 Ziye Zhao
Ziye Zhao Wen Yang
Wen Yang Yixiao Zhai
Yixiao Zhai Yingjian Liang
Yingjian Liang Yuming Zhao
Yuming Zhao- 1College of Information and Computer Engineering, Northeast Forestry University, Harbin, China
- 2International Medical Center, Shenzhen University General Hospital, Shenzhen, China
- 3Department of Obstetrics and Gynecology, The First Affiliated Hospital of Harbin Medical University, Harbin, China
The exploration of DNA-binding proteins (DBPs) is an important aspect of studying biological life activities. Research on life activities requires the support of scientific research results on DBPs. The decline in many life activities is closely related to DBPs. Generally, the detection method for identifying DBPs is achieved through biochemical experiments. This method is inefficient and requires considerable manpower, material resources and time. At present, several computational approaches have been developed to detect DBPs, among which machine learning (ML) algorithm-based computational techniques have shown excellent performance. In our experiments, our method uses fewer features and simpler recognition methods than other methods and simultaneously obtains satisfactory results. First, we use six feature extraction methods to extract sequence features from the same group of DBPs. Then, this feature information is spliced together, and the data are standardized. Finally, the extreme gradient boosting (XGBoost) model is used to construct an effective predictive model. Compared with other excellent methods, our proposed method has achieved better results. The accuracy achieved by our method is 78.26% for PDB2272 and 85.48% for PDB186. The accuracy of the experimental results achieved by our strategy is similar to that of previous detection methods.
Introduction
Organisms contain many macromolecular substances, such as DNA and proteins, which contain the genetic information of organisms and are important components of all cells and tissues that make up an organism. To study the life activities of cells, it is necessary to study DNA and proteins and the interaction between them. Research on DBPs has an extremely important status and significance in related life sciences and plays an important role in DNA replication and recombination, virus infection and proliferation. It is necessary to study the combination of DNA and protein to study the gene expression of organisms at the molecular level. Researchers are paying increasing attention to DBP studies. DBPs are a kind of protein that binds to DNA, and it is critical to determine which of the numerous proteins can attach to DNA (Liu et al., 2019a; Li et al., 2019; Li et al., 2020) However, the traditional use of biochemical methods to find DBP consumes considerable time and money. Based on the above requirements and the development of computer science and ML(Zheng et al., 2019; Zheng et al., 2020; Wang et al., 2021a), relevant researchers have developed many detection methods based on ML algorithms in the hopes of improving the efficiency of detecting DBP and saving manpower and material resources.
ML is frequently utilized in the fields of computational biology (Jiang et al., 2013a; Cheng et al., 2019a; Liu et al., 2019b; Wang et al., 2019; Liu et al., 2020a; Tao et al., 2020a; Wang et al., 2020a; Zhang et al., 2020a; Zhao et al., 2020a; Zhu et al., 2020; Wang et al., 2021b; Wang et al., 2021c; Dao et al., 2021; Yu et al., 2021) to analyze brain disease (Liu et al., 2018a; Cheng et al., 2019b; Bi et al., 2020; Iqubal et al., 2020; Zhang et al., 2021a), lncRNA-miRNA interactions (Cheng et al., 2016; Liu et al., 2020b; Han et al., 2021), protein remote homology (Hong et al., 2020), protein functions (Wei et al., 2018a; Shen et al., 2019a; Shen et al., 2019b; Ding et al., 2019; Wang et al., 2020b; Shen et al., 2020; Tang et al., 2020; Wang et al., 2021d; Shang et al., 2021; Shao and Liu, 2021; Zhao et al., 2021), electron transport proteins (Ru et al., 2019), differential expression (Yu et al., 2020a; Zhao et al., 2020b; Zhai et al., 2020) and protein-protein interconnections (Ding et al., 2016a; Ding et al., 2016b; Yu et al., 2020b).
The protein sequence is very sizeable, and its number far exceeds the number of structures known to researchers (Zuo et al., 2017). Therefore, ML is used in various computer programs that predict DBP. The model IDNA-Prot|dis (Liu et al., 2014) was proposed by Liu et al. and is used to detect DBP based on the pseudo amino acid composition (PseAAC), and it can accurately extract the characteristics of DNA binding proteins. There are two models that use PseACC and physical-chemical distance transformation and support vector machine (SVM) algorithms, named PseDNA-Pro (Liu et al., 2015a) and iDNAPro-PseAAC (Liu et al., 2015b). Lin et al. developed the IDNA-Prot (Lin et al., 2011) prediction model based on the random forest (RF) algorithm through the PseACC feature. Kummar et al. developed two models based on RF and SVM classifiers called DNA-Prot (Kumar et al., 2009) and DNAbinder (Kumar et al., 2007). Dong et al. proposed the Kmer1+ACC (Liu et al., 2016) model based on the SVM algorithms Kmer composition and autocross covariance transformation. The position-specific scoring matrix (PSSM) can be obtained by calculating the protein sequence’s position frequency matrix, which has evolutionary information on the protein (Shao et al., 2021). The Local-DPP (Wei et al., 2017) uses the local pseudo position-specific scoring matrix (Pse-PSSM) and random forest algorithm to detect DBPs. Multiple kernel SVM is a DBP predictor from heuristically kernel alignment, and it is also named MKSVM-HKA (Ding et al., 2020a), which includes a variety of characteristics and was developed by Ding et al. The MSFBinder (Liu et al., 2018b) model proposed by Liu et al. is based on multiview features as well as classifiers. DPP-PseAAC (Rahman et al., 2018) is a model based on Chou’s general PseAAC, and it is used to detect DBPs. Methods have also been developed that combine multiscale features and deep neural networks to predict DBPs, such as MsDBP (Du et al., 2019).Adilina et al. (2019) analyzed protein sequence characteristics and implemented two different feature selection methods to build a DBP predictor.
In recent years, an increasing number of researchers have adopted complex feature extraction methods (Fu et al., 2020; Jin et al., 2021) and classification models to identify DBPs. It is critical to develop a method that uses as few DBP features as possible and includes a simple classification model while also ensuring a good ability to detect DPB. According to previous work, we proposed a DBP identification method based on the XGBoost model. First, several features were extracted from the protein sequence. Second, the features of these sequences were spliced. Third, the dimension of the data was standardized and reduced. Finally, the XGBoost model was used to detect DBPs. We have evaluated the effectiveness of our method on some benchmark data sets. Compared with some current experimental methods, our method achieves a better Matthew’s correlation coefficient (MCC), with a value of 0.713 for PDB186 and 0.5652 for PDB2272.
