
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 20 December 2021
Sec. Neurogenomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.783841
This article is part of the Research Topic Epigenetic Regulation of Gene Expression in Genetic or Acquired Forms of Epilepsy View all 6 articles
 Yueyun Lan1,2
Yueyun Lan1,2 Sheng Yi1,2
Sheng Yi1,2 Mengting Li2,3
Mengting Li2,3 Jinqiu Wang4
Jinqiu Wang4 Qi Yang1,2Shang Yi1,2
Qi Yang1,2Shang Yi1,2 Fei Chen1,2Limei Huang1,2
Fei Chen1,2Limei Huang1,2 Yiyan Ruan1
Yiyan Ruan1 Yiping Shen1,2,5Jingsi Luo1,2*
Yiping Shen1,2,5Jingsi Luo1,2* Zailong Qin1,2*
Zailong Qin1,2*Christianson syndrome (CS) is an X-linked neurodevelopmental syndrome characterized by microcephaly, epilepsy, ataxia, and severe generalized developmental delay. Pathogenic mutations in the SLC9A6 gene, which encodes the Na+/H+ exchanger protein member 6 (NHE6), are associated with CS and autism spectrum disorder in males. In this study, whole exome sequencing (WES) and Sanger sequencing revealed a novel de novo frameshift variant c.1548_1549insT of SLC9A6 in a 14-month-old boy with early-onset seizures. According to The American College of Medical Genetics and Genomics (ACMG)/the Association for Molecular Pathology (AMP) guidelines, the variant was classified as pathogenic. The proband presented with several core symptoms of typical epilepsy, including microcephaly, motor delay, distal muscle weakness, micrognathia, occasional unprovoked laughter, swallowing and speech difficulties. Electroencephalography (EEG) showed spikes-slow waves in frontal pole, frontal, anterior temporal and frontal midline point areas. Gesell development schedules (GDS) indicated generalized developmental delay. We also summarized all the reported variants and analyzed the correlation of genotype and phenotype of CS. Our study extends the mutation spectrum of the SLC9A6 gene, and it might imply that the phenotypes of CS are not correlated with SLC9A6 genotypes.
Christianson syndrome (CS) is induced by sudden abnormal discharge of brain neurons, and is characterized by recurrent seizures due to hypersynchronous discharge (Liu et al., 2018). Symptoms of CS often begin in childhood, with a complex clinical manifestation that is associated with social and learning disabilities. Moreover, severe epilepsy comprises a group of devastating neurological disorders characterized by frequent epileptic seizures associated with developmental delay or regression (McTague et al., 2016). Each type of epilepsy has specific clinical manifestations and distinct underlying pathophysiologic or genetic mechanisms. It is believed that identifying the underlying cause of epilepsy or specific electroclinical syndrome is central to understand the natural history and optimal treatment of the condition (Wirrell, 2016).
To date, multiple genes including Na+/H+ exchanger protein 6 (NHE6, also known as SLC9A6) have been linked to CS (McTague et al., 2016). NHE6 plays a role in both excitatory presynaptic and postsynaptic functions (Deane et al., 2013). Additionally, NHE6 is highly expressed in the brain, and is thought to participate in the targeting of intracellular vesicles and recycling of synaptic vesicles. Therefore, mutations leading loss of function in SLC9A6 protein may damage the development of neurons (Mignot et al., 2013). Furthermore, SLC9A6 has been identified as a pathogenic gene (Ikeda et al., 2020) and its pathogenic variations can lead to an Angelman-like syndrome (AS-like, known as CS) (Cardon et al., 2016), which is characterized by epilepsy, intellectual disability, microcephaly, ophthalmoplegia, craniofacial dysmorphism and progressive cerebellar atrophy (Padmanabha et al., 2017).
Childhood epilepsy, especially the seizures, is manifested in the context of brain development and can change over time. CS in children presents a wide range of treatment challenges that are unique to this age group. Therefore, it is important to distinguish a de novo mutation caused childhood epilepsy or hereditary childhood epilepsy, and effective therapies for different forms of epilepsy should be targeted to the individual child according to the type of seizures. In this study, we identified a novel and de novo SLC9A6 frameshift mutation c.1548_1549insT in the patient with tonic-clonic seizures and generalized developmental delay by whole exome sequencing (WES). We also summarized the pathogenic variants reported previously and widen the spectrum of SLC9A6 mutations.
