 Zhihao Zuo1,2,3†
Zhihao Zuo1,2,3† Yue Lu1,2,3†Minyan Zhu1,2,3
Yue Lu1,2,3†Minyan Zhu1,2,3 Rujia Chen1,2,3Enying Zhang4
Rujia Chen1,2,3Enying Zhang4 Derong Hao5Qianfeng Huang1,2Hanyao Wang1Yanze Su1Zhichao Wang1Yang Xu1,2,3
Derong Hao5Qianfeng Huang1,2Hanyao Wang1Yanze Su1Zhichao Wang1Yang Xu1,2,3 Pengcheng Li1,2,3
Pengcheng Li1,2,3 Chenwu Xu1,2,3*
Chenwu Xu1,2,3* Zefeng Yang1,2,3*
Zefeng Yang1,2,3*- 1Jiangsu Key Laboratory of Crop Genetics and Physiology Key Laboratory of Plant Functional Genomics of the Ministry of Education Jiangsu Key Laboratory of Crop Genomics and Molecular Breeding, Agricultural College of Yangzhou University, Yangzhou, China
- 2Jiangsu Co-Innovation Center for Modern Production Technology of Grain Crops, Yangzhou University, Yangzhou, China
- 3Joint International Research Laboratory of Agriculture and Agri-Product Safety of Ministry of Education of China, Yangzhou University, Yangzhou, China
- 4College of Agronomy, Qingdao Agricultural University, Qingdao, China
- 5Jiangsu Yanjiang Institute of Agricultural Sciences, Nantong, China
The maize (Zea mays L.) ZmCNR13 gene, encoding a protein of fw2.2-like (FWL) family, has been demonstrated to be involved in cell division, expansion, and differentiation. In the present study, the genomic sequences of the ZmCNR13 locus were re-sequenced in 224 inbred lines, 56 landraces and 30 teosintes, and the nucleotide polymorphism and selection signature were estimated. A total of 501 variants, including 415 SNPs and 86 Indels, were detected. Among them, 51 SNPs and 4 Indels were located in the coding regions. Although neutrality tests revealed that this locus had escaped from artificial selection during the process of maize domestication, the population of inbred lines possesses lower nucleotide diversity and decay of linkage disequilibrium. To estimate the association between sequence variants of ZmCNR13 and maize ear characteristics, a total of ten ear-related traits were obtained from the selected inbred lines. Four variants were found to be significantly associated with six ear-related traits. Among them, SNP2305, a non-synonymous mutation in exon 2, was found to be associated with ear weight, ear grain weight, ear diameter and ear row number, and explained 4.59, 4.61, 4.31, and 8.42% of the phenotypic variations, respectively. These results revealed that natural variations of ZmCNR13 might be involved in ear development and can be used in genetic improvement of maize ear-related traits.
Introduction
Maize (Zea mays L.), one of the most important cereal crops, is cultivated worldwide as sources of food, animal feed, and industrial materials. It was suggested that the total global maize production was 1,148.4 million tons in 2019, which was far greater than those of rice (Oryza sativa L.) and wheat (Triticum aestivum L.)1. However, improving kernel yield (KY) is still a primary mission in maize breeding (Li et al., 2018). KY is a complex quantitative trait affected by a variety of genetic and environmental factors and has low heritability (Beavis et al., 1994; Yan et al., 2006). Compared with KY, the heritability of yield components is relatively higher and is less affected by environmental factors (Messmer et al., 2009; Li et al., 2013; Raihan et al., 2016). Therefore, it is more effective to select some yield components than to directly select KY in the breeding process (Sidwell et al., 1976). Among the yield components, kernel size has a crucial effect on kernel weight estimated by kernel length (KL), kernel width (KW), and kernel thickness (KT). In addition, ear length (EL), ear diameter (ED), ear row number (ERN), and kernel number per row (KNR) are essential traits determining the kernel number (Zhang et al., 2017). All of these ear-related traits are quantitative traits regulated by multiple genes and environmental factors (Liu et al., 2012). Many genes regulating maize ear-related traits have been identified, such as fea2 (Bommert et al., 2013), fea3 (Je et al., 2016), KNR1 (Wang et al., 2019) and KNR4 (Liu et al., 2015) for kernel row number, td1 (Bommert et al., 2005), bif2 (McSteen and Hake 2001) and ba1 (Ritter et al., 2002) for tassel morphology.
Tomato fruit-weight 2.2 (fw2.2) was detected as an essential locus in controlling fruit weight and size (Frary et al., 2000; Nesbitt and Tanksley 2001). In plants, the homologs of fw2.2-like (FWL) genes, encoding proteins with a conserved PLAC8 (named after a series of human placental specific protein with unknown function) domain, were suggested to play essential roles in cell division and organ size control (Libault and Stacey 2010). A total of eight members of FWL gene family in rice were detected. Among them, OsFWL1 and OsFWL3 genes modulate rice grain length by regulating cell number, and OsFWL4 gene is a negative regulator of tiller number and plant yield (Xu et al., 2013; Gao et al., 2020). In soybean, the silencing of GmFWL1 expression resulted in a significant decrease in the number of nodules and changes in the structure of cell chromatin (Libault et al., 2010). In maize, a total of 13 FWL gene family members were identified, and named as Cell Number Regulator (CNR) genes (Guo et al., 2010). The ZmCNR1 gene was illustrated to possess a plant-specific cell proliferation function affecting plant and fruit weight. In addition, it was also suggested that the expression of ZmCNR2 negatively correlate with tissue growth activity and hybrid seedling vigor (Guo et al., 2010). These observations revealed that plant FWL/CNR genes play critical roles in plant development.
The maize CNR gene ZmCNR13 was firstly identified through a narrow odd dwarf (nod) mutant (Rosa et al., 2017). Further evidence revealed that the ZmCNR13 gene possessed the function in regulating cell division and differentiation and then affected both vegetative and reproductive development of maize (Rosa et al., 2017). However, the effect of this gene in the regulation of maize ear-related traits remains largely unknown, and there is no association analysis between the nucleotide polymorphisms of the maize ZmCNR13 gene and yield-related traits. In this study, we investigated the nucleotide polymorphism of the maize ZmCNR13 locus, and estimated the association between the sequence polymorphisms of this gene and some ear-related traits.
