
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 17 January 2022
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.784963
 Ligia Pereira Castro1
Ligia Pereira Castro1 Danilo Batista-Vieira1
Danilo Batista-Vieira1 Tiago Antonio de Souza1
Tiago Antonio de Souza1 Ana Rafaela de Souza Timoteo2Jessica Dayanna Landivar Coutinho2Isabel Cristina Pinheiro de Almeida3Sheila Ramos de Miranda Henriques3Fabio Medeiros de Azevedo3
Ana Rafaela de Souza Timoteo2Jessica Dayanna Landivar Coutinho2Isabel Cristina Pinheiro de Almeida3Sheila Ramos de Miranda Henriques3Fabio Medeiros de Azevedo3 Reginaldo Cruz Alves Rosa4,5
Reginaldo Cruz Alves Rosa4,5 Patricia L Kannouche5Alain Sarasin5
Patricia L Kannouche5Alain Sarasin5 Carlos Frederico Martins Menck1
Carlos Frederico Martins Menck1 Tirzah Braz Petta2,3,6,7*
Tirzah Braz Petta2,3,6,7*Xeroderma pigmentosum (XP) is a rare genetic condition in which exposure to sunlight leads to a high tumor incidence due to defective DNA repair machinery. Herein, we investigated seven patients clinically diagnosed with XP living in a small city, Montanhas (Rio Grande do Norte), in the Northeast region of Brazil. We performed high-throughput sequencing and, surprisingly, identified two different mutated genes. Six patients carry a novel homozygote mutation in the POLH/XPV gene, c.672_673insT (p.Leu225Serfs*33), while one patient carries a homozygote mutation in the XPC gene, c.2251-1G>C. This latter mutation was previously described in Southeastern Africa (Comoro Island and Mozambique), Pakistan, and in a high incidence in Brazil. The XP-C patient had the first symptoms before the first year of life with aggressive ophthalmologic tumor progression and a melanoma onset at 7 years of age. The XP-V patients presented a milder phenotype with later onset of the disorder (mean age of 16 years old), and one of the six XP-V patients developed melanoma at 72 years. The photoprotection is minimal among them, mainly for the XP-V patients. The differences in the disease severity between XP-C (more aggressive) and XP-V (milder) patients are obvious and point to the major role of photoprotection in the XPs. We estimate that the incidence of XP patients at Montanhas can be higher, but with no diagnosis, due to poor health assistance. Patients still suffer from the stigmatization of the condition, impairing diagnosis, education for sun protection, and medical care.
Xeroderma pigmentosum is an autosomal recessive disease in which the patients develop a high frequency of skin cancer, internal tumors, and neurological abnormalities (20–30% of the patients) (Menck and Munford, 2014). The tumor susceptibility is due to germline mutations that impair the processing of DNA lesions by nucleotide excision repair (NER) or by the translesion synthesis (TLS) pathway. The initial recognition step distinguishes two subpathways of NER. The transcription-coupled repair (TCR), in which the recognition occurs on the transcribed strand by the arrest of the RNA polymerase, and the nontranscribed genome is repaired by the global genomic repair (GGR) pathway (Hanawalt, 2002). These pathways are responsible for correctly cleaning up the DNA from lesions induced mainly by the ultraviolet radiation (UVR) from sunlight. Deficiency in this process leads to genetic instability, mutagenesis, and tumor progression (Kraemer et al., 1994).
XP has a wide range of symptom severity and clinical features among the eight complementation groups (XP-A to XP-G and XP-V). Patients mutated at one of the seven genes from XPA to XPG are NER-deficient, while mutations at POLH/XPV lead to a deficiency in TLS, a pathway involved with damage tolerance (Menck and Munford, 2014). XP-C cells lack an essential step of the DNA damage recognition at the GGR, and the XP variant (XP-V) type is NER proficient, but lacking the DNA polymerase eta function of bypassing the UV-induced lesions. The difference in which the mechanism is not working is directly linked to the phenotype of the patient. Therefore, molecular diagnosis is critical for the management of their prognosis (Fassihi et al., 2016; Lehmann and Fassihi, 2020). XP-C, XP-E, and XP-V are mainly affected by skin and eye tumors with no neurological abnormalities. XP-C patients are seriously affected with aggressive skin tumors and more than 1,000-fold increased risk of hematological malignancies, while XP-E and XP-V patients present a milder phenotype (Fassihi et al., 2016; Sarasin et al., 2019b; Oetjen et al., 2020).