Written informed consent for participation in this study was collected from the family members of patients. The study was approved by the ethics committees of Maternal and Child Health Hospital of Guangxi Zhuang Autonomous Region.
The patient was a 14-month-old boy who presented with convulsions for a month without apparent trigger. In order to clarify the etiology and clinical diagnosis, the patient underwent physical examination and clinical auxiliary examination during hospitalization, including rehabilitation training treatment, three usual medical practices, blood biochemistry, 24-hour ambulatory electroencephalogram, visual-auditory synchrony evoked potential, abdominal ultrasonography (US), the cerebrospinal fluid (CSF), routine electrocardiogram (ECG), thyroid function teats and genetic testing (WES and Sanger sequencing).
Genomic DNA was isolated from peripheral blood lymphocytes using LabAid DNA kit (Zeesan Biotech Co., Ltd, Xiamen, China). For WES, genomic DNA sample of the patient were captured to create a sequencing library by Agilent SureSelect Clinical Research Exome V2 Kit (Agilent Technologies, Santa Clara, CA), and the prepared libraries were sequenced on a HiSeq2500 (Illumina, San Diego, CA). Sequence alignment and variant calling against the reference human genome (GRCh37) were performed using Burrows-Wheeler Aligner (BWA) and the Genome Analysis Toolkit (GATK) (Van der Auwera et al., 2013). Copy number variants (CNV) analysis based on reads depth method was performed using an inhouse pipeline, and CNVs of significant interest were further visually inspected with the Integrative Genomics Viewer. CNVs and indels were annotated and prioritized by the TGex software (LifeMap Sciences, Alameda, CA). The variant pathogenicity was assessed according to American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) guidelines (Richards et al., 2015). All operations were carried out according to the instructions of the manufacturer.
The patient was delivered to term with a weight of 3200 g at birth, did not undergo suffocation rescue when he was born, and had jaundice but did not receive phototherapy, adopt exclusive breastfeeding after birth and give supplementary food on time. The patient lagged behind his peers in growth and development. At the age of 5 months, he could roll over; at 6 months, he could hold his head up and at 9 months, he could sit up. At the age of 14 months, he could crawl and stand with support. However, tonic-clonic seizures began at 13 months of age and occurred several times a day. Examination revealed microcephaly (43cm, −3SD), motor delay, distal muscle weakness, micrognathia, convulsions, slender fingers, adduction of the thumbs and occasional unprovoked laughter. He also had swallowing and speech difficulties. We used Gesell development schedules (GDS) to assess the level of development. The GDS revealed the patient had delays in adaptation, large motor movement, fine motor movement, personal-social development (mild), and language (moderate). The patient was diagnosed with tonic-clonic seizures and generalized developmental delay. Therefore, it is suggested that the patient should be treated with rehabilitation training. Figure 1 showed the results of sleep EEG using common average montage, EEG demonstrated that 3.5–8 Hz diffuse wave as background activity, spike-slow waves discharged in frontal pole, frontal, anterior temporal and frontal midline point areas. Brain magnetic resonance imaging, brain-stem auditory evoked potential and visual evoked potential showed no abnormal findings.
