 Tao Zhang1,2†
Tao Zhang1,2† Hua Yuan1,2†Hongdan Zhu1,2Yuyi Ying1,2Jinlong Ding1,2Haigang Ding1,2Xiaoliang Shi1,2Yao He1,2Haitao Pan1,2*Yongxing Zhong1,2*
Hua Yuan1,2†Hongdan Zhu1,2Yuyi Ying1,2Jinlong Ding1,2Haigang Ding1,2Xiaoliang Shi1,2Yao He1,2Haitao Pan1,2*Yongxing Zhong1,2*- 1Shaoxing Maternity and Child Health Care Hospital, Shaoxing, China
- 2Obstetrics and Gynecology Hospital of Shaoxing University, Shaoxing, China
Background: Fetal congenital heart disease (CHD) is the most common congenital defect, with an incidence of 0.6–0.8%, accounting for 30–50% of infant congenital disease deaths. The pathogenesis of CHD is still unclear, so an active and effective prenatal diagnosis is very important for the prevention and control of CHD. Herein, a Chinese CHD patient with rare compound heterozygous mutations in the DNAH9 gene was reported, and the 3D structure and functional changes of DNAH9 protein were predicted.
Case presentation: A 23-year-old pregnant woman came to our hospital for prenatal diagnosis at 27 weeks of gestation. Both she and her partner were unaffected. Fetal CHD was detected by ultrasound screening. Copy number variation sequencing (CNV-seq) revealed an 81 kb deletion at chr17p12 (11,486,795–11,568,385), including exons 1–15 of DNAH9 gene, which plays a key role in cardiac development. Then, whole exome sequencing (WES) was used and identified a nonsense mutation (c.10975C>T) in DNAH9, which resulted in the mutation of amino acid 3,659 from glutamine to termination. The 3D mutant protein structures were predicted using SWISS-MODEL and showed structural changes from functional β-sheet and α-helix to termination, respectively.
Conclusion: We describe a case of fetal CHD caused by DNAH9 mutations and provide an effective diagnostic technique for identifying intragenic deletions. This diagnostic process can be implicated in prenatal diagnosis of CHD.
Introduction
Congenital heart diseases (CHD) are the most common birth defects, which can severely affect human health, accounting for 2–8% of newborn children (Hoffman et al., 2004; Lambrechts et al., 2005). The clinical manifestations and severity of CHD are widely variable. Mild cases, such as some small ventricular septal defects, can self-close after birth. Severe cases, such as most instances of tetralogy of Fallot (TOF), seriously affect the function and structure of the heart, with a high mortality. CHD is often accompanied by multiple organ malformations, which is one of the main causes of neonatal death (Pediatric Cardiac Genomics Consortium et al., 2013; Egbe et al., 2014). Traditional thought is that CHD is the combined result of environmental and genetic factors, in which environmental factors are dominant (Kalisch-Smith et al., 2020). However, with the development of technologies, it has been found that the incidence of CHD is significantly higher in monozygotic twins, or when patients have family history of CHD or consanguinity. A variety of chromosomal aberrations is often associated with different types of CHD. It is now highly suggested that genetic factors play an important role in the pathogenesis of CHD (Van der Bom et al., 2011). In particular, the invention and application of chromosome microarray technology (CMA) and high-throughput sequencing technology provide a strong basis for elucidating the extremely important role of genetic factors in the occurrence of CHD (Charron et al., 2010; Rehm, 2013; Aburawi et al., 2015).
Monogenic inherited disease refers to disease caused by a single gene abnormality, also known as Mendelian genetic disease. Monogenic disease can be divided into autosomal dominant or recessive, X-chromosome dominant or recessive genetic diseases. The incidence rate of any individual monogenic disease is not high, but the overall incidence rate of all monogenic diseases combined is 4–5%. This kind of genetic disease can also be seen in various CHD, which can be an isolated CHD phenotype (such as in GATA4 and NKX2.5 gene mutations (Winston et al., 2012)) or a part of complex syndrome, such as Noonan syndrome or Holt-Oram syndrome.
DNAH9 gene associated primary ciliary dyskinesia 40 is characterized by chronic respiratory tract infection, visceral translocation and infertility (Fassad et al., 2018; Loges et al., 2018). Most of the patients with primary ciliary dyskinesia have congenital respiratory diseases. From early childhood, patients will have repeated respiratory infections. However, the respiratory compromise of primary ciliary dyskinesia type 40 is relatively light, and usually does not develop into serious lung disease. About 50% of patients with primary ciliary dyskinesia have mirror image inversion of internal organs, and some patients have been reported to have severe congenital heart malformations.