This work identified a total of 13 patients (9 alive and 4 deceased) with a clinical diagnosis of XP in the city of Montanhas (state of Rio Grande do Norte-RN-, Brazil), leading to an estimated XP frequency of 1/1,120 inhabitants, considering the total population as 11,200 inhabitants in that city (Brazilian Institute of Geography and Statistics). Three patients did not participate in the study, and probably the XP incidence is underestimated due to a stigmatization effect. The denial of the disease impairs diagnosis, medical care, genetic counseling, and prevention practices reducing their quality and life expectancy. By performing the next-generation sequencing (NGS) with a specific panel of DNA repair genes, we identified homozygous mutations in two different genes: POLH (six patients) and XPC (one patient). This is the second XP genetic cluster described in Brazil (Munford et al., 2017), but the first to identify two different mutated XP genes in such a small community.
We collected saliva samples from 16 individuals (14 families), 7 XP patients (mean: 48 years of age, range: 7 to 89 years), and 9 relatives (Table 1). All patients were born in the city of Montanhas (RN) and were followed at the Hospital Universitário Onofre Lopes—HUOL/UFRN. Basal cell carcinoma (BCC) and squamous cell carcinoma (SCC) records were available from 2017 to 2021.
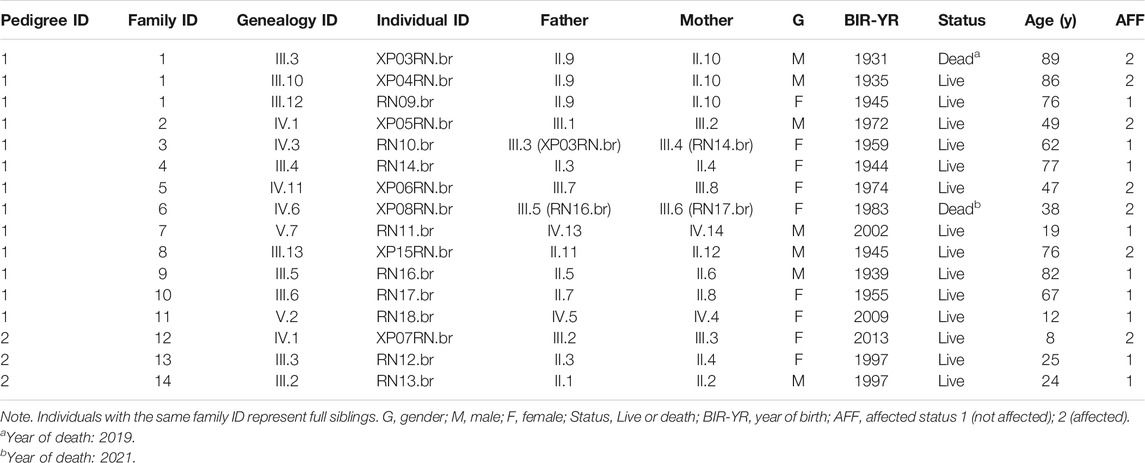
TABLE 1. Family structure information from seven patients and nine relatives.
This study had previous authorization approved by the Ethics Committee of the Institute of Biomedical Science at the University of São Paulo (ICB—USP), approval number #48347515.3.00005467. All saliva samples and pictures were collected with prior written informed consent, complied with legal and ethical rules.
The DNA Repair Laboratory received, from October 2018 to April 2019, saliva samples from 16 individuals (7 patients and 9 relatives) collected using an Oragene DNA self‐collection kit (DNA Genotek Inc., Ottawa, Canada). We extracted the genomic DNA as recommended by the protocols of the manufacturers. DNA quantity and quality were analyzed by Nanodrop-1000 (Wilmington, DE, USA) and 1% agarose gel electrophoresis. We used a customized DNA repair panel to prepare the DNA library for NGS from samples of the patients, as previously described (Menck and Munford, 2014). The alignment and variant calling were performed at Surecall software v3.5.1.46 (Agilent®), using the GRCh37/hg19 version of the reference human genome. Following the assertion criteria established by the DNA repair laboratory, we validated the mutations by Sanger sequencing and classified the variants according to the American College of Medical Genetics (ACMG) guidelines, using the VarSome variants search Engine (Richards et al., 2015; Kopanos et al., 2019). The DNA repair laboratory assertion criteria and the accessions for this submission (SCV001652706 - SCV001652707) can be found on the ClinVar database (ClinVar data access: https://www.ncbi.nlm.nih.gov/clinvar/submitters/500218/). We performed the segregation analysis of the mutation at the XPC (3p25.1; NC_000003.11; NM_004628.4) and POLH (6p21.1; NC_000006.11; NM_006502.2) genes by genotyping the relatives of the patient. Variants were reported at the nucleotide and protein levels as recommended by the Human Genome Variation Society (HGVS) (den Dunnen et al., 2016). The pedigrees were drawn following the Standardized Human Pedigree Nomenclature (Bennett et al., 1995).