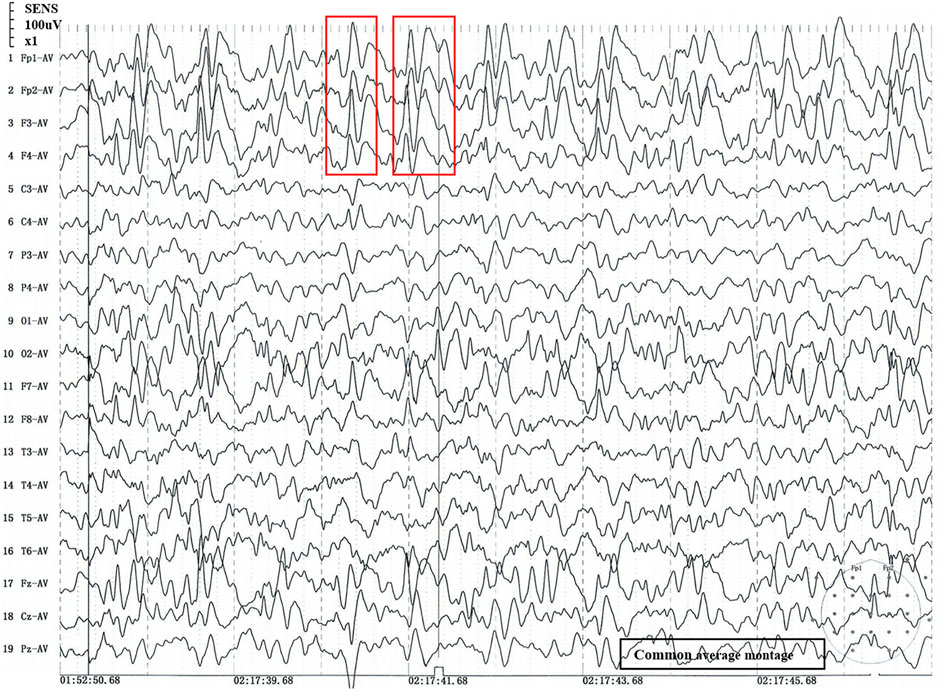
FIGURE 1. Sleep stage II EEG. 1) 1 to 19: Recording electrode. 2) Fp1-AV to Pz-AV: Average montage. 3) Fp1/Fp2: pre frontal lobe, F7/F8: inferior frontal lobe, F3/F4/Fz: frontal lobe, C3/C4/Cz: central lobe, T3/T4: temperal lobe, T5/T6: posterior temperal lobe, P3/P4/Pz: parietal lobe, O1/O2: occipital lobe; The number of the recording electrodes mentioned above is usually odd for the left and even for the right. 4) Red box: abnormal wave. 5) The time window is 2 s, and every 2 s in the signal is an epoch.
Relevant data from the episode of care is shown in Supplementary Table S1: 1) Blood ammonia level [54.0 μmol/L, reference range 11–35 μmol/L]. 2) Routine blood test: red blood cell (RBC) [3.9×109/L, reference range (4.3–5.8) ×1012/L], hemoglobin (Hb) [107 g/L, reference range 180–190 g/L]. 3) Lactic acid [9.41 mg/dL, reference range 12.00–16.00 mg/dL]. 4) Determination of serum immunoglobulin: immunoglobulin A (IgA) [0.30 g/L, reference range 0.70–3.30 g/L]. 5) Thyroid function: thyroxine (T4) [69.70 nmol/L, reference range 66.00–181.00 nmol/L]. 6) Routine CSF: serum sodium valproate concentration [41.14 μg/mL, reference range 50–100 μg/mL], herpes simplex virus type I, herpes simplex virus type II and CSF culture were negative. 7) Routine Conventional electrocardiogram (ECG) examination found that the patient had sinus tachycardia with irregularities. 8) 25-hydroxy vitamin D level, 25-hydroxy vitamin D3, blood gas analysis, ferritin, calcitonin, electrolytes, zinc, iron and Anti-Streptolysin O, stool routine test and abdominal ultrasound were normal. The results of other related testing items involved were normal.
A novel frameshift mutation, c.1548_1549insT (p. Leu517fs*5), was identified in the SLC9A6 gene (NM_006359.2) by WES analysis. This variant was absent in the Clinvar Database, the Single Nucleotide Polymorphism database and the Human Gene Mutation Database. The novel variant was validated by Sanger sequencing. The child’s family history was unremarkable. The pedigree of the family was shown in Figure 2A. SLC9A6 mutation (c.1548_1549insT) was not detected in peripheral blood samples obtained from the parents (Figure 2B). The mutation was considered to be de novo. The paternity was confirmed by short tandem repeat markers. According to the ACMG/AMP guidelines, this novel and de novo frameshift variant was classified as a pathogenic variation (PVS1+PM2_supporting+PS2+PP4): a null variant (frameshift) was confirmed in the gene that loss of function is a known mechanism of disease (PVS1); a de novo variant (both maternity and paternity confirmed) in a patient with the disease and without family history (PS2); absent from controls (or at extremely low frequency if recessive) in the Exome Sequencing Project, 1,000 Genomes Project, Exome Aggregation Consortium or gnomAD databases (PM2).
