
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Genet. , 25 November 2021
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.773177
 Pauline Le Tanno1
Pauline Le Tanno1 Mathilde Folacci2
Mathilde Folacci2 Jean Revilloud2Laurence Faivre3Gabriel Laurent4Lucile Pinson5,6,7Pascal Amedro8
Jean Revilloud2Laurence Faivre3Gabriel Laurent4Lucile Pinson5,6,7Pascal Amedro8 Gilles Millat9
Gilles Millat9 Alexandre Janin9
Alexandre Janin9 Michel Vivaudou2
Michel Vivaudou2 Nathalie Roux-Buisson1*
Nathalie Roux-Buisson1* Julien Fauré1
Julien Fauré1Andersen-Tawil Syndrome (ATS) is a rare disease defined by the association of cardiac arrhythmias, periodic paralysis and dysmorphic features, and is caused by KCNJ2 loss-of-function mutations. However, when extracardiac symptoms are atypical or absent, the patient can be diagnosed with Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT), a rare arrhythmia at high risk of sudden death, mostly due to RYR2 mutations. The identification of KCNJ2 variants in CPVT suspicion is very rare but important because beta blockers, the cornerstone of CPVT therapy, could be less efficient. We report here the cases of two patients addressed for CPVT-like phenotypes. Genetic investigations led to the identification of p. Arg82Trp and p. Pro186Gln de novo variants in the KCNJ2 gene. Functional studies showed that both variants forms of Kir2.1 monomers act as dominant negative and drastically reduced the activity of the tetrameric channel. We characterize here a new pathogenic variant (p.Pro186Gln) of KCNJ2 gene and highlight the interest of accurate cardiologic evaluation and of attention to extracardiac signs to distinguish CPVT from atypical ATS, and guide therapeutic decisions. We also confirm that the KCNJ2 gene must be investigated during CPVT molecular analysis.
Andersen Tawil Syndrome (ATS) is characterized by a triad of symptoms: periodic paralysis (episodic flaccid muscle weakness), distinctive clinical features (such as low-set ears, broad forehead, small palpebral fissures, hypertelorism, broad nasal root, bulbous nose, thin upper lip, maxillary, malar and mandibular hypoplasia, abnormal extremities), and cardiac arrhythmias. Less frequently, skeletal, dental and renal findings are present (Andersen, 1971, Tawil, 1994, Andelfinger et al., 2002; Tristani-Firouzi et al., 2002).
Pathogenic variants in KCNJ2 were first associated with Andersen Tawil Syndrome (ATS) in 2001 (Plaster et al., 2001) and until now account for 80% of the cases (Zhang et al., 2005; Haruna et al., 2007). KCNJ2 encodes the pore-forming alpha subunit of the potassium channel Kir 2.1 that underlies the inward rectifier potassium current IK1stabilizing resting potential during cardiac and skeletal myocytes repolarization (Plaster, et al., 2001; Wible et al., 1995; Sumitomo, 2016). KCNJ2 variants are also associated in rare familial forms of atrial fibrillation type 9, short QT syndrome type 3 and Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT) (Tester et al., 2006).
CPVT is a heritable arrhythmia syndrome characterized by symptoms of palpitations, dizziness, syncope or sudden death triggered by physical activity or sympathetic nervous system stimulation such as acute emotion or stress (Hayashi et al., 2009; Ackerman et al., 2011). While resting ECG is usually normal, exercise stress test may unmask ventricular arrhythmias (VA), arising in the absence of structural cardiac abnormalities. VA range from premature beats to sustained ventricular tachycardia (VT) as workload increases, the most pathognomonic aspect observed being bidirectional VT (BVT) in 1/3 cases (Andersen 1971). In 50–65% of CPVT cases genetic mutations are identified in RYR2, a gene encoding the main intracellular calcium channel of cardiomyocytes, and much less frequently in others genes that are also related to calcium homeostasis [CASQ2, CALM1, CALM2, CALM3, TRDN, TECRL (Priori et al., 2002; Ackerman et al., 2011; Ackerman et al., 2011; Nyegaard et al., 2012; Roux-Buisson et al., 2012; Devalla et al., 2016; Jensen et al., 2018)]. Phenocopies of CPVT have been described in association with mutations in KCNJ2 but to date, only a few mutations of KCNJ2 were clearly showed responsible for a CPVT phenotype. In the case of atypical arrhythmia, ATS or CPVT diagnosis may therefore both be evoked. In such cases the identification of the mutated gene is of uppermost importance because first line treatment of RYR2 mutation-induced CPVT is based on beta blockers that can be less efficient on KCNJ2 mutated cases (Hayashi et al., 2009; Priori et al., 2013; Manzatti et al. 2020).
We report here two cases of atypical ATS initially addressed for suspicion of CPVT for which mutations in KCNJ2 were identified. Molecular characterization of the mutations allowed to clarify the pathophysiology of the two cases and add a new pathogenic KCNJ2 variant in databases. Our report also adds a new description of CPVT patients with KCNJ2 mutations, and specific features of such patients are discussed.
All clinical investigations reported in this study were realized for medical purpose. Molecular genetics analysis was performed after obtaining written informed consent of the patients and/or their legal guardians for children. Written authorization for publication was obtained from every individual and/or their parent/guardian.
Animal handling and experiments fully conformed with European regulations and were approved by the Ethics Committee of the Commissariat à l’Energie Atomique et aux Energies Alternatives (Ethics Approval #12-040). Authorization of the animal facility has been delivered by the regional administration (Préfet de l’Isère, authorization # D 38,185 10,001).
Probands and relatives were referred at the University Hospital of Dijon, France and the University Hospital of Montpellier, France for the families 1 and 2 respectively. Clinical data, resting electrocardiogram, exercise stress test, 24 h Holter monitoring, transthoracic echocardiography and cardiac magnetic resonance (CMR) imaging were performed on each proband. Proband 1 also underwent coronary computed tomography angiography (CCTA).
Genomic DNA was extracted from blood’s leucocytes with an automated technique using the Blood XL kit (Macherey Nalgel). For each proband addressed for CPVT, the analysis of the coding sequence and 10 base pairs of the intron-exon junction for RYR2 (NM_001035.2), CASQ2 (NM_001232.3), TRDN (NM_001256021), CALM1 (NM_006888.4) and KCNJ2 (NM_000891.2) genes were sequenced with Ion Torrent PGM sequencer (Thermo Fisher). Bioinformatics analyses were performed using the Torrent_Suite 5.0, Torrent variant calling v5.0.4. Variants were annotated using two independent softwares, Ion Reporter v5.0.4.0 and a local plug-in. Exon not covered by the gene panel at a minimal depth of 30X and variation of class 3, 4 or 5 according to ACMG recommendations (Richards et al., 2015) were controlled by Sanger sequencing on an ABI 3130xl sequencer (PE Applied Biosystems®, Foster City, United States). Analysis of the chromatograms was performed with SeqScape V (PE Applied Biosystems®, Foster City, United States). The segregation analysis of KCNJ2 variants within the family was performed by targeted Sanger sequencing when the relative DNAs and clinical data were available.