We provided the molecular diagnosis for 7 XP patients, out of 13, at Montanhas, a small city in the south of Rio Grande do Norte (RN), Brazil. Different from the genetic cluster previously reported at Araras (Goiás, Brazil) (Munford et al., 2017), Montanhas is not an isolated community. Natal, the state capital, is 60 miles away from Montanhas (approximately 1 h and a half of paved road by car) where patients receive medical care (Figure 1).
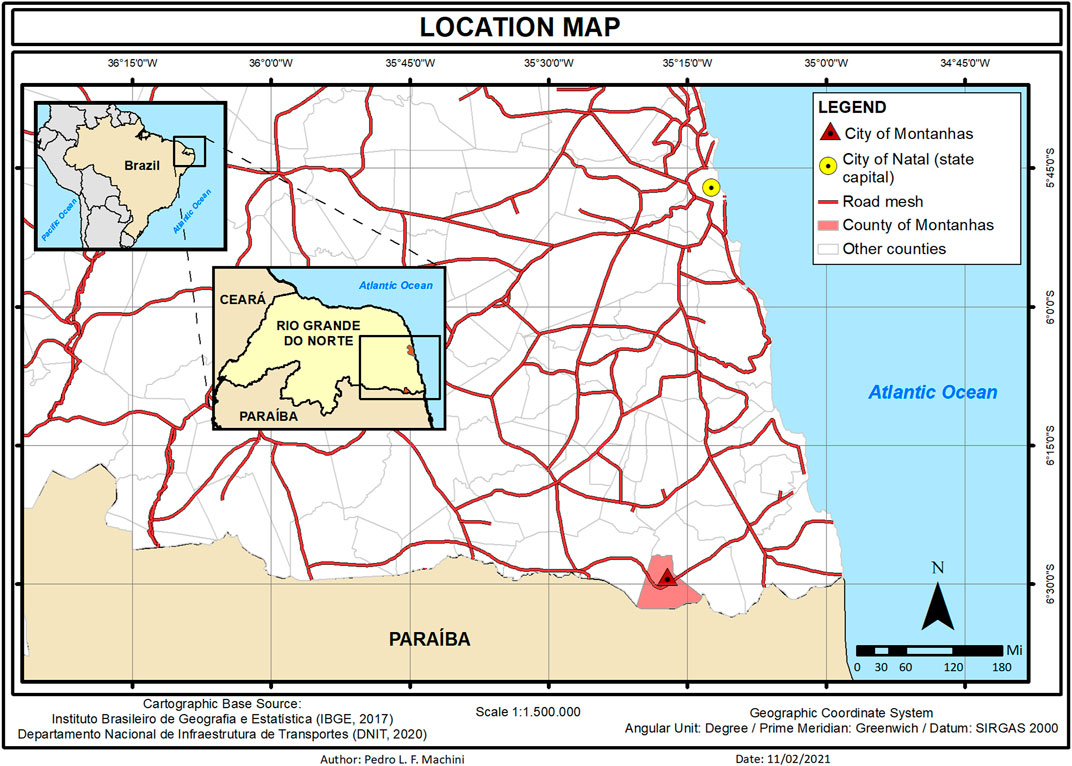
FIGURE 1. Location map from the city of Montanhas in the state of Rio Grande do Norte (RN) in the northeast of Brazil. Patients receive medical care 60 miles away in the city of Natal (capital of the state).
The majority of the patients were born from consanguineous marriages. All of them presented the typical clinical phenotype of XP with no neurological abnormalities: photosensitivity, actinic keratosis, basal and squamous cell carcinomas (BCC and SCC), freckling, hyper- and hypopigmentation, and ophthalmologic abnormalities as ocular lesions and blurred vision (Figure 2 and Table 2). None of them reported sunburn after sun exposure. The different clinical symptoms comparing patient XP07RN.br and the other XPs call attention, as one would expect the same mutation affecting all patients in this small community.