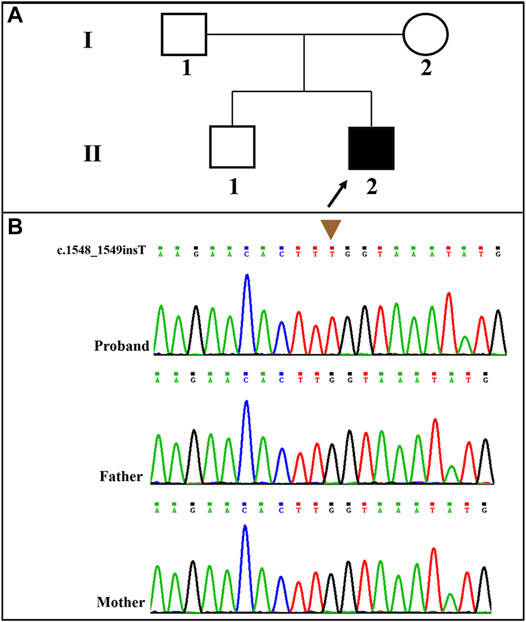
FIGURE 2. (A): Pedigree of the proband; (B): The patient carried a novel and de novo hemizygous SLC9A6 mutation (NM_006359.2: c.1548_1549insT) that was confirmed by Sanger sequencing.
The main clinical manifestations of patients with central nervous system infection are fever, convulsions, disturbance of consciousness and increased intracranial pressure. In addition, these patients had poor mental state and CSF was altered accordingly. Genetic metabolic diseases can lead to feeding difficulty, developmental delay, convulsions, specific facial features, electrolyte abnormalities, damage of important organs. Physical examination showed rash and hepatosplenomegaly. However, no obvious abnormalities were found in nervous system and skin examination of our patient. In view of this, it can be diagnosed that the convulsions of our patient were not caused by central nervous system infection or genetic metabolic diseases. The decrease of glucose content in CSF is the characteristic of glucose transporter type 1 deficiency syndrome, but the results of CSF examination in our patient were normal, so it can be ruled out that seizures are related to glucose transporter-1 deficiency syndrome. Electrolyte disorder, such as low calcium and low magnesium, can cause convulsions, but the diagnosis was not supported because our patient had no abnormal electrolyte after admission. To summarize, epilepsy with tonic-clonic seizure and generalized developmental delay of the patient was caused by SLC9A6 mutation.
CS is a rare but increasingly diagnosed neurodevelopmental and degenerative form of X-linked intellectual disability disorder. NHE6 regulates recycling endosomal pH homeostasis and trafficking (Petitjean et al., 2020). The ion transporter is abundantly expressed in the central nervous system (CNS), which may explain why the neural phenotype of CS is so complex and diverse. Therefore, the identification of NHE6-dependent specific receptors helps to clarify the underlying mechanism of neuronal dysfunction in CS. Endosomal acidification as a result of a non-functional or absent NHE6 caused by SLC9A6 gene mutation may affect the transport of vesicles containing AMPA receptors to and from the postsynaptic membrane (Shepherd and Huganir, 2007). Brain-derived neurotrophic factor (BDNF)/tropomyosin receptor kinase B (TrkB) neurotrophin signaling pathway are necessary for correct dendrite development in many neurons in the CNS (Barford et al., 2017). Coupled with accelerated degradation of TrkB, this over-acidification can lead to a disruption of endosomal BDNF/TrkB signaling, resulting in the failure of neuronal axons and dendritic branches (Fuster and Alexander, 2014).