Human Kir2.1 in the oocyte expression plasmid pXoom was a kind gift from Dr Saïd Bendahhou, from Nice Sophia Antipolis University. The p. Arg82Trp and p. Pro186Gln mutations were inserted by site directed mutagenesis using the QuickChange II Site-Directed Mutagenesis Kit (Agilent, Santa Clara CA, United States). The entire coding sequence was verified by Sanger sequencing.
Xenopus oocytes were prepared as previously reported (Moreau et al., 2017; Vivaudou et al., 2017). The protocols used to record and analyze data are detailed in Supplementary Figure S1. Briefly, after surgical retrieval, the oocytes are defolliculated by type 1A collagenase. Each oocyte was injected with 50 nl of RNAse-free water containing 2 or 50 ng of Kir2.1 mRNA. Microinjected oocytes were incubated for more than 2 days at 19°C in Barth’s solution (in mM: 1 KCl, 0.82 MgSO4, 88 NaCl, 2.4 NaHCO3, 0.41 CaCl2, 16 Hepes, pH 7.4) supplemented with 100 U.ml−1 of penicillin and streptomycin and 0.1 mg ml−1 of gentamycin. Whole-cell currents were recorded with the two-electrode voltage clamp (TEVC) technique using a HiClamp robot (MultiChannels System) (Vivaudou et al., 2017). Microelectrodes were filled with 3 M KCl and oocytes were bathed in solutions all containing in mM: 1.8 CaCl2, 1 MgCl2, 5 HEPES, 3 niflumic acid to block endogenous Cl− currents (pH 7.4). High K+ solution contained also 91 mM KCl, Low K+ solution 91 mM NaCl and 2 mM KCL, and Ba2+ solution 91 mM KCl and 3 mM BaCl2. Currents were recorded at −50 mV while oocytes were bathed sequentially for 10 s in Low K+, High K+ and Ba2+ solutions. Current-Voltage curves were obtained in each solution using a ramp from −50 to +50 mV. These curves were used to verify that the recorded currents were inwardly rectifying as expected from Kir2.1 current. Small outward currents at positive potentials were often observed and are thought to be from endogenous currents because they were not blocked by Ba2+. Oocytes displaying large (>1 µA) outward currents at positive potentials were discarded as too leaky. The values shown in the figures are those recorded at −50 mV.
Binomial analysis of the subunit distribution in a tetrameric channel was performed as previously described (Hosy and Vivaudou, 2014). Oocytes display a notorious variability in terms of expression levels from batch to batch. To minimize this source of error when comparing current amplitudes, currents arising from different coexpression ratios were obtained and normalized by considering only the same batches of oocytes. Curve fitting was performed with a modified version of the eeFit program (Vivaudou, 2019).
Average values are presented as mean ± s.e.m.
All detailed protocols are available upon request.
Patient 1 was a young woman whose first symptoms started at the age of 16. She was referred to the hospital for episodes of palpitations triggered by exercise or emotion, occurring at least once a month. Dizziness with hypotension occurred during physical exercise, without loss of consciousness. With time she developed symptoms of dyspnea, hot flushes, chest pain and asthenia associated with the palpitations. Her resting ECG was normal (QTc of 412 ms) except for asymptomatic monomorphic premature ventricular contractions (PVC) with right bundle branch block (RBBB) pattern. The exercise stress test revealed several RBBB PVCs with left axis, increasing with workload and junctional beating, but no VT (Figures 1A,B). The 24-hour-holter monitor evidenced diurnal PVC mostly isolated with rare doublets, and premature atrial contractions (PAC) spread over the recording. Transthoracic echocardiography showed a moderate dilatation of the right ventricle and CMR did not show other signs. She had no history of periodic paralysis and did not present with any particular morphological or extracardiac features. She was the first daughter of non-consanguineous parents, who were asymptomatic and presented no symptoms upon cardiologic explorations. No significant cardiac event was reported from the family history, other than atrial fibrillation in her paternal grandfather. Altogether, these data lead to a diagnosis of CPVT. Several first-line drugs were administered (propranolol, propafenone, sotalol), with no efficacy. Nadolol was finally introduced and implantable cardioverter defibrillator is under discussion.
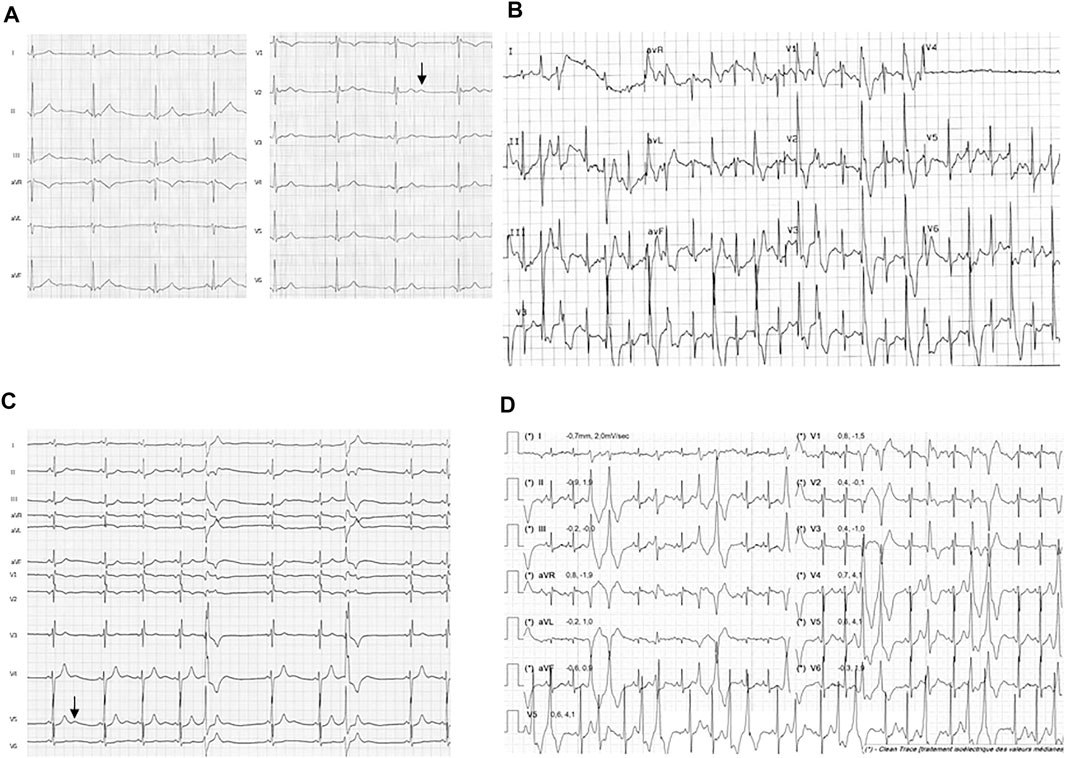
FIGURE 1. Resting ECG and exercise ECG from the probands 1 and 2. Traces from proband 1 shows (A) resting ECG with U wave (arrow) and (B) PVCs during exercise test (120 W workload). Traces from proband 2 shows (C) resting ECG with U wave (arrow) and (D) polymorphic PVCs at exercise (50 W workload) during the first Cardiopulmonary exercise test (CPET) before medical treatment was started.