Methods
Identifying DBPs is a common dichotomy problem. First, we used six different feature extraction models for DBPs sequences to extract the corresponding sequence feature information. Then, the sequence feature information was spliced. Next, dimensionality reduction was performed on the spliced sequence feature information. Finally, the XGBoost model was utilized to identify DBPs. Figure 1 depicts the flowchart of our adopted technique.
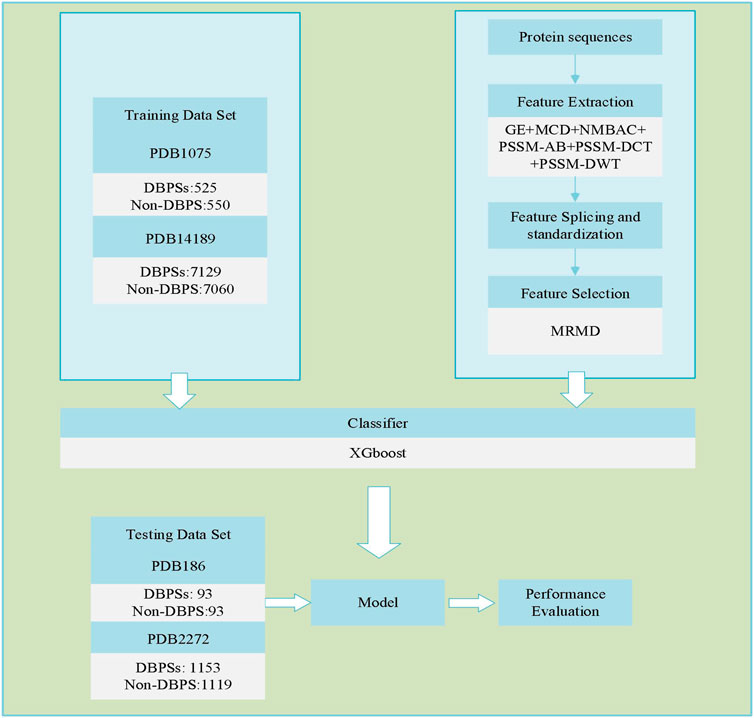
FIGURE 1. Process of predicting DBPs.
Extracting Features
To recognize DBPs, the corresponding features must be extracted. We adopt six feature extraction methods to obtain sequence information: global encoding, GE (Li et al., 2009); multi-scale continuous as well as discontinuous descriptor, MCD (You et al., 2014); normalized Moreau-Broto auto correlation, NMBAC (Ding et al., 2016b; Feng and Zhang, 2000); position specific scoring matrix-based average blocks, PSSM-AB (Jeong et al., 2011; Zhu et al., 2019); PSSM-based discrete cosine transform, PSSM-DCT (Huang et al., 2015); and PSSM-based discrete wavelet transform, PSSM-DWT (Nanni et al., 2012). The abovementioned feature extraction models are all well-known protein sequence extraction algorithm s and commonly used, which could be described in related works (Zou et al., 2021). Table 1 shows the feature dimensions derived by various feature extraction methods. After completing the above work, we used MATLAB to horizontally stitch together (Ding et al., 2020c; Ding et al., 2020d; Yang et al., 2021a) the features extracted from the same protein sequence using different feature extraction methods. The spliced features are represented by

TABLE 1. Dimensional information about the features.
Standardize the Data
To make the data more standardized and unified and to strengthen the relationship between the characteristics of the data and the labels of the data, we use Z-score standardization to process the data.
Z-score standardization is defined as follows:
where N is the total number of samples and
The DBP sequence was processed in three stages: feature extraction, feature information splicing, and data standardization. Following the aforementioned three stages, we can obtain the sequence feature information
Dimensionality Reduction by Max-Relevance-Max-Distance
Zou et al. (Quan et al., 2016; Niu et al., 2020) developed a dimensionality reduction method in 2015 named Max-Relevance-Max-Distance (MRMD), and the user guide and complete runtime program can be obtained and downloaded from the following URL: https://github.com/heshida01/MRMD3.0. It judges data independence through a distance function and completes the dimensionality reduction operation in three steps (Tao et al., 2020b). It first evaluates each feature’s contribution to the classification and then quantifies each feature’s contribution to the classification. Second, the weights of different features are calculated for classification and the selected features are sorted accordingly. Third, the different numbers of features are filtered and classified and the results are recorded. We analyze and compare the results of the previous step to select the most effective group and use the sequence features chosen from this group as the result of dimensionality reduction.
The maximum correlation and the maximum distance are the main bases for the MRMD algorithm to judge the weight of each feature to the prediction result. The Pearson correlation coefficient can be used to quantify the degree of correlation between features and cases, and it can be calculated by the maximum relevance (MR).
The Pearson correlation coefficient is defined as follows:
The ith characteristic from the sequence and the category label to which those sequences belong make up the vectors X and Y. The maximum distance (MD) is used to assess feature redundancy. We calculate the three indices between characteristics in total.
Equations 3A, E3B, E3C represent Euclidean distance, cosine similarity and Tanimoto coefficient, respectively. We can obtain the MD value by calculating the three indicators. Finally, the classification contribution value of each feature is calculated by combining MR and MD in a specific ratio.
After dimensionality reduction, the dimensions of PDB14189 and PDB2272 are 379, and the dimensions of PDB1075 and PDB186 are 1460.
Based on the three steps of feature extraction and splicing, data standardization and dimensionality reduction operations, we obtain the final sequence features.
Extreme Gradient Boosting Algorithm
In 2011, Tianqi Chen and Carlos Guestrin (Chen and Guestrin, 2016) first proposed the XGBoost algorithm, or the extreme gradient boosting algorithm. It is a machine learning model that achieves a stronger learning effect by integrating multiple weak learners. The XGBoost model has many advantages, such as strong flexibility and scalability (Yang et al., 2021b; Zhang et al., 2021b).