In addition, microtubule-associated proteins (MAP) also play a significant role in neurodevelopment and growth. Tau protein is the main microtubule associated protein of mature neurons. The spatiotemporal expression of different tau isoforms is a characteristic during brain development (Avila et al., 2004), suggesting that the regulation of tau isoforms is important during the formation of the brain (Lee et al., 1989). Moreover, deposition of tau may be mediated by the interaction with the mutant SLC9A6 protein (Garbern et al., 2010). After using a conservative genome-wide correction, it was found that the low expression of SLC9A6 was significantly related to increased tau deposition (Pescosolido et al., 2019). The most obvious pathological event in several neurodegenerative diseases is the aggregation of tau subtypes into the filamentous content of neurons, resulting in hyperphosphorylation of tau proteins (Buée et al., 2000). The c.1548_1549insT pathogenic variant in the SLC9A6 gene causes a frameshift starting at codon Leucine 517, which changes this amino acid to a proline residue, and creates a premature stop codon at position 5 of the new reading frame, denoted p.Leu517fs*5 which cause premature termination of NHE6 protein synthesis and may greatly reduce the stability of tau proteins. It can be further speculated that epilepsy and developmental delay in our patient may be due to the inability of the mutant SLC9A6 to compensate in those tissues.
Mutations in SLC9A6 seem to cause clinical symptoms that reflect developmental and progressive pathophysiology (Pescosolido et al., 2014). Slc9a6 knockout mouse models revealed that loss of functions of SLC9A6 leads to altered endosomal-lysosomal function and cholesterol accumulation in some neuronal populations in a manner that is similarly toprimary lysosomal storage diseases (Sinajon et al., 2016). Moreover, Slc9a6−/− mutant female mouse and Slc9a6–/0 mutant male mouse showed an epileptic phenotype and reduced seizure threshold (Gilfillan et al., 2008). One of the causes of cognitive and language impairment in children with SLC9A6 mutations may be the failure of axonal and dendritic branching, leading to impaired neuronal connectivity (Roxrud et al., 2009).
Disruption of SLC9A6 should be considered in male patients with a non-15q11-13-related AS phenotype, particularly when X-linked inheritance in the family is suspected. Moreover, CS and AS share some common clinical features, and part of the patients with AS-like phenotypes carried mutation in SLC9A6 gene. Supplementary Table S2 summarizes the main clinical phenotypes of SLC9A6 mutations. AS-like phenotype was observed in the related families described by case 1 and case 2. Developmental delay, microcephaly, seizures, language retardation were present in most patients. Intellectual disability, behavior disorder and ataxia are seen in several cases. The phenotype of patients in the table is consistent with clinical diagnosis, but genotype and phenotype correlations were confirmed to be negative. Subsequently, SLC9A6 conserved domain was predicted (Figure 3). Total of 28 pathogenic variants were reported in ClinVar Database. However, 22 out of 28 (78.57%) pathogenic variants in SLC9A6 located in predicted functional domains, which include frameshift mutations, missense mutations and splicing mutations. And CS was the most reported clinical phenotype in these variations. Although 6 variants (21.43%,6/28) located in the non-domain region, 5 of them with CS located in non-domain region. It might imply that there was no significant correlation between the type of SLC9A6 gene variant and the clinical phenotype and protein domain.
