz, Slizovskiy, Kaufman and Noyes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Noelle R. Noyes, bm5veWVzQHVtbi5lZHU=