In this study, we reported the case of a Chinese fetal proband, who presented with fetal CHD by ultrasound screening. Copy number variation sequencing (CNV-seq) revealed an 81 kb deletion at chr17p12, including a deletion of exons 1–15 in DNAH9. Then, whole exome sequencing (WES) revealed a nonsense mutation (c.10975C>T) in DNAH9 on the other allele. This base mutation resulted in the codon of amino acid 3,659 from glutamine to termination. To predict the changes of 3D protein structure, we used SWISS-MODEL and PyMO and revealed these two mutations changed functional β-sheet and α-helix structures, respectively.
Materials and Methods
Sample Collection
The study was approved by the institutional ethics committee of Shaoxing Maternity and Child Health Care Hospital. The family members had signed informed consent documents. Parental consent was obtained for collecting the prenatal fetal cord venous blood at 27 weeks of pregnancy. Peripheral blood samples also were collected from proband’s parents.
Copy-Number Variation Sequencing and Whole Exome Sequencing
Genomic DNA was extracted from the proband and parental blood using a DNEasy Blood and Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s procedures, respectively. For WES, the genomic DNA of the proband was enriched for coding exons using Agilent SureSelect Low Input Reagent Kit and sequenced on Illumina HiSeq X Ten platform. The sequencing data captured 99.75% of coding regions across 35,519,957 bp length of 25,701 genes in total. The average sequence depth is 180.347X and 97.87% of targeted regions with average depth >20X. For CNV-seq, the genomic DNA was fragmented using Hieff NGS® Fast-PaceTM DNA Fragmentation Reagent and prepared for the PCR-free library by Hieff NGS® Complete Adapter Kit for Illumina®.
Data Analysis
The AfterQC (Chen et al., 2017) was used to evaluate the sequencing quality of the original sequencing data, and the low quality and contaminated reads were removed. After data were aligned to human reference hg19 by BWA software (Li and Durbin, 2010), the single nucleotide variants (SNV) and indels in genome were called by using the GATK software (Mckenna et al., 2010). Then, we used 1000 Genomes database (1000 human genome dataset), Genome AD (Genome Aggregation Database dataset) 2.1.1, and ExAC (The Exome Aggregation Consortium dataset) to screen the SNV and indels and the OMIM, HGMD, and Clinvar databases to filter the reported mutations. dbNSFP database was used to predict the pathogenicity of missense mutation and splice mutation. All mutation sites were classified by ACMG genetic variation classification criteria and guidelines. Finally, Sanger sequencing method was used to verify all possible pathogenic sites.
Protein Structure Prediction
The protein sequence with 4,486 amino acid residues of DNAH9 was download from NCBI (NP_001363.2). The wild-type and mutant-type 3D structure of the DNAH9 protein was predicted using SWISS-MODEL web server (https://swissmodel.expasy.org/) (Waterhouse et al., 2018). The best model was selected based on QMEANDisCo global score. The final predicted structure was visualized using PyMOL program (https://pymol.org/).
Results
Clinical Features
At 31 weeks of gestation, the parents were fully informed of severe fetal deformity and the significance of the compound DNAH9 mutations. The parents strongly requested the induction of labor. With the approvement of the institutional ethics committee of Shaoxing Maternity and Child Health Care Hospital, the pregnancy was terminated for fetal anomalies. The parents and other families were in normal physical condition, without any family history of genetic diseases. Prior to the proband, her mother had two histories of adverse pregnancies: 1) At 26 weeks of gestation, pregnancy was terminated due to the discovery of fetal single chamber heart and 2) at the third month of another pregnancy, spontaneous abortion occurred. Her mother’s menstruation was irregular in the period of 30–40 days.
There was no early pregnancy reaction, no exposure to poison or radioactive substances, and no history of folic acid supplementation in early pregnancy. At 9 weeks of gestation, her mother came to our hospital for ultrasound examination and found the normal gestational sac and gestational age. At 15 weeks of gestation, her mother came to our hospital for routine prenatal examination and found the normal physical condition, low risk value of non-invasive prenatal testing (NIPT), and normal range of OGTT. However, at 27 weeks of gestation, the result of ultrasound examination showed fetal cardiac abnormalities include single ventricle, pulmonary artery stenosis and visceral inversion (Figure 1). More detailed tests were carried out 2 days later and confirmed that the proband had no ventricular septal structure. CDFI showed mild left atrioventricular regurgitation. Because of the continuous histories of adverse pregnancy outcomes, the parents hoped to clarify the genetic factors and guide the next birth.