Generally, most boosting tree models have difficulty implementing distributed training because when training nth trees, they will be affected by the residuals of the first n-1 trees and only use first-order derivative information. The XGBoost model is different. It performs a second-order Taylor expansion of the loss function and uses a variety of methods to prevent overfitting as much as possible. XGBoost can also automatically use the CPU’s multithreaded parallel computing to speed up the running speed. This feature represents a great advantage of XGBoost over other methods. XGBoost has improved significantly in terms of effect and performance.
The XGBoost algorithm is described in detail as follows:
where M is the number of trees and F represents the basic model of the trees.
The objective function is defined as follows:
The error between the predicted value and the true value is represented by the loss function l, and the regularized function
where the weight and number of leaves of each tree are represented by
After performing the quadratic Taylor expansion on the objective function, the information gain generated after each split of the objective function can be expressed as follows:
We can see that the split threshold
XGBoost also has the following two features:
1. Splitting stops when the threshold is greater than the weight of all samples on the leaf node too prevent the model from learning special training samples.
2. The features are randomly sampled when constructing each tree.
These features can prevent the XGBoost model from overfitting during the experiment.
Experimental Results
In this chapter, we obtain experimental results through experiments on four benchmark data sets, evaluate our methods of identifying DBP and compare our experimental results with that of other methods.
Data Sets
The four benchmark data sets are PDB1075, PDB186, PDB14189, and PDB2272. Liu et al. (2015a) and Lou et al. (2014) provided PDB1075 (training set) and PDB186 (independent testing set), respectively, and Du et al. (2019) provided PDB14189 (training set) and PDB2272 (independent testing set). These data sets are from the Protein Data Bank (PDB), and Table 2 shows the results of their detailed information.

TABLE 2. Basic information about four standard data sets.
Measurement Standard
In this research, the following coefficients are used to evaluate our method: specificity (SP), sensitivity (SN), Matthew correlation coefficient (MCC), accuracy (ACC) and area under the ROC curve (AUC) (Jiang et al., 2013b; Wei et al., 2014; Wei et al., 2018a; Wei et al., 2018b; Cheng et al., 2018; Jin et al., 2019; Zhang et al., 2020b; Cheng et al., 2020; Liu et al., 2020c; Wang et al., 2020c; Guo et al., 2020; Huang et al., 2020; Wei et al., 2020; Zeng et al., 2020; Zhai et al., 2020). The calculation formulas for these coefficients are as follows:
Among them, TN, TP, FP and FN reflect the values of true negatives, true positives, false positives, and false negatives, respectively.
Performance Analysis
On the PDB 1075 data set, the performance of the spliced sequence features and single sequence features is evaluated by randomly extracting 30% of the data as a test set. Figure 2; Table 3 depict the experimental outcomes. PSSM-DWT (MCC: 0.4981) achieved better performance than other single sequence features. The spliced sequence features perform better than the single sequence feature on all parameters. The spliced sequence feature (ROC: 0.81) also gained the best ROC performance.
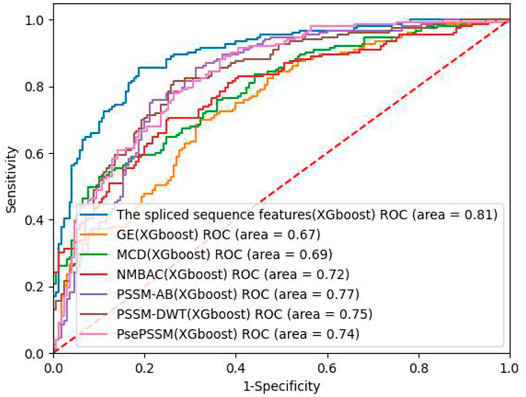
FIGURE 2. ROC curves of different feature extraction methods on PDB1075 data.
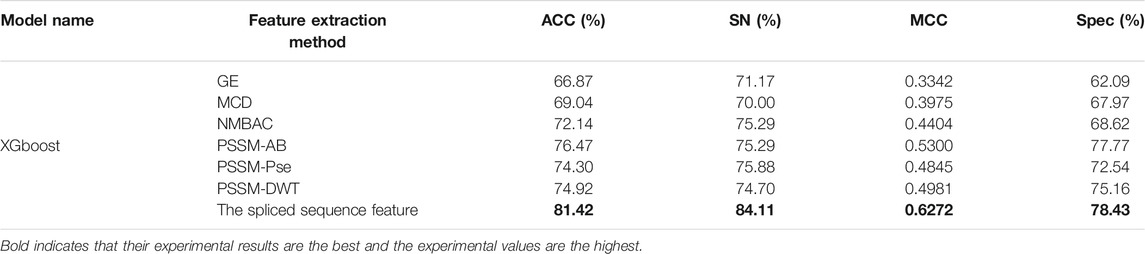
TABLE 3. Performance of PDB1075 using different feature extraction methods in XGBoost.
Independent Data Set of PDB186
In this experiment, different sequence features have different prediction performances. We use PDB1075 as the training set and PDB186 as the test set to evaluate our experimental method and compared the experimental findings of our approach to those of 13 other methods. Table 4 clearly shows the complete experimental outcomes.
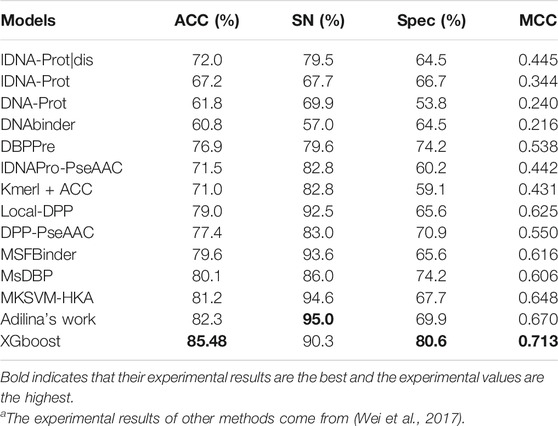
TABLE 4. Comparison between the XGBoost model and other methods on the PDB186 data set.
The MCC values of the five methods are all above 0.6 for MSDBP, MSFBinder, Local-DPP MKSVM-HKA, and Adilina’s work (0.606, 0.616, 0.625, 0.648 and 0.670, respectively). Thus, these methods have excellent performance. Although Adilina’s work (SN: 95.0%) performs best in terms of the value of SN, the results of XGBoost achieve optimal ACC (85.48%), MCC (0.713) and Spec (80.6%). On PDB1075 and PDB186, XGBoost outperforms the other methods.
