 Xiaomei Gu
Xiaomei Gu Lina Guo5†
Lina Guo5† Bo Liao
Bo Liao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 01 December 2021
Sec. Computational Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.796327
Phages have seriously affected the biochemical systems of the world, and not only are phages related to our health, but medical treatments for many cancers and skin infections are related to phages; therefore, this paper sought to identify phage proteins. In this paper, a Pseudo-188D model was established. The digital features of the phage were extracted by PseudoKNC, an appropriate vector was selected by the AdaBoost tool, and features were extracted by 188D. Then, the extracted digital features were combined together, and finally, the viral proteins of the phage were predicted by a stochastic gradient descent algorithm. Our model effect reached 93.4853%. To verify the stability of our model, we randomly selected 80% of the downloaded data to train the model and used the remaining 20% of the data to verify the robustness of our model.
The term bacteriophage is actually a generic name for viruses or microorganisms. Phage virus proteins can be either viruses that invade bacteria or genetic material. According to the literature, phages are the most diverse entities in the ocean and affect biochemical systems around the world (Jahn et al., 2019; Cheng et al., 2020). Phages also affect the development of anticancer drugs because phage fusion proteins can promote the amplification and manufacturing of combinatorial chemistry products and nanotechnology to be applied in clinical trials for cancer treatment (Petrenko and Jayanna, 2016; Cheng et al., 2018; Yu et al., 2021a; Yu et al., 2021b). Phages may also cause acute or chronic skin infections and, in severe cases, may lead to death in patients with multidrug resistance (Al-Wrafy et al., 2019). Phages may play a part in the spread of antibiotic resistance, and thorough investigation must determine whether they contain antibiotic-resistance genes (Lekunberri et al., 2017). Individual glycoside hydrolases have been identified in the phage virion, which may facilitate phage annotation during infection (Yuan and Gao, 2016). However, experimental methods for the identification of phage viral proteins are time-consuming, and the cost is very high. Additionally, the identification of phage viral proteins presents challenges due to the diversity of phages and their abundant physical functions, and databases for phage annotation are rare (Seguritan et al., 2012; Bhakta and Tsukahara, 2020; Cheng et al., 2021). This also increases our difficulties with phage identification, so novel methods are needed to overcome these shortcomings. Therefore, we must develop accurate and affordable methods to predict phage viruses. Meeting these requirements based on the sequence calculation method can overcome these difficulties (Zeng et al., 2017; Hong et al., 2019; Zou et al., 2019; Cai et al., 2020a; Cai et al., 2020b; Fu et al., 2020; Hasan et al., 2020; Hu et al., 2020; Li et al., 2020a; Meng et al., 2020; Naseer et al., 2020; Zhang et al., 2020a; Hu et al., 2021a; Hu et al., 2021b; Wang et al., 2021a), and using bioinformatics methods to identify phage proteins, such as analysing protein and amino acid composition (Wu et al., 2019; Xu et al., 2021a), can facilitate the extraction of features, combined with artificial neural networks (Chen et al., 2020) and the use of random forest (Ao et al., 2020; Chen et al., 2020; Zhang et al., 2020b; Ahmed et al., 2021) integrated indicators to identify protein phages (Zhang et al., 2015; Ba Lachandran et al., 2018; Wu and Yu, 2021). For the development of phage virus protein identification, we need not only an affordable identification method but also the accuracy to judge whether the method can be used.
In this paper, we established a model of Pseudo-188D. The process of establishing this model involved first selecting suitable phage virus protein data and downloading the data from UniProt, which constituted our benchmark dataset, as our database for phage protein identification. Second, we used the pseudoKNC method to extract the digital characteristics of phages. In this process, we selected the appropriate value of ktuple (k) after tuning. Then, to reduce the impact of the dimensional disaster on the experimental results, the AdaBoost tool was used to select the appropriate vector. After selecting the appropriate feature vector, SVMprot-188D (188D) was used to extract the feature vector of the phage protein. After extracting the 188D feature, the features extracted by the two tools were combined. Finally, the random gradient descent (SGD) algorithm was used to predict phage proteins. To establish a model with stability and good robustness, we randomly selected 80% of the data as a test set to train the model and the remaining 20% of the data as a validation set to prove the stability of our model. At the same time, our model not only shows good stability and robustness but also very high accuracy. Readers can refer to Figure 1 for our model-building process, which clearly expresses our ideas.
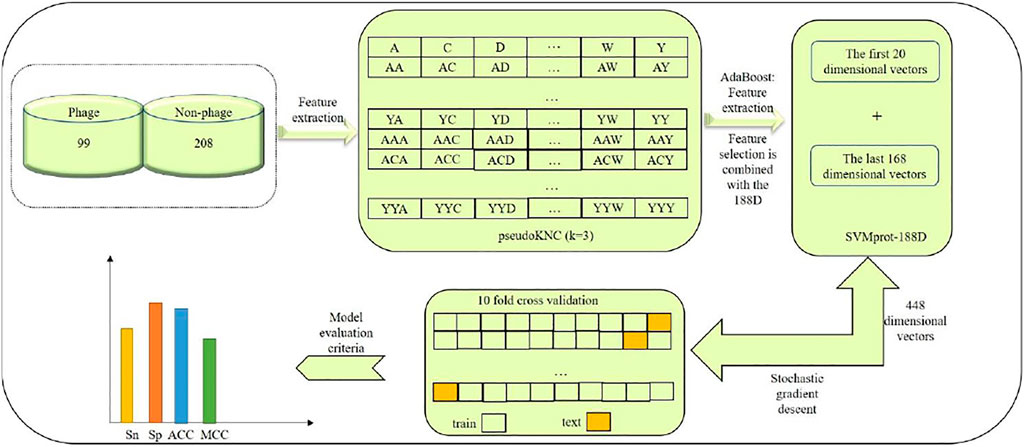
FIGURE 1. Establishing model Pseudo-188D process.