Patient 2 was a teenager female who was referred to pediatric cardiology at the age of 13 since asymptomatic arrhythmia was fortuitously detected on resting ECG. She reported suffering from muscular pain and weakness of lower limbs. She was referred to the clinical genetic department where retro-micrognathia, relative short and low set ears and clinodactyly of the fifth digit were observed. Stature was within normal range (150 cm heigh, −1DS for 38 kg weight, −1DS). In clinical history, a cleft palate surgery was found at the age of 6 months as part of a postnatal Pierre Robin Sequence.
On resting ECG, polymorphic single PVCs were present with normal QTc evaluated at 430 ms and U waves (Figure 1C). Exercise test showed an increase with workload in numbers and polymorphic aspects of PVCs, but no sustained VT (Figure 1D). A 24-h Holter monitoring evidenced numerous polymorphic PVCs and few non-sustained VT (Supplementary Figure S2A). A correlation between the number of PVCs and the heart rate was observed (Supplementary Figure S2B). All cardiac anomalies reported on both 24-h Holter monitoring and exercise test were asymptomatic. Transthoracic echocardiography, CMR and CCTA were all normal. The analysis of a gene panel involved in cleft lip and palate failed to find pathogenic mutations and a high-resolution array CGH was normal.
There was no family history of arrhythmias, and transthoracic echocardiography and 24 h-holter ECG were normal on both parents. There was no other significant family history for both probands.
Additional extracardiac features in proband 2 allowed retrospective diagnosis of Andersen Tawil syndrome with CPVT. Beta-blocking therapy, flecainide were poorly efficient or showed adverse effects and potassium supplementation had positive effects.
Targeted sequencing was performed for both patients with a panel of genes involved in arrhythmia (RYR2, CASQ2, TRDN, CALM1 and KCNJ2). In proband 1, a de novo heterozygous missense variant c.244C > T; p. Arg82Trp of the KCNJ2 gene was identified (accession number: VCV000067568). DNA from her sister was unfortunately not available. In proband 2, a c.557C > A; p. Pro186Gln heterozygous missense variant in KCNJ2 was found de novo (accession number: VCV000652820). The variant was not found in the DNA of her asymptomatic sister. No variants (classe 3, 4 or 5 ACMG) that could be responsible for the proband symptoms were evidenced in other genes for both probands.
Both variants of KCNJ2 are non-conservative amino acid substitutions that impact residues highly conserved across species. They were predicted damaging by five softwares (Polyphen2, Provean, CADD, FatHMM, Mutation Taster), and unreported in the GnomAD database (v2.1.1). Both variations may impact important functional domains of the protein: arginine 82 is located in the first transmembrane segment of the protein while proline 186 belongs to a C-terminal PKKR motif involved in the binding of the channel modulator phospholipid phosphatidyl inositol bisphosphate (PIP2) (Handklo-Jamal et al., 2020). The p. Arg82Trp variant of KCNJ2 had previously been associated to CPVT (Tester et al., 2006), and the p. Arg82Gln variant affecting the same amino acid had been reported several times with ATS or ATS with cardiac phenotype alone (Kimura et al., 2012). To our knowledge, the p. Pro186Gln variant has never been reported in the literature and is absent from the HGMDPro and LOVD databases but is reported once in ClinVar database as Likely Pathogenic in a patient presenting with ATS. However, other variants affecting the same codon and leading to different amino acid transition were reported in ATS (p.Pro186Leu, p. Pro186Ala and p. Pro186Thr) (Soom et al., 2001; Song et al., 2016).
The impact of both variants was analyzed by measuring currents generated after expression of Wild Type (WT) or mutated Kir2.1 in xenopus oocytes. Examples of recording from an oocyte injected with variable amount of KCNJ2 mRNA are detailed in Supplementary Figures S3, S4. Whole cell recordings showed that specific Kir2.1 currents detected in the presence of high extracellular potassium concentration, and abolished by the blocking ion barium, were drastically reduced by the p. Arg82Trp mutation. The p. Pro186Gln mutant was not detectable even when oocytes were injected with high RNA amount (Figure 2). These experiments indicated that both mutations induce a partial (p.Arg82Trp) or total (p.Pro186Gln) loss of channel activity that could be due to alterations of channel function, protein expression or localization. The Kir2.1 channel is a homotetramer and we next tested its function upon expression of various ratios of WT and mutants. As described in Figure 3, co-expression of each mutant with WT Kir2.1 in a 1 to 4 ratio induced a 60% decrease in the current, whereas a 1:1 WT/P186Q ratio resulted in a 95% decrease in the current and a 1:1 WT/R82W resulted in 80% reduction. The 1:1 ratio mimics the phenotype expected in a patient having a heterozygous mutation. These results suggest that both mutants of Kir2.1 were expressed and could assemble with the WT protein, generating inefficient heteromultimers. However the p. Pro186Gln mutant could have a more severe effect on the channel. A model showing the number of heterotetramers of the channel including exactly (Figure 3A) or at least (Figure 3B) 1, 2, 3 or 4 WT subunits was produced as a function of the ratio of WT or mutant mRNAs injected in oocytes, with the reasonable assumption that WT and mutant subunits assemble randomly to produce tetrameric channels. The normalized amount of current produced by the various ratios of WT and mutated Kir2.1 obtained in Figure 3 followed the curve representing the proportion of homotetramers of WT Kir2.1 (Figure 3B). This indicates that in co-expression experiments, the detected current may only come from WT homotetramers of Kir2.1, and that the incorporation of a single mutated subunit is sufficient to confer the mutant phenotype to the whole Kir2.1 tetramer. Because the p. Arg82Trp mutant channel retains some activity, this mutation has less drastic adverse effects than the p. Pro186Gln mutation.