Independent Data Set of PDB2272
Du et al. (2019) removed proteins in PDB2272 that shared more than 40% of their sequence with PDB14189 to avoid homology bias between the two data sets. We conducted experiments on Du’s data set to verify the performance of the XGBoost model. PDB14189 is the training set, and PDB2272 is the test set. We independently tested XGBoost on PDB2272, used PDB14189 as the training set and compared it with five other classification methods. The detailed experimental results can be seen in Table 5. The results clearly show that XGBoost achieves the best ACC, MCC and Spec values of 78.26%, 0.5652 and 76.05%, respectively, compared with the other methods. For PDB2272, XGBoost presents a superior performance relative to the other classification methods.
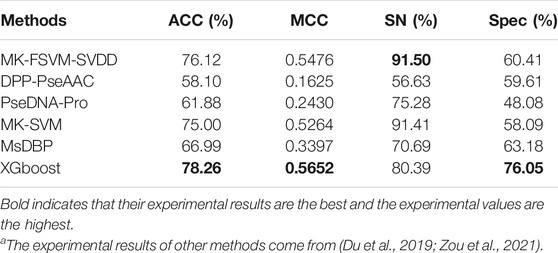
TABLE 5. Experimental findings for the independent data set PDB2272 using the XGBoost algorithm and other models.
Experimental Results With PDB2272 and PDB186 as Test Set
We combined PDB14189 and PDB1075 as the training set, and combined PDB2272 and PDB186 as the test set. After normalization and dimensionality reduction operations, we got an accuracy of 79.09% and the MCC value was 0.5818. It can be seen that this result is between the previous two experimental results.
Discussion and Conclusion
This paper proposes a method of predicting DBPs using the XGBoost algorithm and by splicing sequence feature information. The final sequence feature is built from multiple sequence features and spliced by MATLAB. To make the data more standardized and strengthen the relationship between data characteristics and data tags, the data are processed using Z-Score standardization. During the experiment, we used MRMD to reduce the dimensionality of the data and thus reduce the characteristics of the data. We performed experiments and compared the performance of XGBoost in terms of single sequence feature information and spliced sequence feature information. On the PDB 1075 data set, performance of the spliced sequence feature (MCC: 0.7272) is obviously better than that of the single sequence feature. To further assess our method, we applied the XGBoost model to the PDB186 and PDB2272 data sets. XGBoost produced superior results for PDB186 (MCC: 0.713) and PDB2272 (MCC: 0.5652) compared to available methods.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Author Contributions
ZZ and WY designed, planned and implemented the experiment. ZZ also wrote the main part of the article, and YXZ wrote other parts of the article. YL and YMZ participated in the coordination of the study and reviewed the article. All authors read and approved the final article.
Funding
This work was supported by the National Natural Science Foundation of China (61971119), The Heilongjiang Postdoctoral Fund (LBH-Q20135).The National Natural Science Foundation of China (NSFC)is a sub ministerial institution in charge of NSFC. NSFC operates relatively independently, and is responsible for the organization and implementation of funding plans, project setting and evaluation, project approval, supervision, etc.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adilina, S., Farid, D. M., and Shatabda, S. (2019). Effective DNA Binding Protein Prediction by Using Key Features via Chou's General PseAAC. J. Theor. Biol. 460, 64–78. doi:10.1016/j.jtbi.2018.10.027
Bi, X.-a., Liu, Y., Xie, Y., Hu, X., and Jiang, Q. (2020). Morbigenous Brain Region and Gene Detection with a Genetically Evolved Random Neural Network Cluster Approach in Late Mild Cognitive Impairment. Bioinformatics 36 (8), 2561–2568. doi:10.1093/bioinformatics/btz967
Chen, T., and Guestrin, C. (2016). “XGBoost: A Scalable Tree Boosting System,” in The 22nd ACM SIGKDD International Conference.
Cheng, L., Hu, Y., Sun, J., Zhou, M., and Jiang, Q. (2018). DincRNA: a Comprehensive Web-Based Bioinformatics Toolkit for Exploring Disease Associations and ncRNA Function. Bioinformatics 34 (11), 1953–1956. doi:10.1093/bioinformatics/bty002
Cheng, L., Qi, C., Zhuang, H., Fu, T., and Zhang, X. (2020). gutMDisorder: a Comprehensive Database for Dysbiosis of the Gut Microbiota in Disorders and Interventions. Nucleic Acids Res. 48 (D1), D554–D560. doi:10.1093/nar/gkz843
Cheng, L., Shi, H., Wang, Z., Hu, Y., Yang, H., Zhou, C., et al. (2016). IntNetLncSim: an Integrative Network Analysis Method to Infer Human lncRNA Functional Similarity. Oncotarget 7 (30), 47864–47874. doi:10.18632/oncotarget.10012
Cheng, L., Wang, P., Tian, R., Wang, S., Guo, Q., Luo, M., et al. (2019). LncRNA2Target v2.0: a Comprehensive Database for Target Genes of lncRNAs in Human and Mouse. Nucleic Acids Res. 47 (D1), D140–D144. doi:10.1093/nar/gky1051
Cheng, L., Zhao, H., Wang, P., Zhou, W., Luo, M., Li, T., et al. (2019). Computational Methods for Identifying Similar Diseases. Mol. Ther. - Nucleic Acids 18, 590–604. doi:10.1016/j.omtn.2019.09.019
Dao, F. Y., Lv, H., Su, W., Sun, Z-J., Huang, Q-L., and Lin, H. (2021). iDHS-Deep: an Integrated Tool for Predicting DNase I Hypersensitive Sites by Deep Neural Network. Brief Bioinform 22, bbab047. doi:10.1093/bib/bbab047