To better study phage proteins, we used data mainly from the literature (Meng et al., 2020). The data cited in this paper have been used in most studies for the identification of phage viral proteins because of their reliability and application to compare levels between different identifiers. The positive samples in the data were phages with viruses in subcellular positions, whereas the negative samples were nonphages. The sequences containing unrecognizable characters such as “Z”, “X”, “U”, and “B” were removed from the selected data. Finally, to avoid excessive homology of the data, redundant data were removed to ensure that the consistency between any data was not more than 40%, so our data included 99 phage virus protein-positive samples and 208 nonphage-negative samples. We will deposited the data at the website https://github.com/gxm123456/gxm.
PseudoKNC is a kind of software for extracting the digital features of DNA, RNA, and protein, and the features extracted by this software are all digital features (Muhammod et al., 2019; Yang et al., 2020; Ao et al., 2021a; Cao et al., 2021; Jiao et al., 2021; Sheng et al., 2021). Because the characteristics of protein, DNA, and RNA sequences are different, the dimensions of the extracted features are also different (Zuo et al., 2017; Zheng et al., 2019; Ao et al., 2021b). When vis guaranteed, and when the extracted feature sequence is a DNA or RNA sequence, the extracted digital feature dimension is
When the
When the
When the
Therefore, let us define
When the sequence is DNA,
When the sequence is RNA,
When the sequence is protein,
Figure 2 can be used as an example to show the protein sequences we extracted. There are 8,420 features extracted by us. The first 20 feature styles are
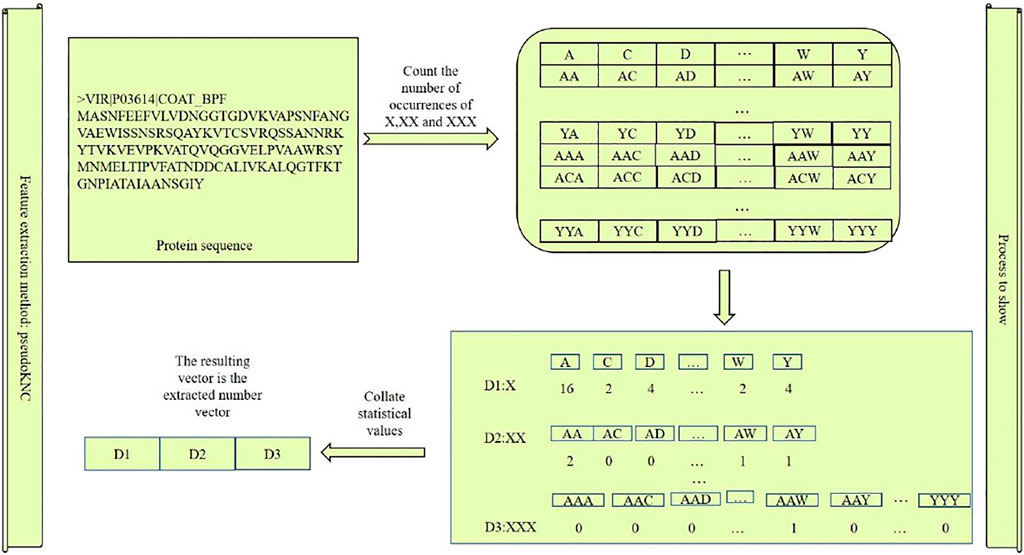
FIGURE 2. Extraction process of vector features by PseudoKNC.
The model AdaBoost is the SCRIT package used in Python, and to avoid any possible overfitting states, RNA and protein data are used as case studies, which can assess the generality of the model (Zhu et al., 2006; Cheng et al., 2016; Chen et al., 2019; Ramzan et al., 2021). After data selection is completed,
This method can extract a total of 188 feature dimensions, so it is also called 188D (Li et al., 2020b). The 188D top 20 extraction dimension vectors were used to calculate the frequency of the arrangement for 20 kinds of natural amino acids (A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, Y) (Zheng et al., 2020). Mainly refer to Formula (1) for calculation:
In Formula (1),
The next 168 features are associated with eight physicochemical properties, all represented by descriptors C (composed of amino acids), T (transition), and D (distribution). These three properties are made up of numbers, where C is composed of 3, representing the frequency of amino acids in a particular class; T is made up of three and represents the percentage of amino acids in the two different categories; D is made up of 15, representing the chain length ratios of the first, quarter, half and last amino acids in a given category, and then expanding the calculation by another hundred times. In this way, we extracted 168-dimensional features later:
This process encompasses the entire process for the extraction of 188 dimension features and the meaning of each feature.