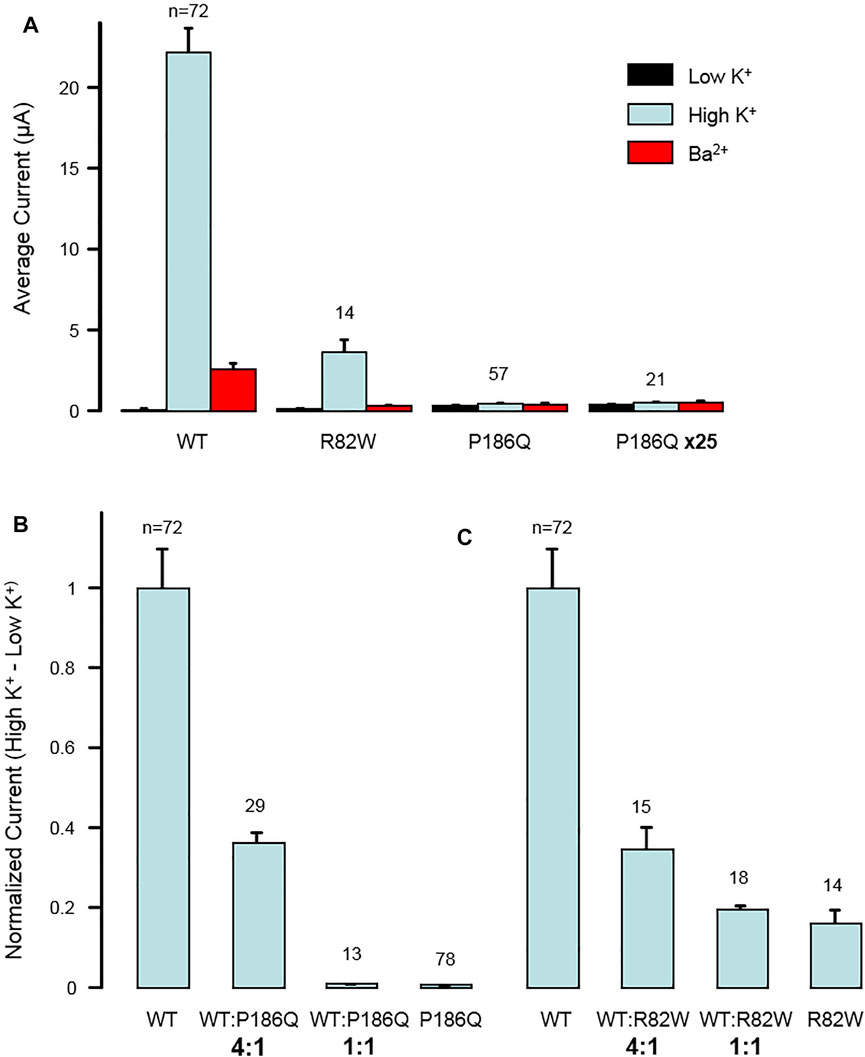
FIGURE 2. Dominant negative effect of mutations p. Arg82Trp (R82W) and p. Pro186Gln (P186Q) on Kir2.1 function. (A) Mutations reduce the activity of homotetrameric channels. The histogram displays the average whole-cell currents recorded in Xenopus oocytes injected with 2 ng of RNA coding for either wild-type Kir2.1 (WT), Kir2.1R82W (R82W), or Kir2.1P186Q (P186Q). Oocytes injected with 50 ng of Kir2.1P186Q were also tested (P186Q x25). Measurements were done successively in extracellular solutions containing 2 mM K+ (Low K+), 96 mM K+ (High K+), and 3 mM Ba2+ and 96 mM K+ (Ba2+). (B,C) Effect of mutations varies gradually with the ratio of WT to mutant subunits. (B) The histogram displays the average whole-cell currents recorded in Xenopus oocytes injected with RNAs coding for wild-type Kir2.1 (WT) or Kir2.1P186Q (P186Q), or with a mixture of RNAs coding for WT and mutant with ratios of 4:1 or 1:1. The currents were calculated as the difference between the current measured in High K+ solution and the current measured in Low K+ solution. They are here normalized to the currents obtained with only WT subunits. (C) Same for mutation R82W. Numbers above bars indicate the number of oocytes included in the averages. Error bars indicate s. e.m.
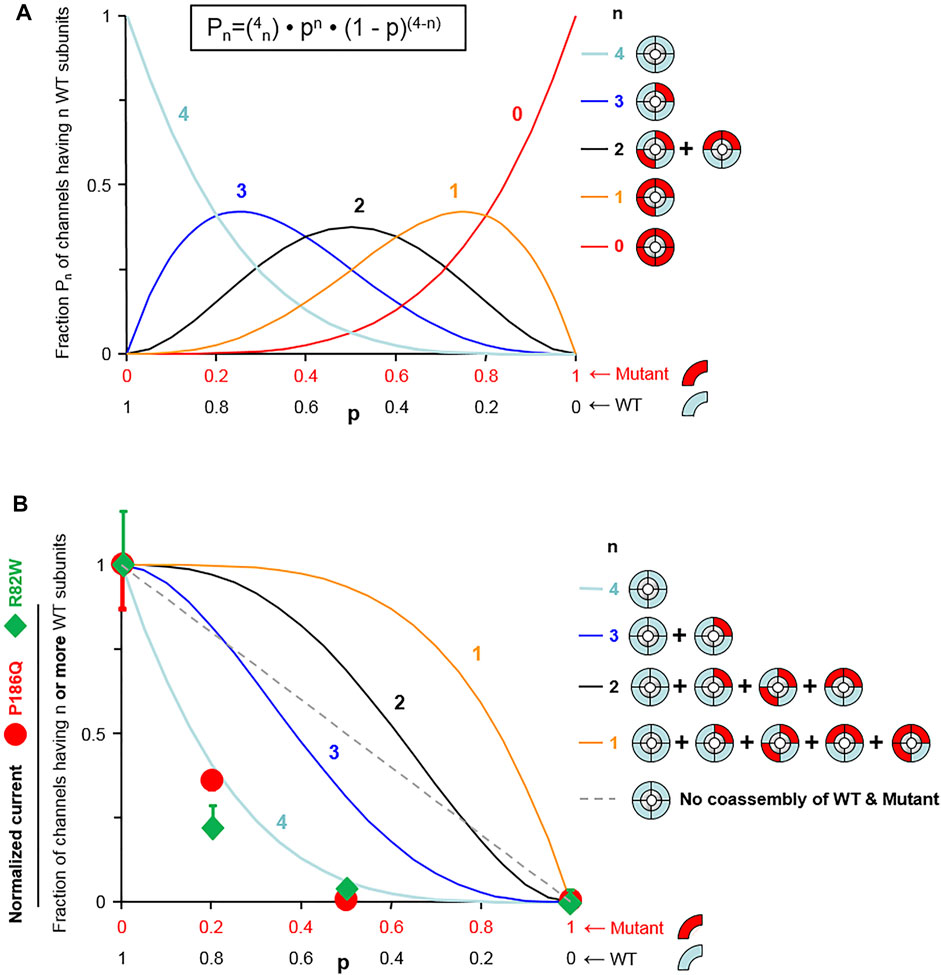
FIGURE 3. Model fitting reveals that one mutant subunit is sufficient to impair the function of Kir2.1 tetrameric channels. (A) Assuming random assembly of WT and mutant subunits, the probability of occurrence of channels having exactly n WT subunits (Pn) follows a binomial distribution (equation shown at top). That probability Pn is shown as a function of the fraction of wild-type subunits (p) for each possible value of n (n indicated next to the curves). (B) Probability of a channel having n or more WT subunits calculated using the distributions of (A) (n indicated next to the curves). The straight dotted line represents the probability of a WT channel if WT subunits and mutant subunits could not co-assemble. The symbols represent normalized K+ current. The percent deviation of the experimental data from each model, calculated from the rmsd (root mean-square deviation) is for p. Pro186Gln: 3.6% (n = 4), 28% (n ≥ 3), 46% (n ≥ 2), 56% (n ≥ 1), and 33% (no WT/mutant mixing; dotted straight line); and for R82W: 9.4% (n = 4), 33% (n ≥ 3), 49% (n ≥ 2), 59% (n ≥ 1), and 37% (no WT/mutant mixing; dotted straight line). For both mutants, the experimental data match best the model n = 4, meaning that channels are fully functional only when they contain no mutant subunits.
Overall we showed that expression of both p. Arg82Trp and p. Pro186Gln mutants in xenopus oocytes produced respectively impaired or non-functional Kir2.1 channels. Co-expression of either mutant with WT protein strongly affected the function of the tetrameric channel, showing a dominant negative effect of the mutations. These functional studies correlated with the dominant mode of inheritance of the CPVT phenotypes in the two families, and confirmed the deleterious effect of the genetic variations of KCNJ2 found in both patients. Considering this, both variants p. Arg82Trp and p. Pro186Gln were classified Pathogenic (class 5) according to ACMG classification (Richards et al., 2015).