Materials and Methods
Plant Materials, Experimental Design, and Analysis of Phenotypic Data
A total of 224 inbred lines (Supplementary Table S1) have been selected for phenotypic observation in this study. These lines had been grown in the field in a randomized block design with two replicates at Sanya (18°23′ N, 109°44′ E) in 2015, 2016 and Yangzhou (32°39′ N, 119°42′ E) in 2017. Each line was planted in a sequential row patterns with 15 plants, 3.5 m long and 0.4 m between adjacent rows. After harvesting and drying, three well-developed ears have been selected to measure ear traits, including ear weight (EW), ear grain weight (EGW), EL, ED, ERN, KNR, hundred kernel weight (HKW), KL, KW, and KT. ANOVA was performed using “aov” function in R software. The “lme4” (Bates et al., 2015) package was used to calculated the broad-sense heritability (h2) for these ear-related traits. The observed values of these traits in three environments were used to calculate the best linear estimated prediction (BLUP) (Piepho et al., 2008) using the package ‘lme4’. The calculation model of BLUP is:
DNA Extraction and ZmCNR13 Re-Sequencing
A total of 224 inbred lines, 56 landraces and 30 teosintes (Supplementary Table S1) were collected for target capture sequencing. Fresh and young leaves were collected from each line at the seeding stage, and a modified cetyl trimethyl ammonium bromide (CTAB) method was used to extract genomic DNA (Allen et al., 2006). DNA sequences of the ZmCNR13 locus in the selected lines were resequenced using the target sequence capture sequencing technology on the NimbleGen platform (Choi et al., 2009) by BGI (Beijing Genomics Institute) Life Tech Co. The genomic sequences and positions of the ZmCNR13 (GRMZM2G027821) locus in the inbred line B73 (AGPv3.21) were used as the references for target capture sequencing.
Analysis of Genotypic Data
The software Clustal X (Larkin et al., 2007) was used for multiple sequence alignment of the ZmCNR13. The nucleotide polymorphisms and allelic diversities of all tested lines were analyzed by DNASP5.0 software (Librado and Rozas 2009). Nucleotide diversity (π) in the ZmCNR13 gene, which is defined as the mean number of nucleotide differences per site between any two DNA sequences, was estimated using R package “PopGenome” (Pfeifer et al., 2014). Linkage disequilibrium (LD) decay was measured by the squares of correlation coefficients (R2) for all pairs of SNPs using the program PopLDdecay v3.41 (Zhang et al., 2019) with default parameters.
Marker-Trait Association Analysis in Inbred Lines
The method of genotyping-by-sequencing (GBS) was used to identify the genotypes of the selected lines (Li et al., 2019a). A total of 361,675 SNPs, which were distributed across the entire maize genome, were used to calculate the population structure and kinship. The population structure was estimated through the method of principal components (PCs). In this analysis, the top five PCs, which can explain 23.56% genetic variation, were used for association mapping. In addition, pair-wise coefficients of relatedness (kinship matrix) was calculated by TASSEL5.0 (Bradbury et al., 2007). In order to increase the accuracy of association analysis, two models were used for marker-trait association analysis. The general linear model (GLM) (Pritchard et al., 2000; Zhao et al., 2007) controlling for population structure (PCs), and mixed linear model (MLM) (Yu et al., 2006) controlling for both population structure (PCs) and kinship, were calculated by TASSEL5.0. A total of 398 markers in ZmCNR13 with minor allele frequency (MAF) higher than 0.05 were used for association analysis. The p-value thresholds were empirically set to 1/398 and 0.05/398 for MLM and GLM, respectively, using the Bonferroni correction method (Bland and Altman 1995).
Results
The Phenotypic Variations Among Maize Inbred Lines
In this study, a total of ten ear-related traits, including EW, EGW, EL, ED, ERN, KNR, HKW, KL, KW, and KT, were obtained in a population of 224 maize inbred lines (Table 1). Coefficients of variation of these traits ranged from 12.11 to 40.79%, suggesting abundant phenotypic diversity among the tested inbred lines. ANOVA analyses also revealed that all these ear-related traits showed significant difference among inbred lines, suggesting that this population hold genetic characteristics for association analysis. In addition, we also noticed that both the environments and genotype-environment interaction had a significant impact on all these traits. The values of broad-sense heritability were further estimated. The results revealed that most of these traits possessed high heritability. The estimated heritability for ED, ERN, HKW, KL, and KW is higher than 40%. To evaluate the correlation relationship among these ten ear-related traits, paired correlation analysis was carried out, and the estimations of correlation coefficient (r) between any two traits were obtained (Figure 1). Significant correlations were observed between most parameters. Among them, EW/EGW had the highest correlation with r = 0.975. Meanwhile, the 12 of 45 pairwise correlations for ear-related traits, including ED/EL, ERN/EL, KNR/ED, KNR/ERN, KW/EW, KW/EGW, KW/EL, KW/ED, KW/KL, KT/EL, KT/ED and KT/KL didn’t reach the significant level. These results indicate that different genetic mechanisms might affect the ear traits of maize.
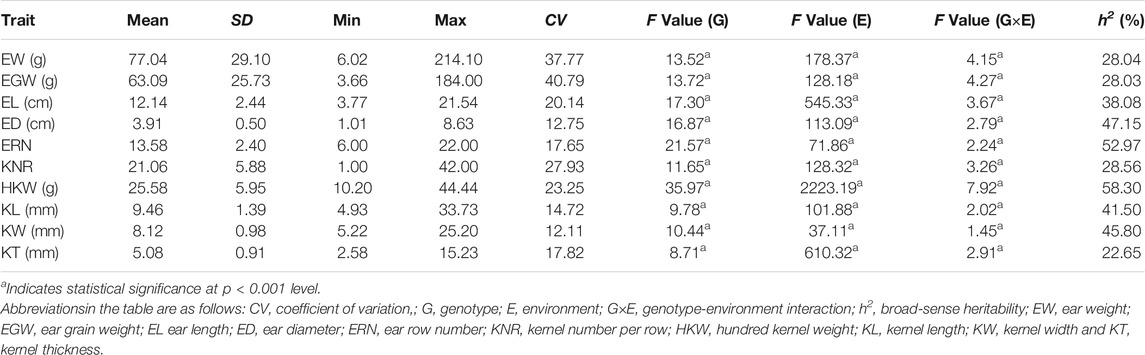
TABLE 1. Descriptive statistics and ANOVA results of the maize ear-related traits.