The stochastic gradient descent algorithm determines an optimal path, and under the selection of this path, the optimal result is achieved by choosing the nearest shortcut. The main process of stochastic gradient descent is as follows:
The stochastic gradient descent algorithm obtains the optimal data by taking partial derivatives of the coefficients many times. The
In this way, the optimal value can be calculated, and the formula of the optimal solution can be calculated as follows:
In Formula (5),
In this paper, sensitivity (Sn), specificity (Sp), accuracy (ACC), and Matthew correlation coefficient (MCC) were still used as indicators to measure the performance of the model (Jiang et al., 2013; Wei et al., 2017a; Wei et al., 2017b; Wei et al., 2017c; Ding et al., 2019; Jin et al., 2019; Manavalan et al., 2019; Riaz and Li, 2019; Shen et al., 2019; Zeng et al., 2019a; Zeng et al., 2019b; Ding et al., 2020; Ding and JijunGuo, 2020; Hasan et al., 2020; Huang et al., 2020; Tao et al., 2020; Wan and Tan, 2020; Wang et al., 2020; Zeng et al., 2020; Zhai et al., 2020; Zhao et al., 2020; Zhang et al., 2020c; An and Yu, 2021; Ao et al., 2021a; Wang et al., 2021b; Xu et al., 2021b; Zhu et al., 2021).
Here, Tp indicates that the model correctly predicts the value of the phage virus protein; Fn represents the value of the model incorrectly predicting phage virus protein as non-phage protein; Fp represents the number of bacteriophage proteins incorrectly predicted by the model as non-phage viral proteins; and Tn indicates that the model correctly predicts the value of non-phage viral proteins.
Phages, although very small in size, have affected our lives, not only in the environment but also in terms of our health. If a phage enters a human, it will take on a bacterial host, live in the human, and even pass on to the next generati on. This requires us to identify phages quickly and accurately, so we built a model, Pseudo-188D, to predict phage proteins. The Pseudo-188D model is roughly the overall content of Chapter 2. First, the required protein digital features were extracted by PseudoKNC software. After the lower dimensional disaster of the model AdaBoost, the features extracted by model 188D are combined with the gradient descent algorithm to predict phage virus proteins.
In this chapter, we will prove the stability and robustness of the Pseudo-188 days model from various perspectives. First, the model that we established is compared with other methods, and the stability of the model is evaluated by Sp, Sn, MCC, and Acc. Second, we used different classifiers to identify phages. By comparing the values of Sp, Sn, MCC, and Acc, it was proven that SGD was a highly correct decision for our model. Finally, we used different cross-validations to more fully prove the accuracy of our model.
This section mainly proves that our model is superior to other methods and models in terms of method performance. We tried many methods to identify phage proteins, but the results were all unsatisfactory, such as those obtained with monoTriKGap (Muhammod et al., 2019), SC-PseaACC (Chou, 2005), and the 188D method for comparison. The performance of our model is stable compared with other methods. Table 1 shows the high accuracy and stability of the Pseudo-188 days model numerically, and the Sp, Sn, MCC, and Acc values are 0.89, 0.96, 0.93, and 0.85, respectively. These data indicate that the model we established is indeed suitable for phage protein identification.

TABLE 1. Performance comparison of different methods under 10-fold cross-validation.
To confirm the accuracy of the classification method we selected, we compared features extracted by the PseudoKNC method at the same time, combined with features extracted by the 188D model AdaBoost with less dimensional disaster, and then verified the accuracy and stability of SGD by using 10-fold cross-validation. Finally, different classification methods were used to verify the accuracy and stability of SGD. We chose several classification methods, such as NaiveBayes (Ahmed et al., 2021), Logistic (Hosmer et al., 2015; Sikandar et al., 2019), and multilayer Perceptron (Lek and Park, 2018; Ahmad et al., 2020). Table 2 fully shows that the classification method we chose is correct. According to comparison with other methods, NaiveBayes algorithm is not stable, MCC value is only 0.49, while the ACC value is 0.94. By comparing ACC value and MCC value, it is found that the NaiveBayes classification algorithm for our model is not stable. Logistic algorithm for processing our data, Sn,Sp, ACC, MCC values are not more than 0.9, accuracy is not as high as SGD classification method; The stability of multi-layer perceptron algorithm is relatively stable, but the accuracy is 0.02 lower than SGD, so we choose SGD as the classification algorithm. Because the classifier we choose has shown its advantages, not only fast but also better accuracy than other methods.

TABLE 2. Performance comparison of the same method in different classifiers.
To further prove that our model can show good performance in the identification of phage protein vector features, we used Pseudo-188D processed features of the model to evaluate with different cross-validations. According to Table 3, the results of 5-fold cross-validation, 6-fold cross-validation and 8-fold cross-validation were all stable. However, it can be seen from Table 3 that when 5-fold cross-validation is selected, MCC value is 0.8, 0.05 smaller than 10-fold cross-validation, and other values are also slightly smaller than 10-fold cross-validation. When selecting the 8-fold cross-validation, the VALUE of MCC was 0.83, 0.02 smaller than the value of 10-fold cross-validation. From various indicators, the actual effect of 10-fold cross-validation was more stable and accurate than that of other methods, so 10-fold cross-validation was selected to evaluate the performance of our model.

TABLE 3. Performance comparison of Pseudo-188D models under different cross-validations.