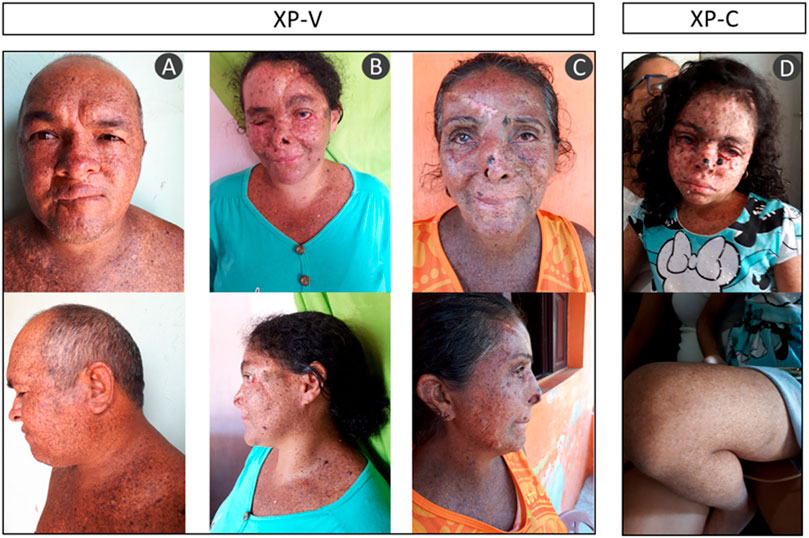
FIGURE 2. Patient phenotype from three XP-V: (A) XP04RN.br, (B) XP08RN.br, (C) XP06RN.br, and one XP-C (D) XP07RN.br. The pictures are shown with the consent of the patients.

TABLE 2. Clinical symptoms from the XP-V and XP-C patients from Montanhas (RN).
Molecular genetic analysis revealed two mutations, each one in two different genes. Most of the patients (six) carry a homozygous insertion that leads to a frameshift mutation in the POLH gene and are, thus, XP-V (OMIM #278750). The map location of the mutation according to GRCh37/hg19 sequence reference is NC_000006.11: g.43568736_43568737insT; NM_006502.2: c.672_673insT; NP_006493.1: p.Leu225Serfs*33. The patient XP07RN.br, however, carries a homozygous mutation at a canonical splice site in the XPC gene (NC_000003.11: g.14190232C > G; NM_004628.4: c.2251-1G>C) and, therefore, is XP-C (OMIM #278720).
The mean age of the XP-V patients was 63 years old (range: 37–89 years), with the first symptoms (skin lesions) at 16 years (range: 12–20 years). Only one of them developed melanoma at 72 years. The oldest XP-V patient (XP03RN.br) died in 2019 at 89 years old. According to his medical record, he went to the Hospital bedridden due to a stroke. Approximately 2 weeks before, he developed edema with nodulation in the right submandibular angle, suspected of melanoma metastasis. He was treated with amoxicillin–clavulanate and died a few months later, probably not related to the XP phenotype. In October of 2021, the patient XP08RN.br died (38 years), and we did not have access to the cause of death.
The XP-C patient, XP07RN.br, presented an aggressive phenotype with the first symptoms before the first year of life with extreme photophobia and ophthalmologic sensitivity. Around 3–4 months of age, her eyes used to water a lot, with difficulties to open in bright environments. At 7 months of age, there was a medical concern about the blood result test from serum thyroid-stimulating hormone level (TSH) of 9.35 mIU/L (reference from 0 to 11 months: 0.8–6.3 mIU/L). However, the follow-up showed that the level decreased 3 months later to 4.95 mIU/L and 1 month later to 3.59 mIU/L. In 2020, at 8 years of age the level is 6.83 mIU/L, in the limit of the normal reference. Freckles got intense after 1 year old, and at 4 years, she had her first tumor onset in the left eye. As her family had to wait 1 year for the surgery due to the lack of medical assistance available in the location, the tumor grew rapidly, and unfortunately, the eye had to be enucleated.
In October 2020 (7 years), she presented a melanoma onset at the glabella region and nasal dorsum. The lymph node was not evaluated. The surgery completely removed the melanoma and she is being treated with interferon (10.000 UI/m2) three times a week for a total of 48 weeks. There was an excellent response to the treatment. An improvement was observed in the behavior of the child, who now is more willing to talk and interact in the clinic.
The POLH insertion c.672_673insT (p.Leu225Serfs*33) was not previously reported in an XP patient. However, a deletion at Lys224, c.672delA (p.Lys224Trpfs229), was reported in two XP patients from Algeria (Opletalova et al., 2014). The patients, XP888VI and XP968VI (siblings), who are compound heterozygote for c.672delA and c.2251-1G>C, presented a mild clinical phenotype, similar to the XP-V patients from Montanhas. Due to their genomic proximity, we used Sanger sequencing to compare these two mutations (from Brazil and Algeria) and confirmed that these are different mutations (Supplementary Figure S1).