Ding, Y., Chen, F., Guo, X., Tang, J., and Wu, H. (2020). Identification of DNA-Binding Proteins by Multiple Kernel Support Vector Machine and Sequence Information. Current Proteomics 17 (4), 302–310. doi:10.2174/1570164616666190417100509
Ding, Y., Tang, J., and Guo, F. (2020). Human Protein Subcellular Localization Identification via Fuzzy Model on Kernelized Neighborhood Representation. Appl. Soft Comput. 96, 106596. doi:10.1016/j.asoc.2020.106596
Ding, Y., Tang, J., and Guo, F. (2020). Identification of Drug-Target Interactions via Dual Laplacian Regularized Least Squares with Multiple Kernel Fusion. Knowledge-Based Syst. 204, 106254. doi:10.1016/j.knosys.2020.106254
Ding, Y., Tang, J., and Guo, F. (2020). Identification of Drug–Target Interactions via Fuzzy Bipartite Local Model. Neural Comput. Appl. 32 (D1), 1–17. doi:10.1007/s00521-019-04569-z
Ding, Y., Tang, J., and Guo, F. (2016). Identification of Protein-Protein Interactions via a Novel Matrix-Based Sequence Representation Model with Amino Acid Contact Information. Int. J. Mol. Sci. 17 (10), 1623. doi:10.3390/ijms17101623
Ding, Y., Tang, J., and Guo, F. (2016). Predicting Protein-Protein Interactions via Multivariate Mutual Information of Protein Sequences. Bmc Bioinformatics 17 (1), 398. doi:10.1186/s12859-016-1253-9
Ding, Y., Tang, J., and Guo, F. (2019). Protein Crystallization Identification via Fuzzy Model on Linear Neighborhood Representation. IEEE/ACM Trans. Comput. Biol. Bioinformatics 18, 1986. doi:10.1109/TCBB.2019.2954826
Du, X., Diao, Y., Liu, H., and Li, S. (2019). MsDBP: Exploring DNA-Binding Proteins by Integrating Multiscale Sequence Information via Chou's Five-step Rule. J. Proteome Res. 18 (8), 3119–3132. doi:10.1021/acs.jproteome.9b00226
Feng, Z.-P., and Zhang, C.-T. (2000). Prediction of Membrane Protein Types Based on the Hydrophobic index of Amino Acids. J. Protein Chem. 19 (4), 269–275. doi:10.1023/a:1007091128394
Fu, X., Cai, L., Zeng, X., and Zou, Q. (2020). StackCPPred: a Stacking and Pairwise Energy Content-Based Prediction of Cell-Penetrating Peptides and Their Uptake Efficiency. Bioinformatics 36 (10), 3028–3034. doi:10.1093/bioinformatics/btaa131
Guo, Z., Wang, P., Liu, Z., and Zhao, Y. (2020). Discrimination of Thermophilic Proteins and Non-thermophilic Proteins Using Feature Dimension Reduction. Front. Bioeng. Biotechnol. 8, 584807. doi:10.3389/fbioe.2020.584807
Han, X., Kong, Q., Liu, C., Cheng, L., and Han, J. (2021). SubtypeDrug: a Software Package for Prioritization of Candidate Cancer Subtype-specific Drugs. Bioinformatics 2021, btab011. doi:10.1093/bioinformatics/btab011
Hong, Z., Zeng, X., Wei, L., and Liu, X. (2020). Identifying Enhancer-Promoter Interactions with Neural Network Based on Pre-trained DNA Vectors and Attention Mechanism. Bioinformatics 36 (4), 1037–1043. doi:10.1093/bioinformatics/btz694
Huang, Y. A., You, Z. H., Gao, X., Wong, L., and Wang, L. (2015). Using Weighted Sparse Representation Model Combined with Discrete Cosine Transformation to Predict Protein-Protein Interactions from Protein Sequence. Biomed. Res. Int. 2015, 902198. doi:10.1155/2015/902198
Huang, Y., Zhou, D., Wang, Y., Zhang, X., Su, M., Wang, C., et al. (2020). Prediction of Transcription Factors Binding Events Based on Epigenetic Modifications in Different Human Cells. Epigenomics 12 (16), 1443–1456. doi:10.2217/epi-2019-0321
Iqubal, A., Iqubal, M. K., Khan, A., Ali, J., Baboota, S., and Haque, S. E. (2020). Gene Therapy, A Novel Therapeutic Tool for Neurological Disorders: Current Progress, Challenges and Future Prospective. Curr. Gene Ther. 20 (3), 184–194. doi:10.2174/1566523220999200716111502
Jeong, J. C., Lin, X., and Chen, X.-W. (2011). On Position-specific Scoring Matrix for Protein Function Prediction. IEEE/ACM Trans. Comput. Biol. Bioinformatics (Tcbb) 8 (2), 308. doi:10.1109/tcbb.2010.93
Jiang, Q., Wang, G., Jin, S., Li, Y., and Wang, Y. (2013). Predicting Human microRNA-Disease Associations Based on Support Vector Machine. Int. J. Data Min Bioinform 8 (3), 282–293. doi:10.1504/ijdmb.2013.056078
Jiang, Q., Wang, G., Jin, S., Li, Y., and Wang, Y. (2013). Predicting Human microRNA-Disease Associations Based on Support Vector Machine. Int. J. Data Min Bioinform 8 (3), 282–293. doi:10.1504/ijdmb.2013.056078
Jin, S., Zeng, X., Fang, J., Lin, J., Chan, S. Y., Erzurum, S. C., et al. (2019). A Network-Based Approach to Uncover microRNA-Mediated Disease Comorbidities and Potential Pathobiological Implications. NPJ Syst. Biol. Appl. 5 (1), 41–11. doi:10.1038/s41540-019-0115-2
Jin, S., Zeng, X., Xia, F., Huang, W., and Liu, X. (2021). Application of Deep Learning Methods in Biological Networks. Brief. Bioinform. 22 (2), 1902–1917. doi:10.1093/bib/bbaa043
Kumar, K. K., Pugalenthi, G., and Suganthan, P. N. (2009). DNA-prot: Identification of DNA Binding Proteins from Protein Sequence Information Using Random Forest. J. Biomol. Struct. Dyn. 26 (6), 679–686. doi:10.1080/07391102.2009.10507281