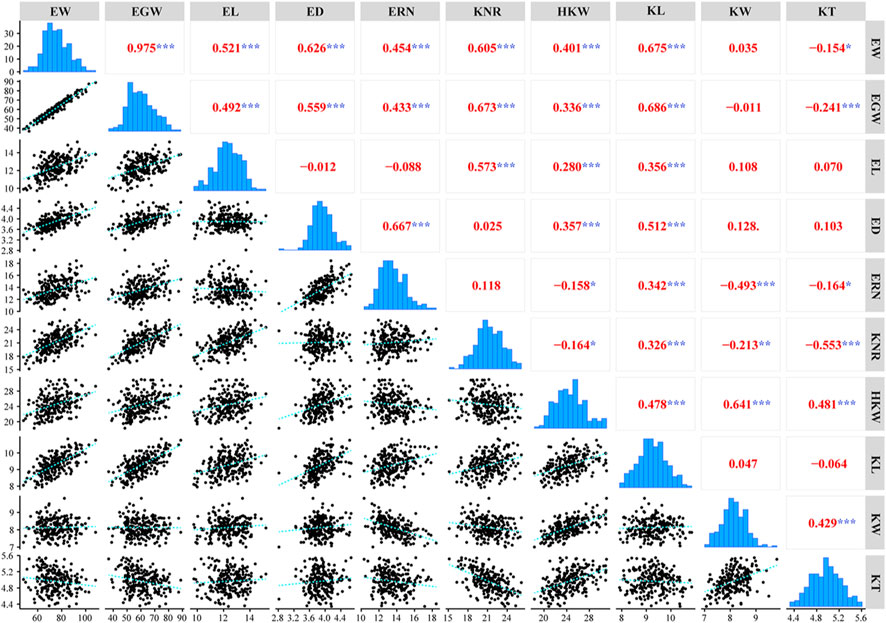
FIGURE 1. Phenotypic distribution and Pearson correlation coefficients for ten ear-related traits (***, p < 0.001; **, p < 0.01; *, p < 0.05).
Sequence Polymorphisms of the Maize ZmCNR13 Gene
To evaluate the sequence polymorphisms of the ZmCNR13 gene, the full-length sequences of this locus were re-sequenced in 224 inbred lines. After multiple sequence alignment, a full-length 6,197 bp sequence was obtained, including 582 bp upstream region, 5,098 bp coding region containing seven exons and seven introns, and 517 bp downstream region. A total of 501 variants were identified in the genomic sequence, including 415 SNPs and 86 indels covering 306 sites. On average, SNPs and Indels occurred per 12.37 bp and 72.06 bp, respectively. The highest frequency of variation sites was detected in the intron region (11.28 bp for SNP and 58.11 bp for InDels). The frequency of variation sites in the exon region was the lowest (23.23 bp for SNP and 321.75 bp for InDels). For all the tested lines, the overall nucleotide diversity (π) of the ZmCNR13 locus was 0.01719, where the estimated π values of intron regions were relatively higher than exon regions. The nucleic acid polymorphisms of exon regions were low (θ = 0.00765), while that of other regions were relatively higher (θ = 0.01702 for intron regions and 0.01544 for downstream) (Table 2). In addition, π and θ were calculated using the sliding window of 200 bp with a step length of 50 bp. Two peaks in the intron3 and intron7 revealed that these regions were more diverse than others (Figure 2).
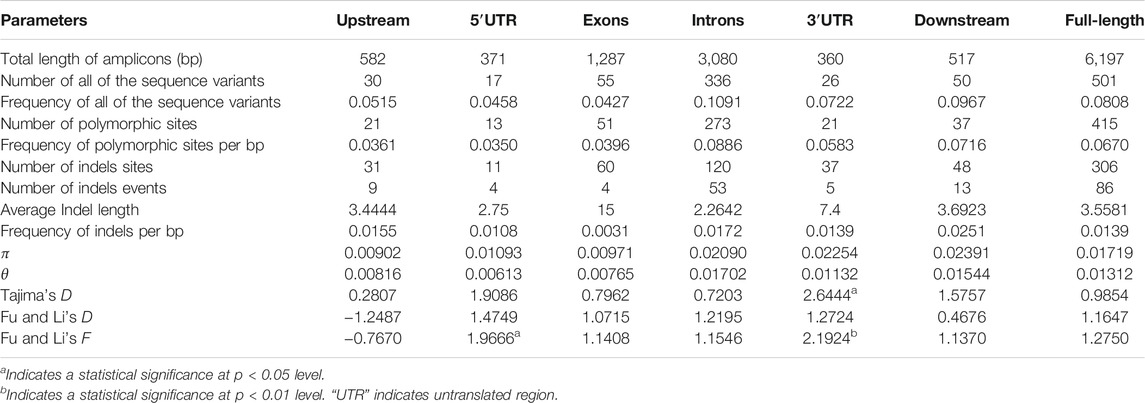
TABLE 2. Summary of parameters for the analysis of nucleotide polymorphisms.

FIGURE 2. Nucleotide diversity (π and θ) estimated along the sequences of maize ZmCNR13. π and θ were calculated using the method of sliding windows of 200 bp with a step of 50 bp.