Previously, we have introduced the influence of ktuple (k) value on the number and style of feature extraction. In this summary, we compare the accuracy and stability when
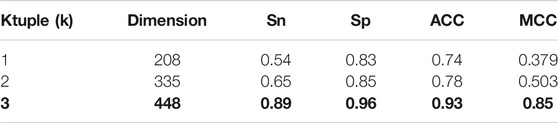
TABLE 4. Performance comparison under different Ktuple (k).
In this chapter, we have compared the monoTriKGap, SC-PSEaAC, and 188D methods and different classification methods. We have also compared different cross-validations, and our model Pseudo-188D shows good performance. To demonstrate Pseudo-188D performance more clearly, we combined the phage proteins extracted by the PseudoKNC method with the features extracted by 188D after AdaBoost treatment of the model. Then, 80% of feature vectors were randomly selected as the test set, and the training model and the remaining 20% of feature vectors were selected as the test set to verify the robustness of our model. The experimental results show that the model pseudo-188 days still shows good performance, and the accuracy of the results reaches 95.082%. Moreover, the values of Sp, Sn, MCC, and Acc also show good stability, reaching 0.94, 0.93, 0.95, and 0.89, respectively. These values fully demonstrate the stability and accuracy of Pseudo-188D.
Phages affect human lives all the time, and some of them are latent and inherited in the human body. Phages can also be used if they are understood. Many years ago, phages successfully prevented Pseudomonas aeruginosa infection in burn patients. Therefore, we need to accurately identify phages so that they can be used for medical research or prevention and control of life inconveniences caused by phages. When establishing the model in this paper, we choose PseudoKNC to extract features. When the k value is 3, a total of 8420-dimensional features are extracted. After processing the AdaBoost model, 260 features with the best performance are selected, and combined with features extracted from 188D, there are 448-dimensional features. The 448-dimensional vectors were classified by SGD, and the accuracy was 93.4853% under 10-fold cross verification. To further improve the rigor of the experiment, we randomly selected 80% of the data as the test set and the remaining 20% as the validation set. After this validation, our model pseudo-188 days still showed stability and accuracy and, most importantly, significantly saved time and cost.
This paper mainly introduces the Pseudo-188D model that we established, which accurately predicts phage proteins and makes a small contribution to phage prediction, improving the accuracy of phage prediction. In addition, our model greatly reduces the time and expense of predicting phage proteins, which saves considerable time and money. The greatest innovation in this paper is the combination of PseudoKNC and 188D, which can improve the predictive accuracy of phages. This will facilitate phage research, whether it is using phages for medical problems, anticancer methods based on phages, or solving environmental problems around us. That is where the value of phage research is realized.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
XG and LG co-experiment and collate papers; BL guided us in the experiment; QJ solved some of the problems in the experiment and shared the literature.
This work was supported by the National Nature Science Foundation of China (Grant Nos. 61863010, 11926205, 11926412, and 61873076), National Key R and D Program of China (No.2020YFB2104400), Natural Science Foundation of Hainan, China (Gran-tNos.121RC538, 119MS036, and 120RC588).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Thanks to the guidance of my tutor and the joint efforts of other authors, the success of this article is the result of everyone’s joint efforts.
Ahmad, F., Farooq, A., Khan, M. U. G., Shabbir, M. Z., Rabbani, M., and Hussain, I. (2020). Identification of Most Relevant Features for Classification of Francisella Tularensis Using Machine Learning. Curr. Bioinformatics 15 (10), 1197–1212. doi:10.2174/1574893615666200219113900
Ahmed, F. F., Khatun, M. S., Mosharaf, M. P., and Mollah, M. N. H. (2021). Prediction of Protein-Protein Interactions in Arabidopsis thaliana Using Partial Training Samples in a Machine Learning Framework. Cbio 16 (6), 865–879. doi:10.2174/1574893616666210204145254