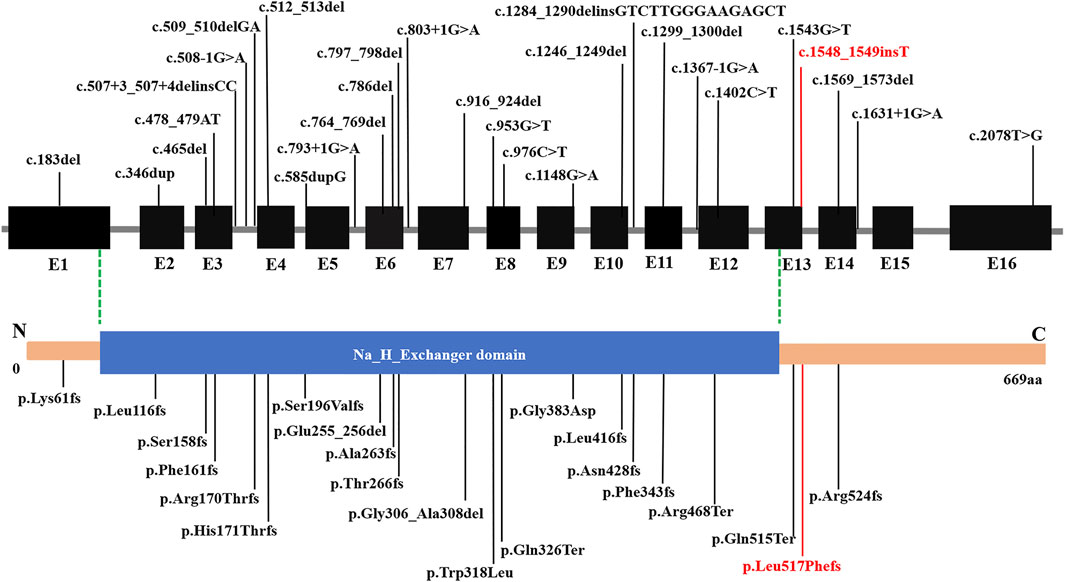
FIGURE 3. Structure of SLC9A6 protein. The SLC9A6 gene is composed of 669 amino acids and has only one domain. Predicted functional domains are shown together with the position of a novel de novo mutation in the report. SLC9A6 conserved domain was predicted by NCBI Conserved Domain Database. The black fonts: Previously reported pathogenic mutations, the red fonts: pathogenic mutations reported in this paper.
Based on the information of SLC9A6 gene variation and clinical phenotype included in ClinVar Database (SLC9A6 [gene]–ClinVar–NCBI (nih.gov)), we summarized the variants that were only caused by SLC9A6 mutations (Supplementary Table S3): 1) All frameshift mutations (N = 13) of SLC9A6 are pathogenic variations. The clinical outcomes of partial variation were related to CS and no phenotype was reported for the remaining variants. 2) Seven cases of single nucleotide variants (SNVs) were pathogenic and one case were likely pathogenic, which were also associated with CS. Most SNVs in SLC9A6 were not pathogenic, but the phenotypes of most patients with SNVs were typical CS. The frameshift mutations seem to be more serious than the SNVs. Taken together, no matter the type of SLC9A6 gene mutation is frameshift mutations or SNVs, the clinical phenotype mainly characterized by CS and developmental disorder. Moreover, the complexity of the neuronal arbors in CS patients was reduced regardless of the nature of the mutation and the deficiency may contribute to postnatal microcephaly in CS (Lizarraga and Ma, 2021). The great majority of CS patients with SLC9A6 mutations appear to be result in loss of function of the relevant proteins, such as putatively protein-truncating due to early frameshift, nonsense or splicing mutations, while some missense or intra-frame deletions may be residual protein (Pescosolido et al., 2014). The most recent research indicates that most CS mutations lead to loss of protein through nonsense-mediated decay (NMD) mechanisms, the loss and expression of wild-type SLC9A6 in cells with an SLC9A6 nonsense mutation could rescue the neuronal arborization defect; But cells with recurrent missense mutation (e.g., c.1148G > A, p.G383D) in the SLC9A6 gene that resulted in a residual, nonfunctional NHE6 protein could not be rescued (Lizarraga and Ma, 2021).
The original contributions presented in the study are publicly available. This data can be found here: PRJNA770360.
Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Conceptualization: ZQ and JL. Data collection: SeY. Data analysis: SeY, SaY, QY, and ML. Funding acquisition: YR and YS. Performed molecular: LH. Writing original draft: YL. Writing-review and editing: YL, SeY, FC, JL, and ZQ. Interpretation of EEG results: JW. All authors have read and approved the manuscript.
The “Guangxi Bagui Scholar” fund (No. 2016A20); the National Key Research and Development Program (2018YFC1002501); Science and Technology Program of Guangxi (No. AB17195011).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank the patient and his parents for approving the reporting of this case.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.783841/full#supplementary-material.
Supplementary Table S1 | A timeline of the patient's hospitalization that relevant data from the episode of care.
Supplementary Table S2 | The main clinical phenotypes of patients with SLC9A6 mutations.