Kumar, M., Gromiha, M. M., and Raghava, G. P. (2007). Identification of DNA-Binding Proteins Using Support Vector Machines and Evolutionary Profiles. Bmc Bioinformatics 8, 463. doi:10.1186/1471-2105-8-463
Li, H., Long, C., Xiang, J., Liang, P., Li, X., and Zuo, Y. (2020). Dppa2/4 as a Trigger of Signaling Pathways to Promote Zygote Genome Activation by Binding to CG-Rich Region. Brief Bioinform 22, bbaa342. doi:10.1093/bib/bbaa342
Li, H., Ta, N., Long, C., Zhang, Q., Li, S., liu, S., et al. (2019). The Spatial Binding Model of the pioneer Factor Oct4 with its Target Genes during Cell Reprogramming. Comput. Struct. Biotechnol. J. 17, 1226–1233. doi:10.1016/j.csbj.2019.09.002
Li, X., Liao, B., Shu, Y., Zeng, Q., and Luo, J. (2009). Protein Functional Class Prediction Using Global Encoding of Amino Acid Sequence. J. Theor. Biol. 261 (2), 290–293. doi:10.1016/j.jtbi.2009.07.017
Lin, W. Z., Fang, J. A., Xiao, X., and Chou, K. C. (2011). iDNA-Prot: Identification of DNA Binding Proteins Using Random Forest with Grey Model. Plos One 6 (9), e24756. doi:10.1371/journal.pone.0024756
Liu, B., Wang, S., and Wang, X. (2015). DNA Binding Protein Identification by Combining Pseudo Amino Acid Composition and Profile-Based Protein Representation. Sci. Rep. 5, 15479. doi:10.1038/srep15479
Liu, B., Gao, X., and Zhang, H. (2019). BioSeq-Analysis2.0: an Updated Platform for Analyzing DNA, RNA and Protein Sequences at Sequence Level and Residue Level Based on Machine Learning Approaches. Nucleic Acids Res. 47 (20), e127. doi:10.1093/nar/gkz740
Liu, B., Wang, S., Dong, Q., Li, S., and Liu, X. (2016). Identification of DNA-Binding Proteins by Combining Auto-Cross Covariance Transformation and Ensemble Learning. IEEE Trans.on Nanobioscience 15 (4), 328–334. doi:10.1109/tnb.2016.2555951
Liu, B., Xu, J., Lan, X., Xu, R., Zhou, J., Wang, X., et al. (2014). iDNA-Prot Vertical Bar Dis: Identifying DNA-Binding Proteins by Incorporating Amino Acid Distance-Pairs and Reduced Alphabet Profile into the General Pseudo Amino Acid Composition. Plos One 9 (9), e106691. doi:10.1371/journal.pone.0106691
Liu, B., Xu, J., Fan, S., Xu, R., Zhou, J., and Wang, X. (2015). PseDNA-Pro: DNA-Binding Protein Identification by Combining Chou's PseAAC and Physicochemical Distance Transformation. Mol. Inf. 34 (1), 8–17. doi:10.1002/minf.201400025
Liu, D., Li, G., and Zuo, Y. (2019). Function Determinants of TET Proteins: the Arrangements of Sequence Motifs with Specific Codes. Brief Bioinform 20 (5), 1826–1835. doi:10.1093/bib/bby053
Liu, G., Jin, S., Hu, Y., and Jiang, Q. (2018). Disease Status Affects the Association between Rs4813620 and the Expression of Alzheimer's Disease Susceptibility geneTRIB3. Proc. Natl. Acad. Sci. USA 115 (45), E10519–E10520. doi:10.1073/pnas.1812975115
Liu, H., Ren, G., Chen, H., Liu, Q., Yang, Y., and Zhao, Q. (2020). Predicting lncRNA-miRNA Interactions Based on Logistic Matrix Factorization with Neighborhood Regularized. Knowledge-Based Syst. 191, 105261. doi:10.1016/j.knosys.2019.105261
Liu, X. J., Gong, X. J., Yu, H., and Xu, J. H. (2018). A Model Stacking Framework for Identifying DNA Binding Proteins by Orchestrating Multi-View Features and Classifiers. Genes (Basel) 9 (8). doi:10.3390/genes9080394
Liu, Y., Huang, Y., Wang, G., and Wang, Y. (2020). A Deep Learning Approach for Filtering Structural Variants in Short Read Sequencing Data. Brief Bioinform 22, bbaa370. doi:10.1093/bib/bbaa370
Liu, Y., Zhang, X., Zou, Q., and Zeng, X. (2020). Minirmd: Accurate and Fast Duplicate Removal Tool for Short Reads via Multiple Minimizers. Bioinformatics 37, 1604–1606. doi:10.1093/bioinformatics/btaa915
Lou, W., Wang, X., Chen, F., Chen, Y., Jiang, B., and Zhang, H. (2014). Sequence Based Prediction of DNA-Binding Proteins Based on Hybrid Feature Selection Using Random Forest and Gaussian Naive Bayes. Plos One 9 (1), 86703. doi:10.1371/journal.pone.0086703
Nanni, L., Brahnam, S., and Lumini, A. (2012). Wavelet Images and Chou's Pseudo Amino Acid Composition for Protein Classification. Amino Acids 43 (2), 657–665. doi:10.1007/s00726-011-1114-9
Niu, M., Zhang, J., Li, Y., Wang, C., Liu, Z., Ding, H., et al. (2020). CirRNAPL: A Web Server for the Identification of circRNA Based on Extreme Learning Machine. Comput. Struct. Biotechnol. J. 18, 834–842. doi:10.1016/j.csbj.2020.03.028
Quan, Z., Zenga, J., Caoa, L., and Jia, R. (2016). A Novel Features Ranking Metric with Application to Scalable Visual and Bioinformatics Data Classification. Neurocomputing 173, 346–354. doi:10.1016/j.neucom.2014.12.123
Rahman, M. S., Shatabda, S., Saha, S., Kaykobad, M., and Rahman, M. S. (2018). DPP-PseAAC: A DNA-Binding Protein Prediction Model Using Chou's General PseAAC. J. Theor. Biol. 452, 22–34. doi:10.1016/j.jtbi.2018.05.006
Ru, X., Li, L., and Zou, Q. (2019). Incorporating Distance-Based Top-N-Gram and Random Forest to Identify Electron Transport Proteins. J. Proteome Res. 18 (7), 2931–2939. doi:10.1021/acs.jproteome.9b00250