We report here two sporadic cases of atypical ATS associated with KCNJ2 mutations, the recurrent p. Arg82Trp and a so far uncharacterized p. Pro186Gln. Both variants occurred de novo in the proband as observed in one third of KCNJ2 cases (Manzatti et al., 2020). Cellular studies performed after expression in xenopus oocytes showed that both variants reduced drastically Kir2.1 channel function.
KCNJ2 encodes the pore-forming subunits of the tetrameric potassium channel Kir 2.1 that plays a major role in the stabilization of the resting potential during cardiac and skeletal myocytes repolarization (Wible et al., 1995; Plaster et al., 2001). Loss-of-function KCNJ2 mutations are associated with ATS whereas gain of function mutations are associated with autosomal dominant AF familial 9 and short QT syndrome 3 (Xia 2005 Priori et al., 2005). A total of 147 variants are listed modifying the coding sequence are reported for KCNJ2 gene in the ClinVar database (missense, nonsense, frameshift). Nine are truncating mutations while the others are missense variations. All, except one, are associated to a cardiac phenotype or to ATS, and forty-two variants considered as probably pathogenic or pathogenic are located on 27 amino acid positions that correspond to hot spots of mutations, including amino acid 82 and 186 that are mutated in our report. Eighty-five variants are classified as variant of unknown significance, what underlines the need of functional study to improve molecular diagnosis for KCNJ2 variants.
Here, using expression in xenopus oocytes, we demonstrated that p. Arg82Trp and the p. Pro186Gln mutations of Kir2.1 reduced drastically the channel activity. Previous data published on the p. Arg82Trp variant showed that expression of the mutant allele alone completely abolished outward and inward currents and that its trafficking to cell membrane was normal (Eckhardt et al., 2007; Kimura et al., 2012). Although all experiments performed with this mutant concluded to a reduction of inward current, the notion that it had a dominant negative effect was more controversial (Eckhardt et al., 2007; Kimura et al., 2012). Our experiments demonstrate that a homotetramer of p. Arg82Trp mutated subunits retain some potassium channel activity, confirming that a least a fraction of the expressed proteins can reach plasma membrane, and the mutation does not fully abolish the function of the channel. In our experiments, the p. Arg82Trp mutant of Kir2.1 also showed a clear dominant negative effect whenever expressed in combination with the WT protein. Of note, another recurrent mutation on the same amino acid, the p. Arg82Gln has also been reported and characterized as having a dominant negative effect (Davies et al., 2005; Kimura et al., 2012).
To our knowledge, the variant p. Pro186Gln of Kir2.1 has never been reported in the literature but is referenced in ClinVar database. Substitutions affecting the same codon and leading to different amino acid variations (p.Pro186Leu and p. Pro186Thr) were however already reported with a dominant negative effect (Soom et al., 2001; Song et al., 2016), and functional studies on the variant p. Pro186Ala demonstrated a significantly reduction of PIP2 binding in vitro (Soom et al., 2001). Our experiments demonstrating a loss of function of channel due to the p. Pro186Gln mutation confirm therefore the critical role of the Proline 186 residue in Kir2.1, as expected from its position within the PIP2 binding site of Kir2 channels (Hansen et al., 2011).
The impact of p. Arg82Trp and p. Pro186Gln mutations was slightly different upon Kir1.2 expression, although no clear distinction on cardiological phenotype was observed between the two affected patients. In this study, proband 1 with the p. Arg82Trp variant presented isolated cardiac symptoms, and proband 2 with the p. Pro186Gln variant showed extra cardiac features that allowed retrospective diagnosis of Andersen Tawil syndrome with CPVT. These data are concordant with the observation that C-terminal mutations (AA 183-428) manifested a higher prevalence of skeletal dysmorphisms than N-terminal or pore domains mutations (Manzatti et al., 2020).
It illustrates the already reported variability of expression of ATS (Tristani-Farouzi et al., 2002) and fit with the current knowledge that there is no clear genotype-phenotype correlation between KCNJ2 variants and symptoms. Indeed, patients harboring the same variants, such as p. Arg82Trp and p. Arg82Gln can present with typical ATS, atypical ATS with cardiac features alone, isolated CPVT, or isolated LQTS (Tan et al., Kimura et al., 2012; Davies et al., 2005; Limberg et al., 2013).
To explain this variability, several hypotheses have been raised. It was suggested that N-terminal mutations could be more frequently associated with atypical ATS while C-terminal mutations with typical ATS (Kimura et al., 2012). Mutation on specific residues such as Arg67, Arg82 and Thr305 could lead to an atypical phenotype (Eckhardt et al., 2007; Kimura et al., 2012). Limberg et al. reported two KCNJ2 mutations (p.Asn318Cys, p. Trp322Cys) associated with an isolated cardiac phenotype and the authors proposed that the mild reduction of native Kir2.1 currents due to the mutations may lead to ATS with an isolated cardiac phenotype (Limberg et al., 2013). A sex-dependent expression was described, since women appear more affected by ventricular arrhythmias whereas periodic paralysis appears more frequent in men (Andelfinger et al., 2002; Donaldson et al., 2003). Noticeably, some features of ATS can be missed, such as discrete dysmorphic features or asymptomatic arrhythmias, what happened for proband 2 for whom ATS diagnosis was retrospective. These gaps in the clinical data could contribute to apparent variability in genotype-phenotype correlation. Owing the clinical overlap between ATS and CPVT, the identification of ventricular tachycardia has to alert about neurological and morphological symptoms. Their identification would guide toward the diagnosis of ATS due to a KCNJ2 variant, which has particular implication in the CPVT treatment strategy. On the contrary, neurological and morphological symptoms suggesting ATS should raise the hypothesis of cardiological manifestations such as CPVT.