Supplementary Table S3 | The variants previously reported are shown in the excel according to Clinvar Database. 1) P=pathogenic, LP=likely pathogenic, B=benign, LB=Likely benign, VUS=Uncertain significance; 2) NR=not record, CS=not specified; 3) The point mutations with Conflicting interpretations of pathogenicity are not listed here.
Avila, J., Lucas, J. J., Pérez, M., and Hernández, F. (2004). Role of Tau Protein in Both Physiological and Pathological Conditions. Physiol. Rev. 84, 361–384. doi:10.1152/physrev.00024.2003
Barford, K., Deppmann, C., and Winckler, B. (2017). The Neurotrophin Receptor Signaling Endosome: Where Trafficking Meets Signaling. Devel Neurobio 77, 405–418. doi:10.1002/dneu.22427
Buée, L., Bussière, T., Buée-Scherrer, V., Delacourte, A., and Hof, P. R. (2000). Tau Protein Isoforms, Phosphorylation and Role in Neurodegenerative disorders11These Authors Contributed Equally to This Work. Brain Res. Rev. 33, 95–130. doi:10.1016/s0165-0173(00)00019-9
Cardon, M., Evankovich, K. D., and Holder, J. L. (2016). Exonic Deletion ofSLC9A9in Autism with Epilepsy. Neurol. Genet. 2, e62. doi:10.1212/nxg.0000000000000062
Deane, E. C., Ilie, A. E., Sizdahkhani, S., Das Gupta, M., Orlowski, J., and Mckinney, R. A. (2013). Enhanced Recruitment of Endosomal Na+/H+ Exchanger NHE6 into Dendritic Spines of Hippocampal Pyramidal Neurons during NMDA Receptor-dependent Long-Term Potentiation. J. Neurosci. 33, 595–610. doi:10.1523/jneurosci.2583-12.2013
Fuster, D. G., and Alexander, R. T. (2014). Traditional and Emerging Roles for the SLC9 Na+/H+ Exchangers. Pflugers Arch. - Eur. J. Physiol. 466, 61–76. doi:10.1007/s00424-013-1408-8
Garbern, J. Y., Neumann, M., Trojanowski, J. Q., Lee, V. M.-Y., Feldman, G., Norris, J. W., et al. (2010). A Mutation Affecting the Sodium/proton Exchanger, SLC9A6, Causes Mental Retardation with Tau Deposition. Brain 133, 1391–1402. doi:10.1093/brain/awq071
Gilfillan, G. D., Selmer, K. K., Roxrud, I., Smith, R., Kyllerman, M., Eiklid, K., et al. (2008). SLC9A6 Mutations Cause X-Linked Mental Retardation, Microcephaly, Epilepsy, and Ataxia, a Phenotype Mimicking Angelman Syndrome. Am. J. Hum. Genet. 82, 1003–1010. doi:10.1016/j.ajhg.2008.01.013
Ikeda, A., Yamamoto, A., Ichikawa, K., Tsuyusaki, Y., Tsuji, M., Iai, M., et al. (2020). Epilepsy in Christianson Syndrome: Two Cases of Lennox-Gastaut Syndrome and a Review of Literature. Epilepsy Behav. Rep. 13, 100349. doi:10.1016/j.ebr.2019.100349
Lee, G., Neve, R. L., and Kosik, K. S. (1989). The Microtubule Binding Domain of Tau Protein. Neuron 2, 1615–1624. doi:10.1016/0896-6273(89)90050-0
Liu, J., Tong, L., Song, S., Niu, Y., Li, J., Wu, X., et al. (2018). Novel and De Novo Mutations in Pediatric Refractory Epilepsy. Mol. Brain 11, 48. doi:10.1186/s13041-018-0392-5
Lizarraga, S. B., Ma, L., Maguire, A. M., van Dyck, L. I., Wu, Q., Ouyang, Q., et al. (2021). Human Neurons from Christianson Syndrome iPSCs Reveal Mutation-specific Responses to rescue Strategies. Science Translational Medicine 13 (580), eaaw0682. doi:10.1126/scitranslmed.aaw0682
McTague, A., Howell, K. B., Cross, J. H., Kurian, M. A., and Scheffer, I. E. (2016). The Genetic Landscape of the Epileptic Encephalopathies of Infancy and Childhood. Lancet Neurol. 15, 304–316. doi:10.1016/s1474-4422(15)00250-1