We genotyped 13 individuals from pedigree 1: 1 wild type, 6 heterozygotes, and 6 homozygotes mutated (Figure 3). Other XP patients are indicated in the figure, based on information from the family, with no genetic analysis for the mutation.
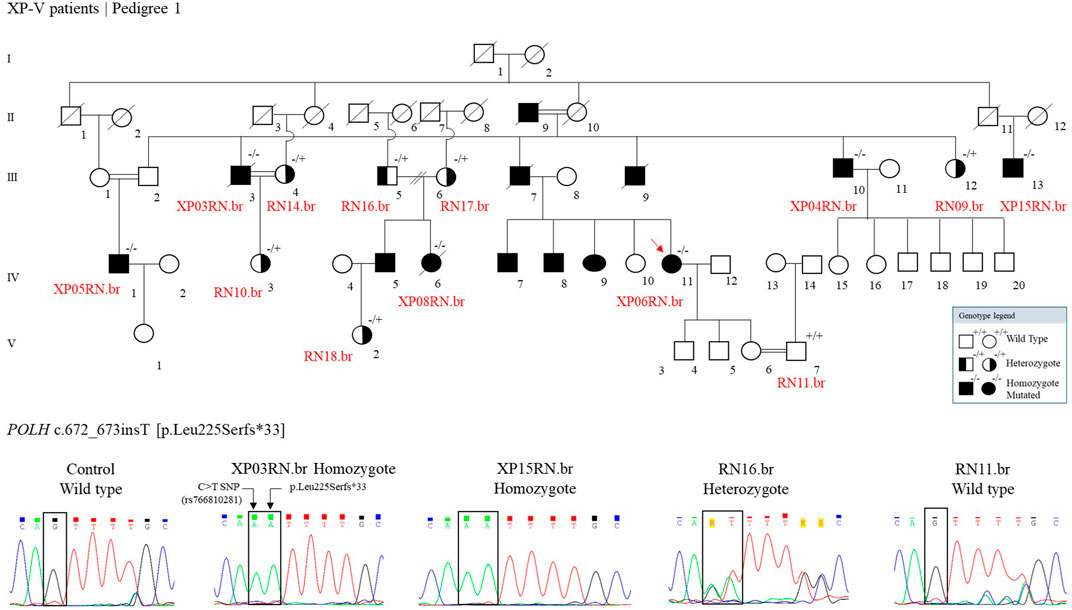
FIGURE 3. The genetic segregation of the p.Leu225Serfs*33 mutation was confirmed in the families from the community. Genealogy and genotype from the XP-V family at Pedigree 1. Electropherogram picture of the Sanger reaction at the POLH mutation site (reverse strand). The p.Leu225Serfs*33 comes alongside a C > T polymorphism (rs766810281) that leads to a synonymous exchange (p.Leu225Leu) and is presented in the Latin American and European populations and is probably segregating with the mutation.
The Genome Aggregation database (gnomAD, https://gnomad.broadinstitute.org/) reported the p.Leu225Serfs*33 (rs772778835) variant in heterozygous state in Latin American (3 out of 34,592 alleles) and European (1 out of 113,736 alleles) populations with a total allele frequency of 0.000008 and 0.000016, respectively. Although the mutation was identified in a low frequency in heterozygosity in Latin American, it was not found in Brazilian databases of germline exonic variants (ABraOM/https://abraom.ib.usp.br/; SELAdb http://intranet.fm.usp.br/sela).
One patient, out of seven, was homozygote for the splicing site mutation, c.2251-1 G>C, in the XPC gene (patient XP07RN.br, Figure 4). This mutation was first described in individuals from the western Indian Ocean in Comoros, IVS 12–1 G>C, the “Comoros mutation” (Cartault et al., 2011), and is also present in the ClinVar database (ClinVar ID 190213). A further ancestry study indicated that these patients were derived from the Bantu group and had a founder effect on the Comoros island about 800 years ago (Sarasin et al., 2019a). At gnomAD and TOPMed databases, this splicing site mutation (rs754673606) was observed in heterozygosity with an allele frequency of 0.00002 and 0.00011, respectively, observed in the African/African-American and Latino/Admixed American population. This mutation has a high incidence in XP patients in Brazil: 15 cases out of 32 (Santiago et al., 2020). The Bantu African people, mainly located on the Atlantic coast of Africa (Congo and Angola) and on the Indian Ocean coast (Mozambique), were one of the major groups (90%) brought to Brazil during the 500 years of the slave trade (Lemos Cardoso and Farias Guerreiro, 2006). Thus, probably, this XPC mutation came with these slaves. Besides Comoros and Brazil, the mutation was also reported in Europe (in a patient with a Middle Eastern origin), Mozambique, and Pakistan (Fassihi et al., 2016; Ijaz et al., 2019; Kgokolo et al., 2019). The information on the ancestry of patients from Montanhas is almost null. When interrogated about the origin of their grandparents and great-grandparents, the patients reported that their families had always been from the region, and we were not able to determine the ethnic origins of these families. This story has probably been lost in the memory of the community.