The patients reported here were females initially referred to our laboratory through a cardiologic gateway, and diagnosed at the age of 16 and 13 with a moderate cardiologic phenotype. Interestingly, they share hallmarks of CPVT patients with KCNJ2 mutations described so far in the literature (Table 1). KCNJ2-mutated patients with CPVT are all females (Andelfinger et al., 2002; Tester et al., 2006; Kukla et al., 2014), for whom the age of onset or diagnosis ranges from 2 to 36, with a mean around 15, as compared to 8 years old for RYR2-related CPVT (Ackerman et al., 2011). In addition, VA reported in patients with KCNJ2 mutations have been described as milder than in RYR2 mutated patient, especially rarely leading to syncope or sudden cardiac arrest (Andelfinger et al., 2002; Tristani-Firouzi et al., 2002; Delannoy et al., 2013; Kukla et al., 2014; Inoue et al., 2018). Reported KCNJ2-mutated patients with CPVT consistently show an abnormal resting ECG, while only exercise stress test triggers detectable ECG signs for other CPVT patients. The resting ECG in KCNJ2-mutated patients may present characteristic T-U wave patterns comprising biphasic and enlarged U-waves increasing in amplitude during tachycardia, prolonged terminal T downslope, wide T-U junctions, fusion of T and U waves after PVC, and U on P sign during sinus tachycardia (Zhang et al., 2005; Kukla et al., 2014; Inoue et al., 2018). Due to these features, QT can appear prolonged explaining why KCNJ2-mutated patients had long been misdiagnosed with LQTS, and ATS considered as a syndromic form of LQTS (named LQT7). In fact, appropriate measurement of the interval with tangent technique shows no QT lengthening in these patients (Zhang et al., 2005; Kukla et al., 2014). Compared to others CPVT patients, KCNJ2-mutated patients also show more PVC and ventricular bigeminy at rest with non-sustained and bidirectional VT (Zhang et al., 2005; Kukla et al., 2014). Noticeably, the ECG during exercise testing is also different for both groups: ventricular arrhythmias appear early, stop at the peak of exercise to finally reappear after the end of exercise for KCNJ2-mutated patients with CPVT, while in other CPVT patients they appear early, increase to reach the maximum at peak and stop at rest. The morphology of PVC is different between the two groups: right BBB pattern is more frequent in KCNJ2-mutated patients with CPVT whereas left BBB pattern if more associated with mutations in other genes (Inoue et al., 2018). Interestingly, proband 2 of our study presented a Pierre Robin sequence. Cleft palate with pathogenic KCNJ2 mutations is rarely described in the literature (Andelfinger et al., 2002; Jakobsen et al., 2007) but it was reported that KCNJ2 knock out homozygous mice present with a complete cleft of the secondary palate (Zaritsky et al., 2000; Dahal et al., 2012). Pierre Robin sequence or cleft palate are not specific of ATS, but our study highlights that it can represent an extracardiac sign associated with a CPVT phenotype that may evoke ATS and KCNJ2 mutations.
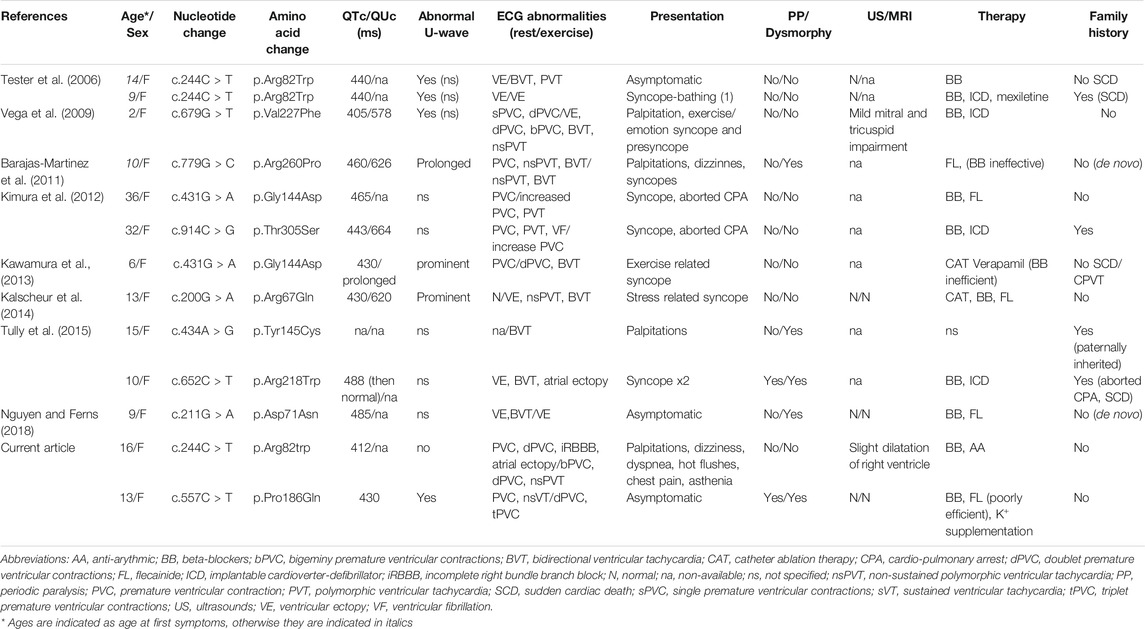
TABLE 1. Clinical and genotype characteristics of KCNJ2 mutation-carrying patients with ventricular arrhythmia diagnosed as CPVT in the literature.
Overall, patients with clinical signs of CPVT and mutations in the KCNJ2 gene can show a specific phenotype, which can distinguish them from individuals with CPVT associated to other genes. This is of particular importance since management can differ between both group of patients, during evaluation following the initial diagnosis, and for treatments and activity recommendations.
We report here 2 novel cases of atypical ATS initially referred for suspicion of CPVT leading to the identification of KCNJ2 pathogenic variants. We confirmed the drastic reduction of function for p. Arg82Trp mutated Kir2.1 channel and described the new loss of function p. Pro186Gln variant in KCNJ2. These two cases add further evidence that patients with KCNJ2 pathogenic variants can present with cardiac arrhythmias that can phenocopy CPVT. On the bases of all published cases, it seems that patients with KCNJ2 mutations can be identified among CPVT cohorts by accurate phenotypical description (ECG features, sex, age and severity of symptoms, associated signs). The diagnosis for these patients can however be difficult and KCNJ2 should be included in the panel of genes used to identify molecular causes of arrhythmias, in particular to adapt CPVT patient care.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
PL, NR-B, and JF contribute to the molecular data and manuscript redaction. GM, AJ contribute to the genetic data and rereading the manuscript MF, JR, and MV contribute to the functionnal studies and manuscript redaction. LF, GL, LP, and PA contribute to the clinical cardiological and genetic data and management and redaction and rereading the manuscript.