Al-Wrafy, F., Brzozowska, E., Górska, S., Drab, M., Strus, M., and Gamian, A. (2019). Identification and Characterization of Phage Protein and its Activity against Two Strains of Multidrug-Resistant Pseudomonas aeruginosa. Sci. Rep. 9 (9), 13487. doi:10.1038/s41598-019-50030-5
An, Q., and Yu, L. (2021). A Heterogeneous Network Embedding Framework for Predicting Similarity-Based Drug-Target Interactions. Brief. Bioinformatics 22 (6), bbab275. doi:10.1093/bib/bbab275
Ao, C., Yu, L., and Zou, Q. (2021). Prediction of Bio-Sequence Modifications and the Associations with Diseases. Brief. Funct. Genomics 20 (1), 1–18. doi:10.1093/bfgp/elaa023
Ao, C., Zhou, W., Gao, L., Dong, B., and Yu, L. (2020). Prediction of Antioxidant Proteins Using Hybrid Feature Representation Method and Random forest. Genomics 112 (6), 4666–4674. doi:10.1016/j.ygeno.2020.08.016
Ao, C., Zou, Q., and Yu, L. (2021). RFhy-m2G: Identification of RNA N2-Methylguanosine Modification Sites Based on Random forest and Hybrid Features. Methods. San Diego, Calif. doi:10.1016/j.ymeth.2021.05.016
Ba Lachandran, M., Shin, T. H., and Gwang, L. (2018). PVP-SVM: Sequence-Based Prediction of Phage Virion Proteins Using a Support Vector Machine. Front. Microbiol. 9, 476. doi:10.3389/fmicb.2018.00476
Bhakta, S., and Tsukahara, T. (2020). Artificial RNA Editing with ADAR for Gene Therapy. Cgt 20 (1), 44–54. doi:10.2174/1566523220666200516170137
Cai, L., Ren, X., Fu, X., Peng, L., Gao, M., Zeng, X., et al. (2020). Interpretable Sequence-Based Enhancers and Their Strength Predictor. Bioinformatics 37 (8), 1060–1067. doi:10.1093/bioinformatics/btaa914
Cai, L., Wang, L., Fu, X., Xia, C., Zeng, X., and Zou, Q. (2020). ITP-pred: an Interpretable Method for Predicting, Therapeutic Peptides with Fused Features Low-Dimension Representation. Brief. Bioinform. 22, bbaa367. doi:10.1093/bib/bbaa367
Cao, Y., Yu, C., Huang, S., Wang, S., Zuo, Y., and Yang, L. (2021). Characterization and Prediction of Presynaptic and Postsynaptic Neurotoxins Based on Reduced Amino Acids and Biological Properties. Cbio 16 (3), 364–370. doi:10.2174/1574893615999200707150512
Chen, P., Shen, T., Zhang, Y., and Wang, B. (2020). A Sequence-Segment Neighbor Encoding Schema for Protein Hotspot Residue Prediction. Cbio 15 (5), 445–454. doi:10.2174/1574893615666200106115421
Chen, X., Shi, W., and Deng, L. (2019). Prediction of Disease Comorbidity Using HeteSim Scores Based on Multiple Heterogeneous Networks. Cgt 19 (4), 232–241. doi:10.2174/1566523219666190917155959
Cheng, L., Hu, Y., Sun, J., Zhou, M., and Jiang, Q. (2018). DincRNA: a Comprehensive Web-Based Bioinformatics Toolkit for Exploring Disease Associations and ncRNA Function. Bioinformatics 34 (11), 1953–1956. doi:10.1093/bioinformatics/bty002
Cheng, L., Qi, C., Yang, H., Lu, M., Cai, Y., Fu, T., et al. (2021). gutMGene: a Comprehensive Database for Target Genes of Gut Microbes and Microbial Metabolites. Nucleic Acids Res., gkab786. doi:10.1093/nar/gkab786
Cheng, L., Qi, C., Zhuang, H., Fu, T., and Zhang, X. (2020). gutMDisorder: a Comprehensive Database for Dysbiosis of the Gut Microbiota in Disorders and Interventions. Nucleic Acids Res. 48 (D1), D554–D560. doi:10.1093/nar/gkz843
Cheng, L., Shi, H., Wang, Z., Hu, Y., Yang, H., Zhou, C., et al. (2016). IntNetLncSim: an Integrative Network Analysis Method to Infer Human lncRNA Functional Similarity. Oncotarget 7 (30), 47864–47874. doi:10.18632/oncotarget.10012
Chou, K. C. (2005). Using Amphiphilic Pseudo Amino Acid Composition to Predict Enzyme Subfamily Classes. Bioinformatics 21 (1), 10–19. doi:10.1093/bioinformatics/bth466
Ding, Y., Tang, J., and Guo, F. (2019). Identification of Drug-Side Effect Association via Multiple Information Integration with Centered Kernel Alignment. Neurocomputing 325, 211–224. doi:10.1016/j.neucom.2018.10.028
Ding, Y., Tang, J., and Guo, F. (2020). Identification of Drug-Target Interactions via Fuzzy Bipartite Local Model. Neural Comput. Applic 32, 10303–10319. doi:10.1007/s00521-019-04569-z
Ding, Y., Jijun, T., and Guo, F. (2020). Identification of Drug-Target Interactions via Dual Laplacian Regularized Least Squares with Multiple Kernel Fusion. Knowledge-Based Syst. 204, 106254. doi:10.1016/j.knosys.2020.106254
Fu, X., Cai, L., Zeng, X., and Zou, Q. (2020). StackCPPred: a Stacking and Pairwise Energy Content-Based Prediction of Cell-Penetrating Peptides and Their Uptake Efficiency. Bioinformatics 36 (10), 3028–3034. doi:10.1093/bioinformatics/btaa131