Nucleotide Diversity and Selection of ZmCNR13 in Inbred Lines, Landraces and Teosintes
To further estimate the genetic diversity of the ZmCNR13 locus, the sequence variation parameters of this gene in three different populations were analyzed and compared. The sequence conservation (C) of the ZmCNR13 gene was similar in landraces (C = 0.903) and inbred lines (C = 0.914), and the lowest value of 0.813 in teosintes. Correspondingly, compared with teosintes, landraces and inbred lines showed lower nucleotide sequence polymorphisms (θ = 42.25 for teosintes, 19.01 for landraces and 13.12 for inbred lines). The neutrality of ZmCNR13 gene was tested by Tajima’s D, Fu and Li’s D*, and Fu and Li’s F*. The Tajima’s D values of the three populations didn’t achieve a significant level. Furthermore, the Tajima’s D was positive in the inbred line population, indicating that the gene was lack of rare alleles in this population (Table 3 and Supplementary Figure S1). There is no prominent LD block of the ZmCNR13 gene in teosintes, and the degree of LD between mutation sites is relatively low. Compared with teosintes, the LD degree of landraces is enhanced to a certain extent, and smaller blocks begin to appear. In inbred lines, further the degree of LD is enhanced and larger blocks appear (Figure 3A). We also estimated the attenuation of LD with physical distance in three populations. The result revealed that the LD attenuation rate of inbred lines was slower than that of landraces and teosintes, and R2 decreased to less than 0.2 after 200 bp, while those of landraces and teosintes decreased to less than 0.2 when it was less than 100 bp (Figure 3B). Taken together, although the gene didn’t escape from neutral evolution, bottleneck effect of population has led to the lower nucleotide polymorphisms and LD decay in inbred lines.
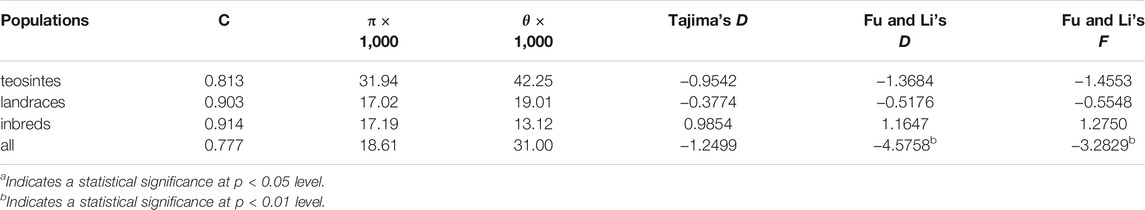
TABLE 3. Summary of nucleotide polymorphisms and neutrality test of ZmCNR13.
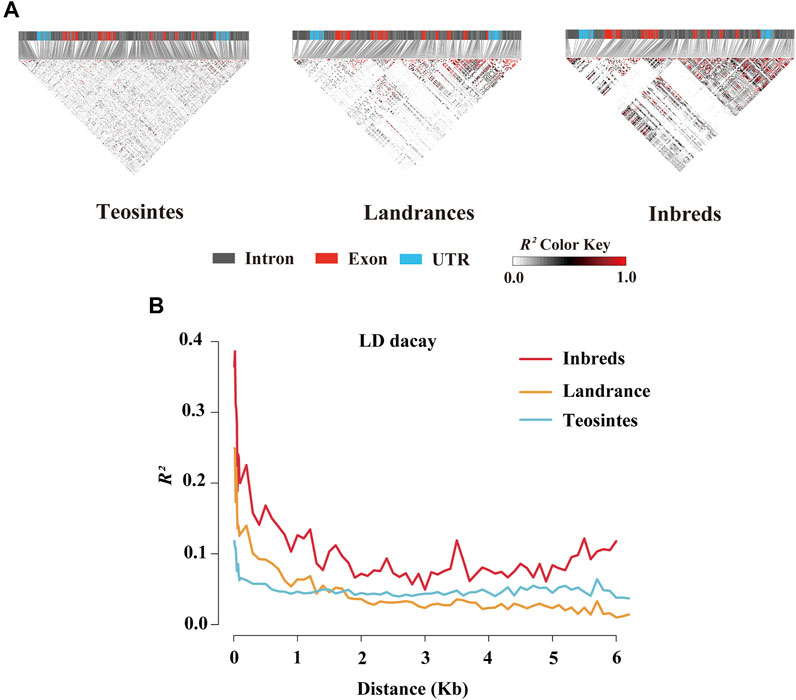
FIGURE 3. Linkage disequilibrium (LD) analysis of ZmCNR13 gene in three populations. (A) LD model of ZmCNR13 gene in three populations. (B) LD decay for three populations measured by R2.
Association Analysis of Ear-Related Traits With ZmCNR13
To identify significant variants associated with ear-related traits, a total of 398 markers in ZmCNR13 with minor allele frequency (MAF) higher than 0.05 were used for association analysis. Both methods of GLM and MLM were used for marker-trait association analysis. Four polymorphic sites (InDel413, SNP2286, SNP2305 and SNP2337) were captured jointly by two models, which were found to be significantly associated with six ear traits (EW, EGW, EL, ED, ERN, and KNR). Among them, two sites (SNP2305 and SNP2337) located in exon 2 jointly controlled four ear traits (EW, EGW ED and ERN). Furthermore, we noticed that SNP2305 belongs to non-synonymous mutation, where the transition of T to A will lead to the changes of amino acid serine to threonine. In addition, SNP2305 were found to be associated with EW, EGW, ED and ERN, and could explain 4.31–8.42% and 7.06–10.41% phenotypic variations under MLM and GLM, respectively (Figure 4 and Supplementary Table S2).