Work in the Vivaudou laboratory was supported by CNRS (Centre National de la Recherche Scientifique), CEA (Commissariat à l’Energie Atomique et aux énergies alternatives), Université Grenoble Alpes, and by grants from the Agence Nationale de la Recherche (NewOptogeneticTools, ANR-15-CE11–0029–01; Viral_Rhodopsins ANR-19-CE11-0026). The Vivaudou laboratory is a member of the French National Laboratory of Excellence “Ion Channel Science and Therapeutics” (LabEX ICST) funded by a network grant from ANR (ANR-11-LABX-0015-01). MF is a recipient of a doctoral fellowships from CEA.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank M. Lamairia and L. Lira for technical expertise. We also thank the Molecular Biology Facility of the “Institut de Biologie et Pathologie”, Grenoble University Hospital, and A. Chatagnon for helpful discussions and management of gene panel sequencing. We are grateful to Saïd Bendahhou from Nice Sophia Antipolis University for providing us with a human Kir2.1 clone in an oocyte vector.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.773177/full#supplementary-material
Ackerman, M. J., Priori, S. G., Willems, S., Berul, C., Brugada, R., Calkins, H., et al. (2011). HRS/EHRA Expert Consensus Statement on the State of Genetic Testing for the Channelopathies and Cardiomyopathies: This Document Was Developed as a Partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace 13, 1077–1109. doi:10.1093/europace/eur245
Andelfinger, G., Tapper, A. R., Welch, R. C., Vanoye, C. G., George, A. L., and Benson, D. W. (2002). KCNJ2 Mutation Results in Andersen Syndrome with Sex-specific Cardiac and Skeletal Muscle Phenotypes. Am. J. Hum. Genet. 71, 663–668. doi:10.1086/342360
Barajas-Martinez, H., Hu, D., Ontiveros, G., Caceres, G., Desai, M., Burashnikov, E., et al. (2011). Biophysical and Molecular Characterization of a Novel De Novo KCNJ2 Mutation Associated with Andersen-Tawil Syndrome and Catecholaminergic Polymorphic Ventricular Tachycardia Mimicry. Circ. Cardiovasc. Genet. 4, 51–57. doi:10.1161/CIRCGENETICS.110.957696
Dahal, G. R., Rawson, J., Gassaway, B., Kwok, B., Tong, Y., Ptáček, L. J., et al. (2012). An Inwardly Rectifying K+ Channel Is Required for Patterning. Development 139, 3653–3664. doi:10.1242/dev.078592
Davies, N. P., Imbrici, P., Fialho, D., Herd, C., Bilsland, L. G., Weber, A., et al. (2005). Andersen-Tawil Syndrome: New Potassium Channel Mutations and Possible Phenotypic Variation. Neurology 65, 1083–1089. doi:10.1212/01.wnl.0000178888.03767.74
Delannoy, E., Sacher, F., Maury, P., Mabo, P., Mansourati, J., Magnin, I., et al. (2013). Cardiac Characteristics and Long-Term Outcome in Andersen-Tawil Syndrome Patients Related to KCNJ2 Mutation. Europace 15, 1805–1811. doi:10.1093/europace/eut160
Devalla, H. D., Gélinas, R., Aburawi, E. H., Beqqali, A., Goyette, P., Freund, C., et al. (2016). TECRL , a New Life‐threatening Inherited Arrhythmia Gene Associated with Overlapping Clinical Features of Both LQTS and CPVT. EMBO Mol. Med. 8, 1390–1408. doi:10.15252/emmm.201505719
Donaldson, M. R., Jensen, J. L., Tristani-Firouzi, M., Tawil, R., Bendahhou, S., Suarez, W. A., et al. (2003). PIP2 Binding Residues of Kir2.1 Are Common Targets of Mutations Causing Andersen Syndrome. Neurology 60, 1811–1816. doi:10.1212/01.wnl.0000072261.14060.47
Eckhardt, L. L., Farley, A. L., Rodriguez, E., Ruwaldt, K., Hammill, D., Tester, D. J., et al. (2007). KCNJ2 Mutations in Arrhythmia Patients Referred for LQT Testing: a Mutation T305A with Novel Effect on Rectification Properties. Heart Rhythm 4, 323–329. doi:10.1016/j.hrthm.2006.10.025
Handklo-Jamal, R., Meisel, E., Yakubovich, D., Vysochek, L., Beinart, R., Glikson, M., et al. (2020). Andersen-Tawil Syndrome Is Associated with Impaired PIP2 Regulation of the Potassium Channel Kir2.1. Front. Pharmacol. 11, 672. doi:10.3389/fphar.2020.00672
Hansen, S. B., Tao, X., and MacKinnon, R. (2011). Structural Basis of PIP2 Activation of the Classical Inward Rectifier K+ Channel Kir2.2. Nature 477, 495–498. doi:10.1038/nature10370
Haruna, Y., Kobori, A., Makiyama, T., Yoshida, H., Akao, M., Doi, T., et al. (20072012). Genotype-phenotype Correlations of KCNJ2 Mutations in Japanese Patients with Andersen-Tawil syndromeGenotype-Phenotype Correlations ofKCNJ2 Mutations in Japanese Patients with Andersen-Tawil Syndrome. Hum. Mutat.Cardiovasc Res. 2893, 208666–209208. doi:10.1002/humu.948310.1093/cvr/cvr329
Hayashi, M., Denjoy, I., Extramiana, F., Maltret, A., Buisson, N. R., Lupoglazoff, J.-M., et al. (2009). Incidence and Risk Factors of Arrhythmic Events in Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 119, 2426–2434. doi:10.1161/CIRCULATIONAHA.108.829267
Hosy, E., and Vivaudou, M. (2014). The Unusual Stoichiometry of ADP Activation of the KATP Channel. Front. Physiol. 5, 11. doi:10.3389/fphys.2014.00011
Inoue, Y. Y., Aiba, T., Kawata, H., Sakaguchi, T., Mitsuma, W., Morita, H., et al. (2018). Different Responses to Exercise between Andersen-Tawil Syndrome and Catecholaminergic Polymorphic Ventricular Tachycardia. Europace 20, 1675–1682. doi:10.1093/europace/eux351
Jakobsen, L. P., Ullmann, R., Christensen, S. B., Jensen, K. E., Molsted, K., Henriksen, K. F., et al. (2007). Pierre Robin Sequence May Be Caused by Dysregulation of SOX9 and KCNJ2. J. Med. Genet. 44, 381–386. doi:10.1136/jmg.2006.046177
Jensen, H. H., Brohus, M., Nyegaard, M., and Overgaard, M. T. (2018). Human Calmodulin Mutations. Front. Mol. Neurosci. 11, 396. doi:10.3389/fnmol.2018.00396
Kalscheur, M. M., Vaidyanathan, R., Orland, K. M., Abozeid, S., Fabry, N., Maginot, K. R., et al. (2014). KCNJ2 Mutation Causes an Adrenergic-dependent Rectification Abnormality with Calcium Sensitivity and Ventricular Arrhythmia. Heart Rhythm 11, 885–894. doi:10.1016/j.hrthm.2014.02.015
Kawamura, M., Ohno, S., Naiki, N., Nagaoka, I., Dochi, K., Wang, Q., et al. (2013). Genetic Background of Catecholaminergic Polymorphic Ventricular Tachycardia in Japan. Circ. J. 77, 1705–1713. doi:10.1253/circj.cj-12-1460
Kimura, H., Zhou, J., Kawamura, M., Itoh, H., Mizusawa, Y., Ding, W.-G., et al. (2012). Phenotype Variability in Patients Carrying KCNJ2 Mutations. Circ. Cardiovasc. Genet. 5, 344–353. doi:10.1161/CIRCGENETICS.111.962316
Kukla, P., Biernacka, E., Baranchuk, A., Jastrzebski, M., and Jagodzinska, M. (2014). Electrocardiogram in Andersen-Tawil Syndrome. New Electrocardiographic Criteria for Diagnosis of Type-1 Andersen-Tawil Syndrome. Ccr 10, 222–228. doi:10.2174/1573403x10666140514102528