Shang, Y., Gao, L., Zou, Q., and Yu, L. (2021). Prediction of Drug-Target Interactions Based on Multi-Layer Network Representation Learning. Neurocomputing 434, 80–89. doi:10.1016/j.neucom.2020.12.068
Shao, J., and Liu, B. (2021). ProtFold-DFG: Protein Fold Recognition by Combining Directed Fusion Graph and PageRank Algorithm. Brief. Bioinform. 22, bbaa192. doi:10.1093/bib/bbaa192
Shao, J., Yan, K., and Liu, B. (2021). FoldRec-C2C: Protein Fold Recognition by Combining Cluster-To-Cluster Model and Protein Similarity Network. Brief. Bioinform. 22, bbaa144. doi:10.1093/bib/bbaa144
Shen, Y., Ding, Y., Tang, J., Zou, Q., and Guo, F. (2019). Critical Evaluation of Web-Based Prediction Tools for Human Protein Subcellular Localization. Brief. Bioinformatics 21, 1628. doi:10.1093/bib/bbz106
Shen, Y., Ding, Y., Tang, J., Zou, Q., and Guo, F. (2020). Critical Evaluation of Web-Based Prediction Tools for Human Protein Subcellular Localization. Brief. Bioinform. 21 (5), 1628–1640. doi:10.1093/bib/bbz106
Shen, Y., Tang, J., and Guo, F. (2019). Identification of Protein Subcellular Localization via Integrating Evolutionary and Physicochemical Information into Chou's General PseAAC. J. Theor. Biol. 462, 230–239. doi:10.1016/j.jtbi.2018.11.012
Tang, Y.-J., Pang, Y.-H., and Liu, B. (2020). IDP-Seq2Seq: Identification of Intrinsically Disordered Regions Based on Sequence to Sequence Learning. Bioinformaitcs 36 (21), 5177–5186. doi:10.1093/bioinformatics/btaa667
Tao, Z., Li, Y., Teng, Z., and Zhao, Y. (2020). A Method for Identifying Vesicle Transport Proteins Based on LibSVM and MRMD. Comput. Math. Methods Med. 2020, 8926750. doi:10.1155/2020/8926750
Tao, Z., Li, Y., Teng, Z., and Zhao, Y. (2020). A Method for Identifying Vesicle Transport Proteins Based on LibSVM and MRMD. Comput. Math. Methods Med. 2020, 8926750. doi:10.1155/2020/8926750
Wang, H., Ding, Y., Tang, J., and Guo, F. (2020). Identification of Membrane Protein Types via Multivariate Information Fusion with Hilbert-Schmidt Independence Criterion. Neurocomputing 383, 257–269. doi:10.1016/j.neucom.2019.11.103
Wang, H., Jijun, T., Ding, Y., and Guo, F. (2021). Exploring Associations of Non-coding RNAs in Human Diseases via Three-Matrix Factorization with Hypergraph-Regular Terms on center Kernel Alignment. Brief. Bioinform. 22, bbaa409. doi:10.1093/bib/bbaa409
Wang, H., Liang, P., Zheng, L., Long, C. S., Li, H. S., Zuo, Y., et al. (2021). eHSCPr Discriminating the Cell Identity Involved in Endothelial to Hematopoietic Transition. Bioinformatics 37, 2157. doi:10.1093/bioinformatics/btab071
Wang, H., Yijie, D., Tang, J., Zou, Q., and Guo, F. (2021). Identify RNA-Associated Subcellular Localizations Based on Multi-Label Learning Using Chou's 5-steps Rule. BMC Genomics 22 (56), 1. doi:10.1186/s12864-020-07347-7
Wang, J., Wang, H., Wang, X., and Chang, H. (2020). Predicting Drug-Target Interactions via FM-DNN Learning. Curr. Bioinformatics 15 (1), 68–76. doi:10.2174/1574893614666190227160538
Wang, S., Wang, Y., Yu, C., Cao, Y., Yu, Y., Pan, Y., et al. (2020). Characterization of the Relationship between FLI1 and Immune Infiltrate Level in Tumour Immune Microenvironment for Breast Cancer. J. Cel Mol Med 24 (10), 5501–5514. doi:10.1111/jcmm.15205
Wang, Y., Ding, Y., Tang, J., Dai, Y., and Guo, F. (2021). CrystalM: A Multi-View Fusion Approach for Protein Crystallization Prediction. Ieee/acm Trans. Comput. Biol. Bioinform 18 (1), 325–335. doi:10.1109/TCBB.2019.2912173
Wang, Y., Shi, F., Cao, L., Dey, N., Wu, Q., Ashour, A. S., et al. (2019). Morphological Segmentation Analysis and Texture-Based Support Vector Machines Classification on Mice Liver Fibrosis Microscopic Images. Curr. Bioinformatics 14 (4), 282–294. doi:10.2174/1574893614666190304125221
Wei, L., Chen, H., and Su, R. (2018). M6APred-EL: A Sequence-Based Predictor for Identifying N6-Methyladenosine Sites Using Ensemble Learning. Mol. Ther. - Nucleic Acids 12, 635–644. doi:10.1016/j.omtn.2018.07.004
Wei, L., Ding, Y., Su, R., Tang, J., and Zou, Q. (2018). Prediction of Human Protein Subcellular Localization Using Deep Learning. J. Parallel Distributed Comput. 117, 212–217. doi:10.1016/j.jpdc.2017.08.009
Wei, L., Hu, J., Li, F., Song, J., Su, R., and Zou, Q. (2020). Comparative Analysis and Prediction of Quorum-sensing Peptides Using Feature Representation Learning and Machine Learning Algorithms. Brief. Bioinform. 21 (1), 106–119. doi:10.1093/bib/bby107
Wei, L., Liao, M., Gao, Y., Ji, R., He, Z., and Zou, Q. (2014). Improved and Promising Identification of Human MicroRNAs by Incorporating a High-Quality Negative Set. Ieee/acm Trans. Comput. Biol. Bioinf. 11 (1), 192–201. doi:10.1109/tcbb.2013.146
Wei, L., Tang, J., and Zou, Q. (2017). Local-DPP: An Improved DNA-Binding Protein Prediction Method by Exploring Local Evolutionary Information. Inf. Sci. 384, 135–144. doi:10.1016/j.ins.2016.06.026
Yang, C., Ding, Y., Meng, Q., Tang, J., and Guo, F. (2021). Granular Multiple Kernel Learning for Identifying RNA-Binding Protein Residues via Integrating Sequence and Structure Information. Neural Comput. Appl. 33, 11387. doi:10.1007/s00521-020-05573-4