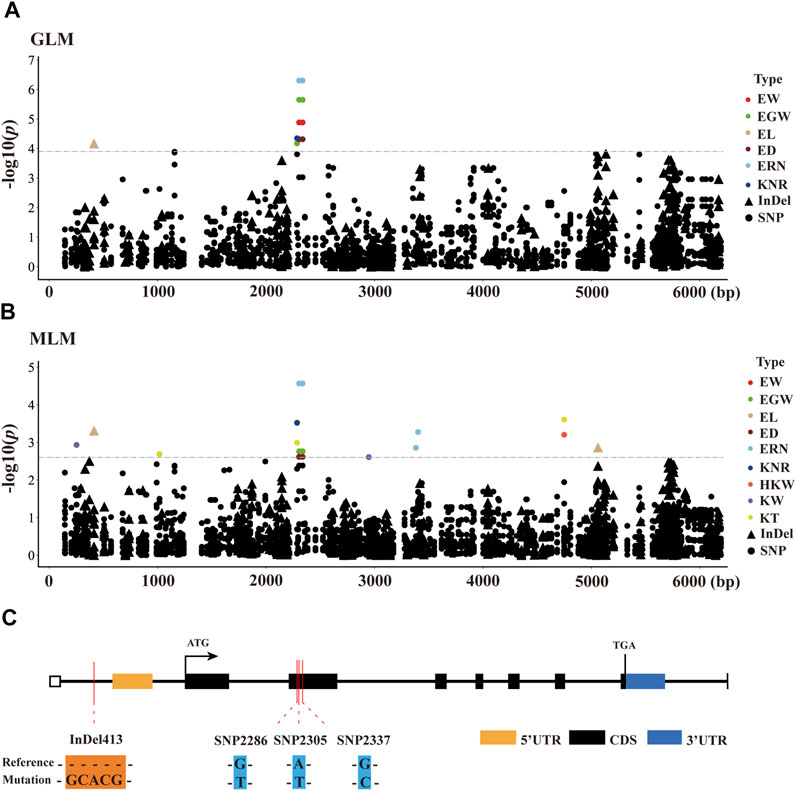
FIGURE 4. Candidate-gene based association of ZmCNR13 gene in inbred lines. (A,B) Manhattan plot using the MLM and GLM model. The p-value threshold was set at 1/398 (MLM) and 0.05/398 (GLM). Triangles and dots represent InDels and SNPs, respectively. (C) Schematic of the ZmCNR13 gene structure and allelic variation.
LD analysis showed that SNP2286, SNP2305 and SNP2337 had relatively high linkage across inbred lines. Interestingly, SNP2305 and SNP2337 had complete linkage (R2 = 1) (Figure 5A). We further classified haplotypes based on the variation of SNP2305, and divided the inbred lines into two major groups. T-test revealed that EW, EGW, ED and ERN showed significant difference between two groups. The allele A group possessed significantly higher values of EW, EGW, ED and ERN than the allele T (Figures 5B–E). On this basis, combined with correlation analysis, we noticed that the changes among the four traits were synergistic. Therefore, we can infer that allele A of SNP2305 has positive regulatory effects on EW, EGW, ED and ERN.

FIGURE 5. Natural variations in ZmCNR13 were significantly associated with four ear-related traits. (A) LD heatmap for four significant variants associated with ear traits. (B–E) Comparison of EW, EGW, ED and ERN between different alleles of SNP2305. p value for t test comparing two groups carrying different alleles were indexed on the top (**, p < 0.01; *, p < 0.05).
Discussion
In the present study, association analyses were employed to illustrate the relationship between the maize ZmCNR13 and ear-related traits. Association analysis, also known as linkage disequilibrium mapping or association mapping, is a method based on linkage disequilibrium to identify the association between genetic polymorphisms and phenotypic variations (Mackay and Powell 2007). Compared with linkage analysis based on parental population, association analysis has higher resolution, and it can be directly used to analyze more than two alleles in natural populations (Salvi and Tuberosa 2005; Mackay and Powell 2007). As an extension of genome-wide association mapping, candidate gene association analysis is mainly used to identify genetic variations and excellent haplotypes significantly associated with target traits (Yan et al., 2011). Maize is a typical cross-pollinated plant with a high recombination rate, rich genetic diversity and LD decay distance of about 1Kb, so it is an ideal material for association analysis (Tenaillon et al., 2001; Whitt et al., 2002). Many loci affecting maize traits have been identified by this method, such as Zmisa2 (Yang et al., 2014) and ZmBT1 (Xu et al., 2014) for starch properties, ZmYS1 (Yang et al., 2015) for kernel mineral concentrations, ZmCKX5 (Wang et al., 2021) and ZmMADS60 (Li et al., 2020) for root morphology, ZmHKT1 (Li et al., 2019a) and ZmPGP1 (Li et al., 2019b) for plant architecture.
Abundant genetic diversity is the basis for crop improvement (Yan et al., 2011). In this study, nucleotide polymorphisms of the ZmCNR13 gene were analyzed in inbred lines through re-sequencing. A total of 501 variations including 415 SNPs and 86 InDels were detected, and most of them concentrated in the intron regions. The exon region had one SNP per 25.71 bp, while the intron region reached one SNP per 11.28 bp. The decrease of nucleotide polymorphism in exon region suggested that these regions might be influenced by greater selection pressure. Moreover, it is worth noted that the LD decays faster in landraces and teosintes than inbred lines, suggesting that there were genetic bottleneck effects (Wang et al., 1998) in the population of inbred lines, although no obvious selection signature was detected through neutral test.
Ear-related traits are the most important components of kernel yield of maize. Illustrating the genetic background of the genes related to ear traits and digging their elite variations will be of great importance in high-yield breeding maize (Zhu et al., 2018). In the present study, we revealed that a non-synonymous mutation in exon 2 (SNP2305) of the ZmCNR13 gene was found to be significantly associated with four ear-related traits, including EW, EGW, ED and ERN. In addition, we further noticed that the inbred lines carrying SNP2305A possess higher EW, EGW, ED and ERN than those carrying SNP2305T. The validity of candidate gene association analysis has been repeatedly confirmed (Yan et al., 2011). For instance, the association between polymorphisms of dwarf8 and flowering time was detected by some independent works (Thornsberry et al., 2001; Andersen et al., 2005; Camus-Kulandaivelu et al., 2006). To confirm the results of association analysis, we compared the phenotypes between the inbred lines carrying SNP2305A and SNP2305T in three environments. The results revealed that there are statistical differences between them for all these traits in different environments (Supplementary Figure S2), suggestive the genetical credibility of the association results. These observations revealed that the superior allelic variations of ZmCNR13 possess potential application values in maize genetic improvement.