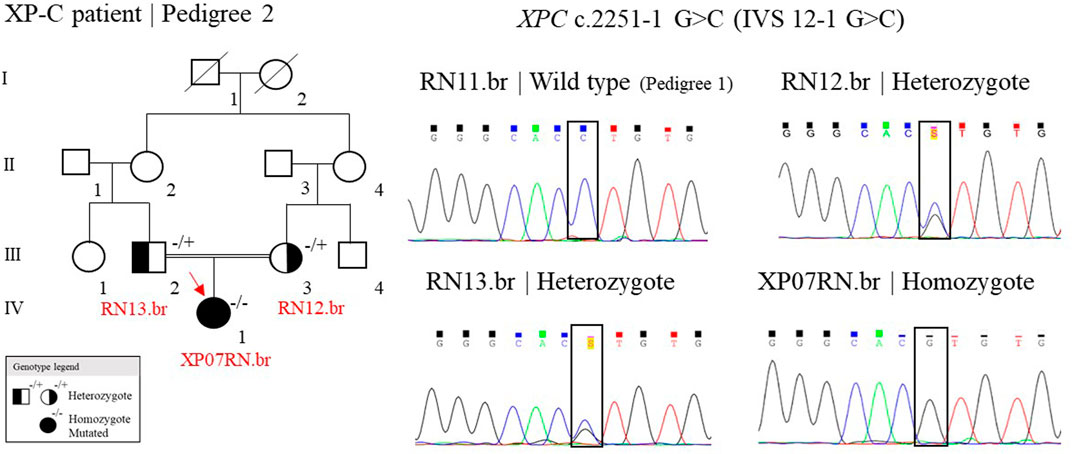
FIGURE 4. Genealogy and genotype from the XP-C family at Pedigree 2 and the electropherogram picture of the Sanger reaction at the XPC mutation site. Samples genotyped by Sanger have their ID. The red arrow indicates the patient sequenced by NGS. (+/+) represent wild type, (−/+) heterozygote, and (−/−) homozygote mutated.
This is the first report of an XP genetic cluster with two different mutated genes. The approach of NGS to carry out molecular diagnosis streamlined precision medicine care. After 50 years of molecular diagnosis and clinical findings on XP, we now know that the XP-C complementation group (NER-deficient) is particularly more prone to develop internal tumors than the other XP groups (Sarasin et al., 2019b). Cells from XP-V patients are NER-proficient, which would explain their milder phenotype, and the tumorigenesis is due to a defective mechanism of TLS during DNA replication. The difference in the molecular mechanism is directly linked to the clinical phenotype and prognosis of the patient. Establishing the culture of primary fibroblasts for functional assays, mainly from the patients with the novel insertion at POLH, could bring a better understanding of the molecular mechanisms involved with the clinic phenotype of these patients.
In Montanhas, none of the patients received educational materials to learn how to perform the appropriate photoprotection. Without financial conditions and government support, none of them protects themselves from sun exposure adequately. Even in this extreme condition, where the XP patients have lived under a high amount of sunlight exposure, only one XP-V (out of six) developed melanoma at 73 years of age, which is different from the XP-C patient, who was diagnosed with a melanoma at 7 years of age, indicating that the variation in clinical phenotypes observed in the XP patients might be due to the different mutated genes involved in their prognosis.
In this work, we reported a small city in the Northeast region of Brazil (Montanhas, RN), with patients mutated at the XPC or POLH gene. The prognosis of the disease varies according to the mutated gene, the affected repair pathway, and the level of individual photoprotection. The XP-C patient had the most severe symptoms, which appeared before the first year of life, and a melanoma onset at 7 years old. For the XP-V patients, the mean age for the first symptoms was 16 years, and one developed melanoma at 73 years. The patients report minimal photoprotection using sporadic sunscreen when they have it. The mother of the XP-C is the most informed and worried about protection and prevention, mainly due to the high photosensitivity of the child. The difficulty in accessing medical care at the locality exacerbates the worst prognosis of a disease such as XP, especially the XP-C group, with an urgent need for continuous care and tumor excision. It should be noted that the low latitude of the location, near the equator line (approximately 445 miles), implicates high levels of DNA damage by sunlight (Schuch et al., 2012). There is an imperative demand on investing in prevention, providing them physical photoprotection and educational support, more importantly, facilitating access to medical care and prioritizing urgency for these patients.