Limberg, M. M., Zumhagen, S., Netter, M. F., Coffey, A. J., Grace, A., Rogers, J., et al. (2013). Non Dominant-Negative KCNJ2 Gene Mutations Leading to Andersen-Tawil Syndrome with an Isolated Cardiac Phenotype. Basic Res. Cardiol. 108, 353. doi:10.1007/s00395-013-0353-1
Mazzanti, A., Guz, D., Trancuccio, A., Pagan, E., Kukavica, D., Chargeishvili, T., et al. (2020). Natural History and Risk Stratification in Andersen-Tawil Syndrome Type 1. J. Am. Coll. Cardiol. 2175 (15), 1772–1784. doi:10.1016/j.jacc.2020.02.033
Moreau, C. J., Revilloud, J., Caro, L. N., Dupuis, J. P., Trouchet, A., Estrada-Mondragón, A., et al. (2017). Tuning the Allosteric Regulation of Artificial Muscarinic and Dopaminergic Ligand-Gated Potassium Channels by Protein Engineering of G Protein-Coupled Receptors. Sci. Rep. 7, 41154. doi:10.1038/srep41154
Nguyen, D., and Ferns, S. J. (2018). Asymptomatic Ventricular Tachycardia: Diagnostic Pitfalls of Andersen-Tawil Syndrome-A Case Report. Eur. Heart J. Case Rep. 2 (3), yty083. doi:10.1093/ehjcr/yty083
Nyegaard, M., Overgaard, M. T., Søndergaard, M. T., Vranas, M., Behr, E. R., Hildebrandt, L. L., et al. (2012). Mutations in Calmodulin Cause Ventricular Tachycardia and Sudden Cardiac Death. Am. J. Hum. Genet. 91, 703–712. doi:10.1016/j.ajhg.2012.08.015
Plaster, N. M., Tawil, R., Tristani-Firouzi, M., Canún, S., Bendahhou, S., Tsunoda, A., et al. (2001). Mutations in Kir2.1 Cause the Developmental and Episodic Electrical Phenotypes of Andersen's Syndrome. Cell 105, 511–519. doi:10.1016/s0092-8674(01)00342-7
Priori, S. G., Napolitano, C., Memmi, M., Colombi, B., Drago, F., Gasparini, M., et al. (2002). Clinical and Molecular Characterization of Patients with Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 106, 69–74. doi:10.1161/01.cir.0000020013.73106.d8
Priori, S. G., Pandit, S. V., Rivolta, I., Berenfeld, O., Ronchetti, E., Dhamoon, A., et al. (2005). A Novel Form of Short QT Syndrome (SQT3) Is Caused by a Mutation in the KCNJ2 Gene. Circ. Res. 96, 800–807. doi:10.1161/0110.1161/01.res.0000162101.76263.8c
Priori, S. G., Wilde, A. A., Horie, M., Cho, Y., Behr, E. R., Berul, C., et al. (2013). HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes. Heart Rhythm 10 (12), 1932–1963. doi:10.1016/j.hrthm.2013.05.014
Richards, S., Aziz, N., Aziz, N., Bale, S., Bick, D., Das, S., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423. doi:10.1038/gim.2015.30
Roux-Buisson, N., Cacheux, M., Fourest-Lieuvin, A., Fauconnier, J., Brocard, J., Denjoy, I., et al. (2012). Absence of Triadin, a Protein of the Calcium Release Complex, Is Responsible for Cardiac Arrhythmia with Sudden Death in Human. Hum. Mol. Genet. 21, 2759–2767. doi:10.1093/hmg/dds104
Song, J., Luo, S., Cheng, X., Yue, D., Zhu, W., Lin, J., et al. (2016). Clinical Features and Long Exercise Test in Chinese Patients with Andersen-Tawil Syndrome. Muscle Nerve 54, 1059–1063. doi:10.1002/mus.25169
Soom, M., Schönherr, R., Kubo, Y., Kirsch, C., Klinger, R., and Heinemann, S. H. (2001). Multiple PIP2 Binding Sites in Kir2.1 Inwardly Rectifying Potassium Channels. FEBS Lett. 490, 49–53. doi:10.1016/s0014-5793(01)02136-6
Sumitomo, N. (2016). Current Topics in Catecholaminergic Polymorphic Ventricular Tachycardia. J. Arrhythmia 32, 344–351. doi:10.1016/j.joa.2015.09.008
Tester, D. J., Arya, P., Will, M., Haglund, C. M., Farley, A. L., Makielski, J. C., et al. (2006). Genotypic Heterogeneity and Phenotypic Mimicry Among Unrelated Patients Referred for Catecholaminergic Polymorphic Ventricular Tachycardia Genetic Testing. Heart Rhythm 3, 800–805. doi:10.1016/j.hrthm.2006.03.025
Tristani-Firouzi, M., Jensen, J. L., Donaldson, M. R., Sansone, V., Meola, G., Hahn, A., et al. (2002). Functional and Clinical Characterization of KCNJ2 Mutations Associated with LQT7 (Andersen Syndrome). J. Clin. Invest. 110, 381–388. doi:10.1172/JCI15183
Tully, I., Atherton, J., Hunt, L., Ingles, J., Semsarian, C., and McGaughran, J. (2015). Rarity and Phenotypic Heterogeneity Provide Challenges in the Diagnosis of Andersen-Tawil Syndrome: Two Cases Presenting with ECGs Mimicking Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT). Int. J. Cardiol. 201, 473–475. doi:10.1016/j.ijcard.2015.07.069
Vega, A. L., Tester, D. J., Ackerman, M. J., and Makielski, J. C. (2009). Protein Kinase A-dependent Biophysical Phenotype for V227F-KCNJ2 Mutation in Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Arrhythm Electrophysiol. 2, 540–547. doi:10.1161/CIRCEP.109.872309
Vivaudou, M. (2019). eeFit: a Microsoft Excel-embedded Program for Interactive Analysis and Fitting of Experimental Dose-Response Data. BioTechniques 66, 186–193. doi:10.2144/btn-2018-0136
Vivaudou, M., Todorov, Z., Reyes-Mejia, G. C., and Moreau, C. (2017). Ion Channels as Reporters of Membrane Receptor Function: Automated Analysis in Xenopus Oocytes. Methods Mol. Biol. 1635, 283–301. doi:10.1007/978-1-4939-7151-0_15
Wible, B. A., De Biasi, M., Majumder, K., Taglialatela, M., and Brown, A. M. (1995). Cloning and Functional Expression of an Inwardly Rectifying K + Channel from Human Atrium. Circ. Res. 76, 343–350. doi:10.1161/01.res.76.3.343
Zaritsky, J. J., Eckman, D. M., Wellman, G. C., Nelson, M. T., and Schwarz, T. L. (2000). Targeted Disruption of Kir2.1 and Kir2.2 Genes Reveals the Essential Role of the Inwardly Rectifying K + Current in K + -Mediated Vasodilation. Circ. Res. 87, 160–166. doi:10.1161/01.res.87.2.160
Keywords: catecholaminergic polymorphic ventricular tachycardia, Andersen-Tawil syndrome, Kir2.1 channel, Pierre Robin sequence, functionnal characterization, KCNJ2 variants
Citation: Le Tanno P, Folacci M, Revilloud J, Faivre L, Laurent G, Pinson L, Amedro P, Millat G, Janin A, Vivaudou M, Roux-Buisson N and Fauré J (2021) Characterization of Loss-Of-Function KCNJ2 Mutations in Atypical Andersen Tawil Syndrome. Front. Genet. 12:773177. doi: 10.3389/fgene.2021.773177
Received: 09 September 2021; Accepted: 03 November 2021;
Published: 25 November 2021.
Edited by:
Zahurul A. Bhuiyan, Centre Hospitalier Universitaire Vaudois (CHUV), SwitzerlandReviewed by:
Oscar Campuzano, University of Girona, SpainCopyright © 2021 Le Tanno, Folacci, Revilloud, Faivre, Laurent, Pinson, Amedro, Millat, Janin, Vivaudou, Roux-Buisson and Fauré. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nathalie Roux-Buisson, TlJvdXhCdWlzc29uQGNodS1ncmVub2JsZS5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.