In conclusion, the maize ZmCNR13 gene was re-sequenced in 224 inbred lines, 56 landraces and 30 teosintes. Although this gene was escaped from artificial selection during maize genetic improvement, a non-synonymous mutation in exon2 was found to be associated with ear-related traits, including EW, EGW, ED and ERN. The superior allelic variations of ZmCNR13 has potential application values in maize genetic improvement.
Data Availability Statement
The original contributions presented in the study are publicly available. This data can be found here:PRJNA764471.
Author Contributions
ZZ, CX, and ZY. conceived and designed the research. ZZ, YL, MZ, RC, EZ, DH, QH, and HW. conducted the experiments. ZZ, YL, YS, ZW, YX, and PL analyzed the data. ZZ, CX, and ZY wrote the manuscript. All authors read and approved the manuscript.
Funding
This work was supported by the grant from the National Natural Science Foundation of China (32070558, 32061143030, 31801028, 31972487, 32100448), a project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and Natural Science Foundation of Jiangsu Province (BK20210799).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.773597/full#supplementary-material
Footnotes
1http://www.fao.org/faostat/zh/#data/QCL.2019
References
Allen, G. C., Flores-Vergara, M. A., Krasynanski, S., Kumar, S., and Thompson, W. F. (2006). A Modified Protocol for Rapid DNA Isolation from Plant Tissues Using Cetyltrimethylammonium Bromide. Nat. Protoc. 1 (5), 2320–2325. doi:10.1038/nprot.2006.384
Andersen, J. R., Schrag, T., Melchinger, A. E., Zein, I., and Lübberstedt, T. (2005). Validation of Dwarf8 Polymorphisms Associated with Flowering Time in Elite European Inbred Lines of maize (Zea mays L.). Theor. Appl. Genet. 111 (2), 206–217. doi:10.1007/s00122-005-1996-6
Bates, D., Mächler, M., Bolker, B., and Walker, S. (2015). Fitting Linear Mixed-Effects Models Usinglme4. J. Stat. Soft. 67 (1):1–48. doi:10.18637/jss.v067.i01
Beavis, W. D., Smith, O. S., Grant, D., and Fincher, R. (1994). Identification of Quantitative Trait Loci Using a Small Sample of Topcrossed and F4 Progeny from maize. Crop Sci. 34 (4), 882–896. doi:10.2135/cropsci1994.0011183X003400040010x
Bland, J. M., and Altman, D. G. (1995). Statistics Notes: Multiple Significance Tests: the Bonferroni Method. BMJ 310 (6973), 170. doi:10.1136/bmj.310.6973.170
Bommert, P., Lunde, C., Nardmann, J., Vollbrecht, E., Running, M., Jackson, D., et al. (2005). Thick Tassel Dwarf1 Encodes a Putative maize Ortholog of the Arabidopsis CLAVATA1 Leucine-Rich Repeat Receptor-like Kinase. Development 132 (6), 1235–1245. doi:10.1242/dev.01671
Bommert, P., Nagasawa, N. S., and Jackson, D. (2013). Quantitative Variation in maize Kernel Row Number Is Controlled by the FASCIATED EAR2 Locus. Nat. Genet. 45 (3), 334–337. doi:10.1038/ng.2534
Bradbury, P. J., Zhang, Z., Kroon, D. E., Casstevens, T. M., Ramdoss, Y., and Buckler, E. S. (2007). TASSEL: Software for Association Mapping of Complex Traits in Diverse Samples. Bioinformatics 23 (19), 2633–2635. doi:10.1093/bioinformatics/btm308
Camus-Kulandaivelu, L., Veyrieras, J.-B., Madur, D., Combes, V., Fourmann, M., Barraud, S., et al. (2006). Maize Adaptation to Temperate Climate: Relationship between Population Structure and Polymorphism in the Dwarf8 Gene. Genetics 172 (4), 2449–2463. doi:10.1534/genetics.105.048603
Choi, M., Scholl, U. I., Ji, W., Liu, T., Tikhonova, I. R., Zumbo, P., et al. (2009). Genetic Diagnosis by Whole Exome Capture and Massively Parallel DNA Sequencing. Pnas 106 (45), 19096–19101. doi:10.1073/pnas.0910672106
Frary, A., Nesbitt, T. C., Frary, A., Grandillo, S., Knaap, E. v. d., Cong, B., et al. (2000). fw2.2 : A Quantitative Trait Locus Key to the Evolution of Tomato Fruit Size. Science 289 (5476), 85–88. doi:10.1126/science.289.5476.85
Gao, Q., Li, G., Sun, H., Xu, M., Wang, H., Ji, J., et al. (2020). Targeted Mutagenesis of the rice FW 2.2-Like Gene Family Using the CRISPR/Cas9 System Reveals OsFWL4 as a Regulator of Tiller Number and Plant Yield in rice. Ijms 21 (3), 809. doi:10.3390/ijms21030809
Guo, M., Rupe, M. A., Dieter, J. A., Zou, J., Spielbauer, D., Duncan, K. E., et al. (2010). Cell Number Regulator1 Affects Plant and Organ Size in Maize: Implications for Crop Yield Enhancement and Heterosis. Plant Cell 22 (4), 1057–1073. doi:10.1105/tpc.109.073676
Je, B. I., Gruel, J., Lee, Y. K., Bommert, P., Arevalo, E. D., Eveland, A. L., et al. (2016). Signaling from maize Organ Primordia via FASCIATED EAR3 Regulates Stem Cell Proliferation and Yield Traits. Nat. Genet. 48 (7), 785–791. doi:10.1038/ng.3567
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X Version 2.0. Bioinformatics 23 (21), 2947–2948. doi:10.1093/bioinformatics/btm404