The mutations were reported at ClinVar, accession number VCV001119983.1 (NM_006502.3(POLH):c.672_673insT (p.Leu225fs), and SCV001652707.1 (NM_004628.5(XPC):c.2251-1G>C).
The studies involving human participants were reviewed and approved by the Ethics Committee of the Institute of Biomedical Science at the University of São Paulo (ICB-USP) 48347515.3.0000.5467. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s) and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
LC performed the experiments, analysis, and interpretation of data and drafted the manuscript. TS and DV performed the experiments and data analysis. RR performed the experiments, analysis and, critically revised the manuscript. AT, JC, and IA contributed to the recruiting of patients and sample collection. SH and FA were the physicians responsible for patient follow-up and contributed to the clinical investigation of the patients. PK contributed to the mutation analysis and manuscript revision. AS critically revised the manuscript. CM supervised the study and critically revised the manuscript. TBP contributed to the recruiting of patients, sample collection, supervised the study, and critically revised the manuscript.
This work was supported under the International Collaboration Research Funding from São Paulo Research Foundation (FAPESP, SP, Brazil) and Netherlands Organization for Scientific Research (NWO, Netherlands), FAPESP Grants # 2019/19435-3 and #2013/08028-1), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Grant # 308868/2018-8), and Coordenação de Aperfeiçoamento de Pessoal do Ensino Superior (CAPES, Brasília, DF, Brazil, financial code 001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are especially grateful to the XP patients from Montanhas and to the ABRAXP, the Brazilian XP support group, for their contribution to this research. We are grateful to Veridiana Munford, who enables and coordinates the research at the DNA Repair laboratory at ICB-USP. We also thank the Core Facility for Scientific Research—University of São Paulo (CEFAP-USP/GENIAL) for providing NGS and Pedro L. F. Machini for providing the map figure.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.784963/full#supplementary-material
Bennett, R. L., Steinhaus, K. A., Uhrich, S. B., O’sullivan, C. K., Resta, R. G., Lochner-Doyle, D., et al. (1995). Recommendations for Standardized Human Pedigree Nomenclature. J. Genet. Couns. 56 (3), 745–752. doi:10.1007/BF01408073
Cartault, F., Nava, C., Malbrunot, A.-C., Munier, P., Hebert, J.-C., N’guyen, P., et al. (2011). A New XPC Gene Splicing Mutation Has lead to the Highest Worldwide Prevalence of Xeroderma Pigmentosum in Black Mahori Patients. DNA Repair 10, 577–585. doi:10.1016/j.dnarep.2011.03.005
den Dunnen, J. T., Dalgleish, R., Maglott, D. R., Hart, R. K., Greenblatt, M. S., McGowan-Jordan, J., et al. (2016). HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 37 (6), 564–569. doi:10.1002/humu.22981
Fassihi, H., Sethi, M., Fawcett, H., Wing, J., Chandler, N., Mohammed, S., et al. (2016). Deep Phenotyping of 89 Xeroderma Pigmentosum Patients Reveals Unexpected Heterogeneity Dependent on the Precise Molecular Defect. Proc. Natl. Acad. Sci. U.S.A. 113, E1236–E1245. doi:10.1073/pnas.1519444113
Hanawalt, P. C. (2002). Subpathways of Nucleotide Excision Repair and Their Regulation. Oncogene 1621 (58), 8949–8956. doi:10.1038/sj.onc.1206096
Ijaz, A., Basit, S., Gul, A., Batool, L., Hussain, A., Afzal, S., et al. (2019). XPC Gene Mutations in Families with Xeroderma Pigmentosum from Pakistan; Prevalent Founder Effect. Congenit. Anom. 59, 18–21. doi:10.1111/cga.12281
Kgokolo, M., Morice‐Picard, F., Rezvani, H. R., Austerlitz, F., Cartault, F., Sarasin, A., et al. (2019). Xeroderma Pigmentosum in South Africa: Evidence for a Prevalent Founder Effect. Br. J. Dermatol. 181, 1070–1072. doi:10.1111/bjd.18030