Mignot, C., Héron, D., Bursztyn, J., Momtchilova, M., Mayer, M., Whalen, S., et al. (2013). Novel Mutation in SLC9A6 Gene in a Patient with Christianson Syndrome and Retinitis Pigmentosum. Brain Dev. 35, 172–176. doi:10.1016/j.braindev.2012.03.010
Padmanabha, H., Saini, A. G., Sahu, J. K., and Singhi, P. (2017). Syndrome of X Linked Intellectual Disability, Epilepsy, Progressive Brain Atrophy and Large Head Associated with SLC9A6 Mutation. BMJ Case Rep. 2017, bcr2017222050. doi:10.1136/bcr-2017-222050
Pescosolido, M. F., Kavanaugh, B. C., Pochet, N., Schmidt, M., Jerskey, B. A., Rogg, J. M., et al. (2019). Complex Neurological Phenotype in Female Carriers of NHE6 Mutations. Mol. Neuropsychiatry 5, 98–108. doi:10.1159/000496341
Pescosolido, M. F., Stein, D. M., Schmidt, M., El Achkar, C. M., Sabbagh, M., Rogg, J. M., et al. (2014). Genetic and Phenotypic Diversity ofNHE6mutations in Christianson Syndrome. Ann. Neurol. 76, 581–593. doi:10.1002/ana.24225
Petitjean, H., Fatima, T., Mouchbahani-Constance, S., Davidova, A., Ferland, C. E., Orlowski, J., et al. (2020). Loss of SLC9A6/NHE6 Impairs Nociception in a Mouse Model of Christianson Syndrome. Pain 161, 2619–2628. doi:10.1097/j.pain.0000000000001961
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Roxrud, I., Raiborg, C., Gilfillan, G. D., Strømme, P., and Stenmark, H. (2009). Dual Degradation Mechanisms Ensure Disposal of NHE6 Mutant Protein Associated with Neurological Disease. Exp. Cell Res. 315, 3014–3027. doi:10.1016/j.yexcr.2009.07.012
Shepherd, J. D., and Huganir, R. L. (2007). The Cell Biology of Synaptic Plasticity: AMPA Receptor Trafficking. Annu. Rev. Cell Dev. Biol. 23, 613–643. doi:10.1146/annurev.cellbio.23.090506.123516
Sinajon, P., Verbaan, D., and So, J. (2016). The Expanding Phenotypic Spectrum of Female SLC9A6 Mutation Carriers: a Case Series and Review of the Literature. Hum. Genet. 135, 841–850. doi:10.1007/s00439-016-1675-5
Van Der Auwera, G. A., Carneiro, M. O., Hartl, C., Poplin, R., Del Angel, G., Levy-Moonshine, A., et al. (2013). From FastQ Data to High Confidence Variant Calls: the Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinformatics 43, 11–33. doi:10.1002/0471250953.bi1110s43
Keywords: christianson syndrome, epilepsy, SLC9A6 gene, Na + /H + exchanger 6, electroencephalography, developmental delay
Citation: Lan Y, Yi S, Li M, Wang J, Yang Q, Yi S, Chen F, Huang L, Ruan Y, Shen Y, Luo J and Qin Z (2021) Case Report: Christianson Syndrome Caused by SLC9A6 Mutation: From Case to Genotype-Phenotype Analysis. Front. Genet. 12:783841. doi: 10.3389/fgene.2021.783841
Received: 27 September 2021; Accepted: 30 November 2021;
Published: 20 December 2021.
Edited by:
Katja Kobow, University Hospital Erlangen, GermanyReviewed by:
Petia Dimova, University Hospital St. Ivan Rilski, BulgariaCopyright © 2021 Lan, Yi, Li, Wang, Yang, Yi, Chen, Huang, Ruan, Shen, Luo and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingsi Luo, SmluZ3NpX0x1b0Bob3RtYWlsLmNvbQ==; Zailong Qin, cWluemFpbG9uZ0Bob3RtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.