Li, C., Li, Y., Sun, B., Peng, B., Liu, C., Liu, Z., et al. (2013). Quantitative Trait Loci Mapping for Yield Components and Kernel-Related Traits in Multiple Connected RIL Populations in maize. Euphytica 193 (3), 303–316. doi:10.1007/s10681-013-0901-7
Li, M., Zhong, W., Yang, F., and Zhang, Z. (2018). Genetic and Molecular Mechanisms of Quantitative Trait Loci Controlling maize Inflorescence Architecture. Plant Cel Physiol 59 (3), 448–457. doi:10.1093/pcp/pcy022
Li, P., Ge, Z., Wang, H., Wei, J., Wang, Y., Xu, Y., et al. (2020). Nucleotide Diversity and Association Analysis of ZmMADS60 with Root Length in the maize Seedling Stage. Agronomy 10 (3), 342. doi:10.3390/agronomy10030342
Li, P., Pan, T., Wang, H., Wei, J., Chen, M., Hu, X., et al. (2019a). Natural Variation of ZmHKT1 Affects Root Morphology in maize at the Seedling Stage. Planta 249 (3), 879–889. doi:10.1007/s00425-018-3043-2
Li, P., Wei, J., Wang, H., Fang, Y., Yin, S., Xu, Y., et al. (2019b). Natural Variation and Domestication Selection of ZmPGP1 Affects Plant Architecture and Yield-Related Traits in maize. Genes 10 (9), 664. doi:10.3390/genes10090664
Libault, M., and Stacey, G. (2010). Evolution of FW2.2-like (FWL) and PLAC8 Genes in Eukaryotes. Plant Signaling Behav. 5 (10), 1226–1228. doi:10.4161/psb.5.10.12808
Libault, M., Zhang, X.-C., Govindarajulu, M., Qiu, J., Ong, Y. T., Brechenmacher, L., et al. (2010). A Member of the Highly Conserved FWL (Tomato FW2.2-like) Gene Family Is Essential for Soybean Nodule Organogenesis. Plant J. 62 (5), 852–864. doi:10.1111/j.1365-313X.2010.04201.x
Librado, P., and Rozas, J. (2009). DnaSP V5: a Software for Comprehensive Analysis of DNA Polymorphism Data. Bioinformatics 25 (11), 1451–1452. doi:10.1093/bioinformatics/btp187
Liu, L., Du, Y., Shen, X., Li, M., Sun, W., Huang, J., et al. (2015). KRN4 Controls Quantitative Variation in maize Kernel Row Number. Plos Genet. 11 (11), e1005670. doi:10.1371/journal.pgen.1005670
Liu, R., Jia, H., Cao, X., Huang, J., Li, F., Tao, Y., et al. (2012). Fine Mapping and Candidate Gene Prediction of a Pleiotropic Quantitative Trait Locus for Yield-Related Trait in Zea mays. PLoS One 7 (11), e49836. doi:10.1371/journal.pone.0049836
Mackay, I., and Powell, W. (2007). Methods for Linkage Disequilibrium Mapping in Crops. Trends Plant Sci. 12 (2), 57–63. doi:10.1016/j.tplants.2006.12.001
McSteen, P., and Hake, S. (2001). Barren Inflorescence2 Regulates Axillary Meristem Development in the maize Inflorescence. Development 128 (15), 2881–2891. doi:10.1242/dev.128.15.2881
Messmer, R., Fracheboud, Y., Bänziger, M., Vargas, M., Stamp, P., and Ribaut, J.-M. (2009). Drought Stress and Tropical maize: QTL-By-Environment Interactions and Stability of QTLs across Environments for Yield Components and Secondary Traits. Theor. Appl. Genet. 119 (5), 913–930. doi:10.1007/s00122-009-1099-x
Nesbitt, T. C., and Tanksley, S. D. (2001). Fw2.2 Directly Affects the Size of Developing Tomato Fruit, with Secondary Effects on Fruit Number and Photosynthate Distribution. Plant Physiol. 127 (2), 575–583. doi:10.1104/pp.010087
Pfeifer, B., Wittelsbürger, U., Ramos-Onsins, S. E., and Lercher, M. J. (2014). PopGenome: an Efficient Swiss Army Knife for Population Genomic Analyses in R. Mol. Biol. Evol. 31 (7), 1929–1936. doi:10.1093/molbev/msu136
Piepho, H. P., Möhring, J., Melchinger, A. E., and Büchse, A. (2008). BLUP for Phenotypic Selection in Plant Breeding and Variety Testing. Euphytica 161 (1), 209–228. doi:10.1007/s10681-007-9449-8
Pritchard, J. K., Stephens, M., Rosenberg, N. A., and Donnelly, P. (2000). Association Mapping in Structured Populations. Am. J. Hum. Genet. 67 (1), 170–181. doi:10.1086/302959
Raihan, M. S., Liu, J., Huang, J., Guo, H., Pan, Q., and Yan, J. (2016). Multi-environment QTL Analysis of Grain Morphology Traits and fine Mapping of a Kernel-Width QTL in Zheng58 × SK maize Population. Theor. Appl. Genet. 129 (8), 1465–1477. doi:10.1007/s00122-016-2717-z
Ritter, M. K., Padilla, C. M., and Schmidt, R. J. (2002). The maize Mutant Barren Stalk1 Is Defective in Axillary Meristem Development. Am. J. Bot. 89 (2), 203–210. doi:10.3732/ajb.89.2.203
Rosa, M., Abraham-Juárez, M. J., Lewis, M. W., Fonseca, J. P., Tian, W., Ramirez, V., et al. (2017). The Maize MID-Complementing Activity Homolog Cell Number Regulator13/Narrow odd Dwarf Coordinates Organ Growth and Tissue Patterning. Plant Cell 29 (3), 474–490. doi:10.1105/tpc.16.00878
Salvi, S., and Tuberosa, R. (2005). To Clone or Not to Clone Plant QTLs: Present and Future Challenges. Trends Plant Sci. 10 (6), 297–304. doi:10.1016/j.tplants.2005.04.008
Sidwell, R. J., Smith, E. L., and McNew, R. W. (1976). Inheritance and Interrelationships of Grain Yield and Selected Yield‐Related Traits in a Hard Red Winter Wheat Cross 1. Crop Sci. 16 (5), 650–654. doi:10.2135/cropsci1976.0011183X001600050013x