Hasan, M. A. M., Ben Islam, M. K., Rahman, J., and Ahmad, S. (2020). Citrullination Site Prediction by Incorporating Sequence Coupled Effects into PseAAC and Resolving Data Imbalance Issue. Cbio 15 (3), 235–245. doi:10.2174/1574893614666191202152328
Hong, Z., Zeng, X., Wei, L., and Liu, X. (2019). Identifying Enhancer-Promoter Interactions with Neural Network Based on Pre-trained DNA Vectors and Attention Mechanism. Bioinformatics 36 (4), 1037–1043. doi:10.1093/bioinformatics/btz694
Hosmer, D. W., Hosmer, T., Le Cessie, S., and Lemeshow, S. (2015). A Comparison of Goodness-Of-Fit Tests for the Logistic Regression Model. Stat. Med. 16 (9), 965–980. doi:10.1002/(sici)1097-0258(19970515)16:9<965:aid-sim509>3.0.co;2-o
Hu, Y., Qiu, S., and Cheng, L. (2021). Integration of Multiple-Omics Data to Analyze the Population-specific Differences for Coronary Artery Disease. Comput. Math. Methods Med. 2021, 7036592. doi:10.1155/2021/7036592
Hu, Y., Sun, J. Y., Zhang, Y., Zhang, H., Gao, S., Wang, T., et al. (2021). rs1990622 Variant Associates with Alzheimer's Disease and Regulates TMEM106B Expression in Human Brain Tissues. BMC Med. 19 (1), 11. doi:10.1186/s12916-020-01883-5
Hu, Y., Zhang, H., Liu, B., Gao, S., Wang, T., Han, Z., et al. (2020). rs34331204 Regulates TSPAN13 Expression and Contributes to Alzheimer's Disease with Sex Differences. Brain 143 (11), e95. doi:10.1093/brain/awaa302
Huang, Y., Zhou, D., Wang, Y., Zhang, X., Su, M., Wang, C., et al. (2020). Prediction of Transcription Factors Binding Events Based on Epigenetic Modifications in Different Human Cells. Epigenomics 12 (16), 1443–1456. doi:10.2217/epi-2019-0321
Jahn, M. T., Arkhipova, K., Markert, S. M., Stigloher, C., Lachnit, T., Pita, L., et al. (2019). A Phage Protein Aids Bacterial Symbionts in Eukaryote Immune Evasion. Cell Host Microbe 26 (4), 542. doi:10.1016/j.chom.2019.08.019
Jiang, Q., Wang, G., Jin, S., Li, Y., and Wang, Y. (2013). Predicting Human microRNA-Disease Associations Based on Support Vector Machine. Ijdmb 8 (3), 282–293. doi:10.1504/ijdmb.2013.056078
Jiao, S., Zou, Q., Guo, H., and Shi, L. (2021). iTTCA-RF: a Random forest Predictor for Tumor T Cell Antigens. J. Transl Med. 19 (1), 449. doi:10.1186/s12967-021-03084-x
Jin, Q., Meng, Z., Pham, T. D., Chen, Q., Wei, L., and Su, R. (2019). DUNet: A Deformable Network for Retinal Vessel Segmentation. Knowledge-Based Syst. 178, 149–162. doi:10.1016/j.knosys.2019.04.025
Lek, S., and Park, Y. S. (2018). Multilayer Perceptron. Alphascript Publishing 6 (2), 131–139. doi:10.1016/b978-008045405-4.00162-2
Lekunberri, I., Subirats, J., Borrego, C. M., and Balcázar, J. (2017). Exploring the Contribution of Bacteriophages to Antibiotic Resistance. Environ. Pollut. 220, 981–984. doi:10.1016/j.envpol.2016.11.059
Li, J., Pu, Y., Tang, J., Zou, Q., and Guo, F. (2020). DeepATT: a Hybrid Category Attention Neural Network for Identifying Functional Effects of DNA Sequences. Brief Bioinform 22 (3), bbaa159. doi:10.1093/bib/bbaa159
Li, Y., Zhang, Z., Teng, Z., and Liu, X. (2020). PredAmyl-MLP: Prediction of Amyloid Proteins Using Multilayer Perceptron. Comput. Math. Methods Med. 2020 (1), 1–12. doi:10.1155/2020/8845133
Manavalan, B., Basith, S., Shin, T. H., Wei, L., and Lee, G. (2019). mAHTPred: a Sequence-Based Meta-Predictor for Improving the Prediction of Anti-hypertensive Peptides Using Effective Feature Representation. Bioinformatics 35 (16), 2757–2765. doi:10.1093/bioinformatics/bty1047
Meng, C., Zhang, J., Ye, X., Guo, F., and Zou, Q. (2020). Review and Comparative Analysis of Machine Learning-Based Phage Virion Protein Identification Methods. Biochim. Biophys. Acta (Bba) - Proteins Proteomics 1868 (6), 140406. doi:10.1016/j.bbapap.2020.140406
Muhammod, R., Ahmed, S., Farid, D. M. D., Shatabda, S., Alok, S., and Dehzangi, A. (2019). A Python-Based Effective Feature Generation Tool for DNA, RNA, and Protein Sequences. Bioinformatics 35 (19), 3831–3833. doi:10.1093/bioinformatics/btz165
Naseer, S., Hussain, W., Khan, Y. D., and Rasool, N. (2020). Sequence-based Identification of Arginine Amidation Sites in Proteins Using Deep Representations of Proteins and PseAAC. Curr. Bioinformatics 15 (8), 937–948. doi:10.2174/1574893615666200129110450
Petrenko, V. A., and Jayanna, P. K. (2016). Phage Protein-Targeted Cancer Nanomedicines. FEBS Lett. 588 (2), 341–349. doi:10.1016/j.febslet.2013.11.011