Kopanos, C., Tsiolkas, V., Kouris, A., Chapple, C. E., Albarca Aguilera, M., Meyer, R., et al. (2019). VarSome: the Human Genomic Variant Search Engine. Bioinformatics 35 (11), 1978–1980. doi:10.1093/bioinformatics/bty897
Kraemer, K. H., Lee, M. M., Andrews, A. D., and Lambert, W. C. (1994). The Role of Sunlight and DNA Repair in Melanoma and Nonmelanoma Skin Cancer. Arch. Dermatol. 130, 1018–1021. doi:10.1001/archderm.1994.01690080084012
Lehmann, A. R., and Fassihi, H. (2020). Molecular Analysis Directs the Prognosis, Management and Treatment of Patients with Xeroderma Pigmentosum. DNA Repair 93, 102907. doi:10.1016/j.dnarep.2020.102907
Lemos Cardoso, G., and Farias Guerreiro, J. (2006). African Gene Flow to North Brazil as Revealed by HBB*S Gene Haplotype Analysis. Am. J. Hum. Biol. 18 (1), 93–98. doi:10.1002/ajhb.20467
Menck, C. F., and Munford, V. (2014). DNA Repair Diseases: What Do They Tell Us about Cancer and Aging? Genet. Mol. Biol. 37, 220–233. doi:10.1590/s1415-47572014000200008
Munford, V., Castro, L. P., Souto, R., Lerner, L. K., Vilar, J. B., Quayle, C., et al. (2017). A Genetic Cluster of Patients with Variant Xeroderma Pigmentosum with Two Different Founder Mutations. Br. J. Dermatol. 176 (5), 1270–1278. doi:10.1111/bjd.15084
Oetjen, K. A., Levoska, M. A., Tamura, D., Ito, S., Douglas, D., Khan, S. G., et al. (2020). Predisposition to Hematologic Malignancies in Patients with Xeroderma Pigmentosum. Haematologica 105 (4), e144–e146. doi:10.3324/haematol.2019.223370
Opletalova, K., Bourillon, A., Yang, W., Pouvelle, C., Armier, J., Despras, E., et al. (2014). Correlation of Phenotype/Genotype in a Cohort of 23 Xeroderma Pigmentosum-Variant Patients Reveals 12 New Disease-CausingPOLHMutations. Hum. Mutat. 35, 117–128. doi:10.1002/humu.22462
Richards, S., Aziz, N., Aziz, N., Bale, S., Bick, D., Das, S., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–423. doi:10.1038/gim.2015.30
Santiago, K. M., Castro, L. P., Neto, J. P. D., Nóbrega, A. F., Pinto, C. A. L., Ashton‐Prolla, P., et al. (2020). Comprehensive Germline Mutation Analysis and Clinical Profile in a Large Cohort of Brazilian Xeroderma Pigmentosum Patients. J. Eur. Acad. Dermatol. Venereol. 34 (10), 2392–2401. doi:10.1111/jdv.16405
Sarasin, A., Munier, P., and Cartault, F. (2019a). How History and Geography May Explain the Distribution in the Comorian Archipelago of a Novel Mutation in DNA Repair-Deficient Xeroderma Pigmentosum Patients. Genet. Mol. Biol. 43, e20190046. doi:10.1590/1678-4685-GMB-2019-0046
Sarasin, A., Quentin, S., Droin, N., Sahbatou, M., Saada, V., Auger, N., et al. (2019b). Familial Predisposition to TP53/complex Karyotype MDS and Leukemia in DNA Repair-Deficient Xeroderma Pigmentosum. Blood 133, 2718–2724. doi:10.1182/blood-2019-01-895698
Keywords: genetic cluster, xeroderma pigmentosum, molecular diagnosis, DNA repair, skin cancer
Citation: Castro LP, Batista-Vieira D, de Souza TA, Timoteo ARdS, Coutinho JDL, Pinheiro de Almeida IC, Henriques SRdM, Azevedo FMd, Rosa RCA, Kannouche PL, Sarasin A, Menck CFM and Petta TB (2022) XPC and POLH/XPV Genes Mutated in a Genetic Cluster of Xeroderma Pigmentosum Patients in Northeast Brazil. Front. Genet. 12:784963. doi: 10.3389/fgene.2021.784963
Received: 29 September 2021; Accepted: 16 November 2021;
Published: 17 January 2022.
Edited by:
Lawrence Todd Reiter, University of Tennessee Health Science Center (UTHSC), United StatesReviewed by:
Natalie Saini, Medical University of South Carolina, United StatesCopyright © 2022 Castro, Batista-Vieira, de Souza, Timoteo, Coutinho, Pinheiro de Almeida, Henriques, Azevedo, Rosa, Kannouche, Sarasin, Menck and Petta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tirzah Braz Petta, dGlyemFoLnBldHRhQHVzYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.