Yang, H., Luo, Y., Ren, X., Wu, M., He, X., Peng, B., et al. (2021). Risk Prediction of Diabetes: Big Data Mining with Fusion of Multifarious Physical Examination Indicators. Inf. Fusion 75, 140–149. doi:10.1016/j.inffus.2021.02.015
You, Z. H., Zhu, L., Zheng, C. H., Yu, H. J., Deng, S. P., and Ji, Z. (2014). Prediction of Protein-Protein Interactions from Amino Acid Sequences Using a Novel Multi-Scale Continuous and Discontinuous Feature Set. Bmc Bioinformatics 15 (Suppl. 15), S9. doi:10.1186/1471-2105-15-S15-S9
Yu, L., Shi, Y., Zou, Q., Wang, S., Zheng, L., and Gao, L. (2020). Exploring Drug Treatment Patterns Based on the Action of Drug and Multilayer Network Model. Int. J. Mol. Sci. 21 (14), 5014. doi:10.3390/ijms21145014
Yu, L., Wang, M., Yang, Y., Xu, F., Zhang, X., Xie, F., et al. (2021). Predicting Therapeutic Drugs for Hepatocellular Carcinoma Based on Tissue-specific Pathways. Plos Comput. Biol. 17 (2), e1008696. doi:10.1371/journal.pcbi.1008696
Yu, L., Zhou, D., Gao, L., and Zha, Y. (2020). Prediction of Drug Response in Multilayer Networks Based on Fusion of Multiomics Data. Methods 192, 85. doi:10.1016/j.ymeth.2020.08.006
Zeng, X., Zhu, S., Lu, W., Liu, Z., Huang, J., Zhou, Y., et al. (2020). Target Identification Among Known Drugs by Deep Learning from Heterogeneous Networks. Chem. Sci. 11 (7), 1775–1797. doi:10.1039/c9sc04336e
Zhai, Y., Chen, Y., Teng, Z., and Zhao, Y. (2020). Identifying Antioxidant Proteins by Using Amino Acid Composition and Protein-Protein Interactions. Front. Cel Dev. Biol. 8, 591487. doi:10.3389/fcell.2020.591487
Zhang, C.-H., Li, M., Lin, Y.-P., and Gao, Q. (2020). Systemic Therapy for Hepatocellular Carcinoma: Advances and Hopes. Curr. Gene Ther. 20 (2), 84–99. doi:10.2174/1566523220666200628014530
Zhang, D., Chen, H. D., Zulfiqar, H., Yuan, S. S., Huang, Q. L., Zhang, Z. Y., et al. (2021). iBLP: An XGBoost-Based Predictor for Identifying Bioluminescent Proteins. Comput. Math. Methods Med. 2021, 6664362. doi:10.1155/2021/6664362
Zhang, J., Zhang, Z., Pu, L., Tang, J., and Guo, F. (2020). AIEpred: an Ensemble Predictive Model of Classifier Chain to Identify Anti-inflammatory Peptides. Ieee/acm Trans. Comput. Biol. Bioinform 18, 1831. doi:10.1109/TCBB.2020.2968419
Zhang, Z., Ding, J., Xu, J., Tang, J., and Guo, F. (2021). Multi-Scale Time-Series Kernel-Based Learning Method for Brain Disease Diagnosis. IEEE J. Biomed. Health Inform. 25 (1), 209–217. doi:10.1109/jbhi.2020.2983456
Zhao, T., Hu, Y., Peng, J., and Cheng, L. (2020). DeepLGP: a Novel Deep Learning Method for Prioritizing lncRNA Target Genes. Bioinformatics 36, 4466. doi:10.1093/bioinformatics/btaa428
Zhao, X., Jiao, Q., Li, H., Wu, Y., Wang, H., Huang, S., et al. (2020). ECFS-DEA: an Ensemble Classifier-Based Feature Selection for Differential Expression Analysis on Expression Profiles. BMC Bioinformatics 21 (1), 43. doi:10.1186/s12859-020-3388-y
Zhao, X., Wang, H., Li, H., Wu, Y., and Wang, G. (2021). Identifying Plant Pentatricopeptide Repeat Proteins Using a Variable Selection Method. Front. Plant Sci. 12, 506681. doi:10.3389/fpls.2021.506681
Zheng, L., Huang, S., Mu, N., Zhang, H., Zhang, J., Chang, Y., et al. (2019). RAACBook: a Web Server of Reduced Amino Acid Alphabet for Sequence-dependent Inference by Using Chou's Five-step Rule. Database (Oxford) 2019, baz131. doi:10.1093/database/baz131
Zheng, L., Liu, D., Yang, W., Yang, L., and Zuo, Y. (2020). RaacLogo: a New Sequence Logo Generator by Using Reduced Amino Acid Clusters. Brief Bioinform 22, bbaa096. doi:10.1093/bib/bbaa096
Zhu, X.-J., Feng, C.-Q., Lai, H.-Y., Chen, W., and Hao, L. (2019). Predicting Protein Structural Classes for Low-Similarity Sequences by Evaluating Different Features. Knowledge-Based Syst. 163, 787–793. doi:10.1016/j.knosys.2018.10.007
Zhu, Y., Li, F., Xiang, D., Akutsu, T., Song, J., and Jia, C. (2020). Computational Identification of Eukaryotic Promoters Based on Cascaded Deep Capsule Neural Networks. Brief. Bioinform. 22, bbaa299. doi:10.1093/bib/bbaa299
Zou, Y., Wu, H., Guo, X., Peng, L., Ding, Y., Tang, J., et al. (2021). MK-FSVM-SVDD: A Multiple Kernel-Based Fuzzy SVM Model for Predicting DNA-Binding Proteins via Support Vector Data Description. Curr. Bioinformatics 16 (2), 274–283. doi:10.2174/1574893615999200607173829
Keywords: DNA-binding protein prediction, machine learning, feature extraction, dimensionality reduction, XGBoost model
Citation: Zhao Z, Yang W, Zhai Y, Liang Y and Zhao Y (2022) Identify DNA-Binding Proteins Through the Extreme Gradient Boosting Algorithm. Front. Genet. 12:821996. doi: 10.3389/fgene.2021.821996
Received: 25 November 2021; Accepted: 07 December 2021;
Published: 28 January 2022.
Edited by:
Juan Wang, Inner Mongolia University, ChinaCopyright © 2022 Zhao, Yang, Zhai, Liang and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yingjian Liang, Z2Vub21lbGlhbmdAaG90bWFpbC5jb20=; Yuming Zhao, enltQG5lZnUuZWR1LmNu
†These authors have contributed equally to this work