Ramzan, Z., Hassan, M. A., Asif, H. M. S., and Farooq, A. (2021). A Machine Learning-Based Self-Risk Assessment Technique for Cervical Cancer. Cbio 16 (2), 315–332. doi:10.2174/1574893615999200608130538
Riaz, F., and Li, D. (2019). Non-coding RNA Associated Competitive Endogenous RNA Regulatory Network: Novel Therapeutic Approach in Liver Fibrosis. Cgt 19 (5), 305–317. doi:10.2174/1566523219666191107113046
Seguritan, V., Alves, N., Arnoult, M., Raymond, A., Lorimer, D., Burgin, A. B., et al. (2012). Artificial Neural Networks Trained to Detect Viral and Phage Structural Proteins. Plos Comput. Biol. 8 (8), e1002657. doi:10.1371/journal.pcbi.1002657
Shen, Y., Tang, J., and Guo, F. (2019). Identification of Protein Subcellular Localization via Integrating Evolutionary and Physicochemical Information into Chou's General PseAAC. J. Theor. Biol. 462, 230–239. doi:10.1016/j.jtbi.2018.11.012
Sheng, Y., Jiang, Y., Yang, Y., Li, X., Qiu, J., Wu, J., et al. (2021). CNA2Subpathway: Identification of Dysregulated Subpathway Driven by Copy Number Alterations in Cancer. Brief Bioinform 22 (5), bbaa413. doi:10.1093/bib/bbaa413
Sikandar, A., Anwar, W., and Sikandar, M. (2019). Combining Sequence Entropy and Subgraph Topology for Complex Prediction in Protein Protein Interaction (PPI) Network. Cbio 14 (6), 516–523. doi:10.2174/1574893614666190103100026
Tao, Z., Li, Y., Teng, Z., and Zhao, Y. (2020). A Method for Identifying Vesicle Transport Proteins Based on LibSVM and MRMD. Comput. Math. Methods Med. 2020, 8926750. doi:10.1155/2020/8926750
Wan, X., and Tan, X. (2020). A Simple Protein Evolutionary Classification Method Based on the Mutual Relations between Protein Sequences. Curr. Bioinformatics 15 (10), 1113–1129. doi:10.2174/1574893615666200305090055
Wang, H., Ding, Y., Tang, J., and Guo, F. (2020). Identification of Membrane Protein Types via Multivariate Information Fusion with Hilbert-Schmidt Independence Criterion. Neurocomputing 383, 257–269. doi:10.1016/j.neucom.2019.11.103
Wang, H., Jijun, T., Ding, Y., and Guo, F. (2021). Exploring Associations of Non-coding RNAs in Human Diseases via Three-Matrix Factorization with Hypergraph-Regular Terms on center Kernel Alignment. Brief. Bioinform. 22 (5), bbaa409. doi:10.1093/bib/bbaa409
Wang, X., Yang, Y., Liu, J., and Wang, G. (2021). The Stacking Strategy-Based Hybrid Framework for Identifying Non-coding RNAs. Brief Bioinform 22 (5), bbab023. doi:10.1093/bib/bbab023
Wei, L., Tang, J., and Zou, Q. (2017). Local-DPP: An Improved DNA-Binding Protein Prediction Method by Exploring Local Evolutionary Information. Inf. Sci. 384 (384), 135–144. doi:10.1016/j.ins.2016.06.026
Wei, L., Wan, S., Guo, J., and Wong, K. K. (2017). A Novel Hierarchical Selective Ensemble Classifier with Bioinformatics Application. Artif. Intelligence Med. 83, 82–90. doi:10.1016/j.artmed.2017.02.005
Wei, L., Xing, P., Zeng, J., Chen, J., Su, R., and Guo, F. (2017). Improved Prediction of Protein-Protein Interactions Using Novel Negative Samples, Features, and an Ensemble Classifier. Artif. Intelligence Med. 83, 67–74. doi:10.1016/j.artmed.2017.03.001
Wu, N., Wang, L., Hu, J., Zhao, S., Liu, B., Li, Y., et al. (2019). A Recurrent Rare SOX9 Variant (M469V) Is Associated with Congenital Vertebral Malformations. Cgt 19 (4), 242–247. doi:10.2174/1566523219666190924120307
Wu, X., and Yu, L. (2021). EPSOL: Sequence-Based Protein Solubility Prediction Using Multidimensional Embedding. Bioinformatics, btab463. Oxford, England. doi:10.1093/bioinformatics/btab463
Xu, B., Liu, D., Wang, Z., Tian, R., and Zuo, Y. (2021). Multi-substrate Selectivity Based on Key Loops and Non-homologous Domains: New Insight into ALKBH Family. Cell. Mol. Life Sci. 78 (1), 129–141. doi:10.1007/s00018-020-03594-9
Xu, Z., Luo, M., Lin, W., Xue, G., Wang, P., Jin, X., et al. (2021). DLpTCR: an Ensemble Deep Learning Framework for Predicting Immunogenic Peptide Recognized by T Cell Receptor. Brief Bioinform 22, bbab335. doi:10.1093/bib/bbab335
Yang, X.-F., Zhou, Y.-K., Zhang, L., Gao, Y., and Du, P.-F. (2020). Predicting LncRNA Subcellular Localization Using Unbalanced Pseudo-k Nucleotide Compositions. Cbio 15 (6), 554–562. doi:10.2174/1574893614666190902151038