Tenaillon, M. I., Sawkins, M. C., Long, A. D., Gaut, R. L., Doebley, J. F., and Gaut, B. S. (2001). Patterns of DNA Sequence Polymorphism along Chromosome 1 of maize (Zea mays Ssp. Mays L.). Proc. Natl. Acad. Sci. 98 (16), 9161–9166. doi:10.1073/pnas.151244298
Thornsberry, J. M., Goodman, M. M., Doebley, J., Kresovich, S., Nielsen, D., and Buckler, E. S. (2001). Dwarf8 Polymorphisms Associate with Variation in Flowering Time. Nat. Genet. 28 (3), 286–289. doi:10.1038/90135
Wang, H., Sun, H., Xia, H., Wu, T., Li, P., Xu, C., et al. (2021). Natural Variation and Domestication Selection of ZmCKX5 with Root Morphological Traits at the Seedling Stage in maize. Plants 10 (1), 1. doi:10.3390/plants10010001
Wang, J., Caballero, A., and Hill, W. G. (1998). The Effect of Linkage Disequilibrium and Deviation from Hardy-Weinberg Proportions on the Changes in Genetic Variance with Bottlenecking. Heredity 81 (2), 174–186. doi:10.1046/j.1365-2540.1998.00390.x
Wang, J., Lin, Z., Zhang, X., Liu, H., Zhou, L., Zhong, S., et al. (2019). krn1 , a Major Quantitative Trait Locus for Kernel Row Number in maize. New Phytol. 223 (3), 1634–1646. doi:10.1111/nph.15890
Whitt, S. R., Wilson, L. M., Tenaillon, M. I., Gaut, B. S., and Buckler, E. S. (2002). Genetic Diversity and Selection in the maize Starch Pathway. Proc. Natl. Acad. Sci. 99 (20), 12959–12962. doi:10.1073/pnas.202476999
Xu, J., Xiong, W., Cao, B., Yan, T., Luo, T., Fan, T., et al. (2013). Molecular Characterization and Functional Analysis of "fruit-weight2.2-like" Gene Family in rice. Planta 238, 643–655. doi:10.1007/s00425-013-1916-y
Xu, S., Yang, Z., Zhang, E., Jiang, Y., Pan, L., Chen, Q., et al. (2014). Nucleotide Diversity of maize ZmBT1 Gene and Association with Starch Physicochemical Properties. PLoS One 9 (8), e103627. doi:10.1371/journal.pone.0103627
Yan, J.-b., Tang, H., Huang, Y.-q., Zheng, Y.-l., and Li, J.-s. (2006). Quantitative Trait Loci Mapping and Epistatic Analysis for Grain Yield and Yield Components Using Molecular Markers with an Elite maize Hybrid. Euphytica 149 (1), 121–131. doi:10.1007/s10681-005-9060-9
Yan, J., Warburton, M., and Crouch, J. (2011). Association Mapping for Enhancing maize (Zea mays L.) Genetic Improvement. Crop Sci. 51 (2), 433–449. doi:10.2135/cropsci2010.04.0233
Yang, Z., Ma, S., Hu, Y., Zhang, E., Xie, Z., Xu, S., et al. (2015). Association Analysis of the maize Gene ZmYS1 with Kernel mineral Concentrations. Plant Mol. Biol. Rep. 33 (5), 1327–1335. doi:10.1007/s11105-014-0836-8
Yang, Z., Zhang, E., Jiang, Y., Xu, S., Pan, L., Chen, Q., et al. (2014). Sequence Polymorphisms in Zmisa2 Gene Are Significantly Associated with Starch Pasting and Gelatinization Properties in maize (Zea mays L.). Mol. Breed. 34 (4), 1833–1842. doi:10.1007/s11032-014-0142-z
Yu, J., Pressoir, G., Briggs, W. H., Vroh Bi, I., Yamasaki, M., Doebley, J. F., et al. (2006). A Unified Mixed-Model Method for Association Mapping that Accounts for Multiple Levels of Relatedness. Nat. Genet. 38 (2), 203–208. doi:10.1038/ng1702
Zhang, C., Dong, S.-S., Xu, J.-Y., He, W.-M., and Yang, T.-L. (2019). PopLDdecay: a Fast and Effective Tool for Linkage Disequilibrium Decay Analysis Based on Variant Call Format Files. Bioinformatics 35 (10), 1786–1788. doi:10.1093/bioinformatics/bty875
Zhang, C., Zhou, Z., Yong, H., Zhang, X., Hao, Z., Zhang, F., et al. (2017). Analysis of the Genetic Architecture of maize Ear and Grain Morphological Traits by Combined Linkage and Association Mapping. Theor. Appl. Genet. 130 (5), 1011–1029. doi:10.1007/s00122-017-2867-7
Zhao, K., Aranzana, M. J., Kim, S., Lister, C., Shindo, C., Tang, C., et al. (2007). An Arabidopsis Example of Association Mapping in Structured Samples. Plos Genet. 3 (1), e4. doi:10.1371/journal.pgen.0030004
Keywords: maize, nucleotide polymorphism, ear-related traits, ZmCNR13, association analysis
Citation: Zuo Z, Lu Y, Zhu M, Chen R, Zhang E, Hao D, Huang Q, Wang H, Su Y, Wang Z, Xu Y, Li P, Xu C and Yang Z (2021) Nucleotide Diversity of the Maize ZmCNR13 Gene and Association With Ear Traits. Front. Genet. 12:773597. doi: 10.3389/fgene.2021.773597
Received: 10 September 2021; Accepted: 14 October 2021;
Published: 26 October 2021.
Edited by:
Ahmed Sallam, Assiut University, EgyptReviewed by:
Zhikai Liang, University of Minnesota Twin Cities, United StatesKaransher Singh Sandhu, Washington State University, United States
Copyright © 2021 Zuo, Lu, Zhu, Chen, Zhang, Hao, Huang, Wang, Su, Wang, Xu, Li, Xu and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zefeng Yang, emZ5YW5nQHl6dS5lZHUuY24=; Chenwu Xu, Y3d4dUB5enUuZWR1LmNu
†These authors have contributed equally to this work