Yu, L., Wang, M., Yang, Y., Xu, F., Zhang, X., Xie, F., et al. (2021). Predicting Therapeutic Drugs for Hepatocellular Carcinoma Based on Tissue-specific Pathways. Plos Comput. Biol. 17 (2), e1008696. doi:10.1371/journal.pcbi.1008696
Yu, L., Xia, M., and An, Q. (2021). A Network Embedding Framework Based on Integrating Multiplex Network for Drug Combination Prediction. Brief. Bioinformatics. doi:10.1093/bib/bbab364
Yuan, Y., and Gao, M. (2016). Proteomic Analysis of a Novel Bacillus Jumbo Phage Revealing Glycoside Hydrolase as Structural Component. Front. Microbiol. 7, 745. doi:10.3389/fmicb.2016.00745
Zeng, X., Lin, W., Guo, M., and Zou, Q. (2017). A Comprehensive Overview and Evaluation of Circular RNA Detection Tools. Plos Comput. Biol. 13 (6), e1005420. doi:10.1371/journal.pcbi.1005420
Zeng, X., Zhong, Y., Lin, W., and Zou, Q. (2019). Predicting Disease-Associated Circular RNAs Using Deep Forests Combined with Positive-Unlabeled Learning Methods. Brief. Bioinform. 21, 1425–1436. doi:10.1093/bib/bbz080
Zeng, X., Zhu, S., Liu, X., Zhou, Y., Nussinov, R., and Cheng, F. (2019). deepDR: a Network-Based Deep Learning Approach to In Silico Drug Repositioning. Bioinformatics 35 (24), 5191–5198. doi:10.1093/bioinformatics/btz418
Zeng, X., Zhu, S., Lu, W., Liu, Z., Huang, J., Zhou, Y., et al. (2020). Target Identification Among Known Drugs by Deep Learning from Heterogeneous Networks. Chem. Sci. 11 (7), 1775–1797. doi:10.1039/c9sc04336e
Zhai, Y., Chen, Y., Teng, Z., and Zhao, Y. (2020). Identifying Antioxidant Proteins by Using Amino Acid Composition and Protein-Protein Interactions. Front. Cel Dev. Biol. 8, 591487. doi:10.3389/fcell.2020.591487
Zhang, J., Zhang, Z., Pu, L., Tang, J., and Guo, F. (2020). AIEpred: an Ensemble Predictive Model of Classifier Chain to Identify Anti-inflammatory Peptides. Ieee/acm Trans. Comput. Biol. Bioinform 18 (5), 1831–1840. doi:10.1109/TCBB.2020.2968419
Zhang, L., He, Y., Song, H., Wang, X., Lu, N., Sun, L., et al. (2020). Elastic Net Regularized Softmax Regression Methods for Multi-Subtype Classification in Cancer. Cbio 15 (3), 212–224. doi:10.2174/1574893613666181112141724
Zhang, L., Xiao, X., and Xu, Z.-C. (2020). iPromoter-5mC: A Novel Fusion Decision Predictor for the Identification of 5-Methylcytosine Sites in Genome-wide DNA Promoters. Front. Cel Dev. Biol. 8, 614. doi:10.3389/fcell.2020.00614
Zhang, L., Zhang, C., Gao, R., and Yang, R. (2015). An Ensemble Method to Distinguish Bacteriophage Virion from Non-virion Proteins Based on Protein Sequence Characteristics. Ijms 16 (9), 21734–21758. doi:10.3390/ijms160921734
Zhao, X., Jiao, Q., Li, H., Wu, Y., Wang, H., Huang, S., et al. (2020). ECFS-DEA: an Ensemble Classifier-Based Feature Selection for Differential Expression Analysis on Expression Profiles. BMC Bioinformatics 21 (1), 43. doi:10.1186/s12859-020-3388-y
Zheng, L., Huang, S., Mu, N., Zhang, H., Zhang, J., Chang, Y., et al. (2019). RAACBook: a Web Server of Reduced Amino Acid Alphabet for Sequence-dependent Inference by Using Chou's Five-step Rule. Database (Oxford) 2019, baz131. doi:10.1093/database/baz131
Zheng, L., Liu, D., Yang, W., Yang, L., and Zuo, Y. (2020). RaacLogo: a New Sequence Logo Generator by Using Reduced Amino Acid Clusters. Brief Bioinform 22, bbaa096 doi:10.1093/bib/bbaa096
Zhu, J., Arbor, A., and Hastie, T. (2006). Multi-class AdaBoost. Stat. Its Interf. 2 (3), 349–360. doi:10.4310/sii.2009.v2.n3.a8
Zhu, Y., Li, F., Xiang, D., Akutsu, T., Song, J., and Jia, C. (2021). Computational Identification of Eukaryotic Promoters Based on Cascaded Deep Capsule Neural Networks. Brief Bioinform 22 (4), 1–11. doi:10.1093/bib/bbaa299
Zou, Q., Lin, G., Jiang, X., Liu, X., and Zeng, X. (2019). Sequence Clustering in Bioinformatics: an Empirical Study. Brief. Bioinformatics 21 (1), 1–10. doi:10.1093/bib/bby090
Keywords: model pseudo-188D, phage, stochastic gradient descent, dimensional disaster, digital characteristics
Citation: Gu X, Guo L, Liao B and Jiang Q (2021) Pseudo-188D: Phage Protein Prediction Based on a Model of Pseudo-188D. Front. Genet. 12:796327. doi: 10.3389/fgene.2021.796327
Received: 21 October 2021; Accepted: 15 November 2021;
Published: 01 December 2021.
Edited by:
Quan Zou, University of Electronic Science and Technology of China, ChinaCopyright © 2021 Gu, Guo, Liao and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo Liao, ZHJhZ29uYndAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.