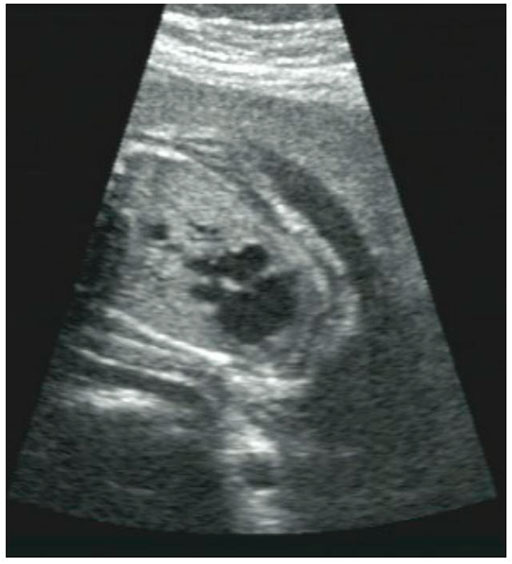
FIGURE 1. The fetus with congenital heart disease. Ultrasound scans of the fetus showed abnormal heart development, a single ventricle, pulmonary artery stenosis and situs inversus.
Molecular Analysis
Using CNV-seq, we identified an 81 kb deletion at chr17p12 (11,486,795–11,568,385) in the fetus (Figure 2A). This deletion region includes 1–15 exons of the DNAH9 gene. After verified in her parents by CNV-seq, it is found that the mother had a 79 kb deletion at chr17p12 (11,486,795–11,565,741) (Figure 2B). The result implied the deletion at chr17p12 of the fetus was inherited from her mother. Moreover, we also performed the WES in the fetus and revealed another mutation at c.10975C>T (p.Q3659*) in the exon 57 of DNAH9 gene (Figure 3A). Subsequently, sanger sequencing was used to verify this nonsense mutation in all family members and showed c.10975C>T DNAH9 mutation was inherited from her father (Figure 3B). According to the American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015), both the 81 kb deletion at chr17p12 and c.10975C>T in DNAH9 gene were predicted to be pathogenic mutations because of the evidence chain (PVS1+PM2+PP3).
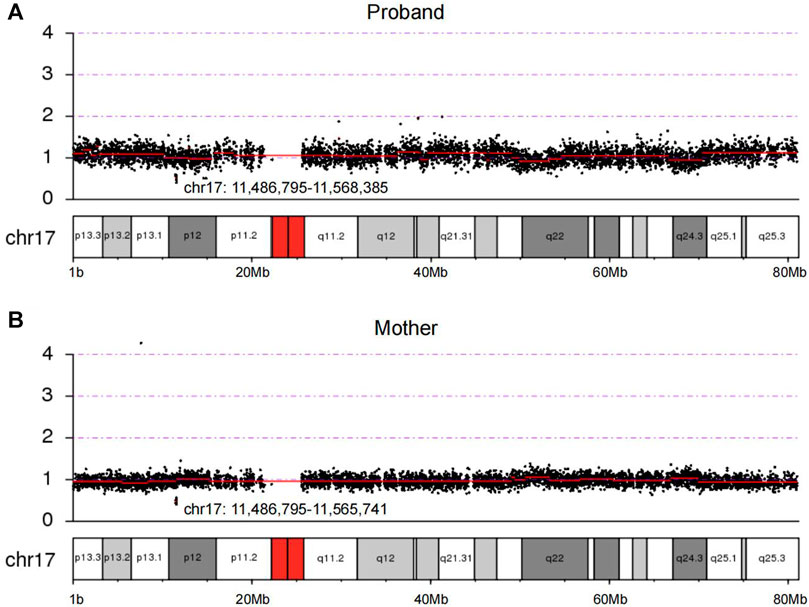
FIGURE 2. The loss of heterozygosity at 17p12 in the proband and the mother were detected by CNV-seq. (A) Copy number of chromosome 17 in the proband by sequencing. (B) Copy number of chromosome 17 in the proband’s mother by sequencing. The chr17:11,486,795–11,568,385 and chr17:11,486,795–11,565,741 showed in (A) and (B) were regions of deletion in the proband and her mother, respectively.
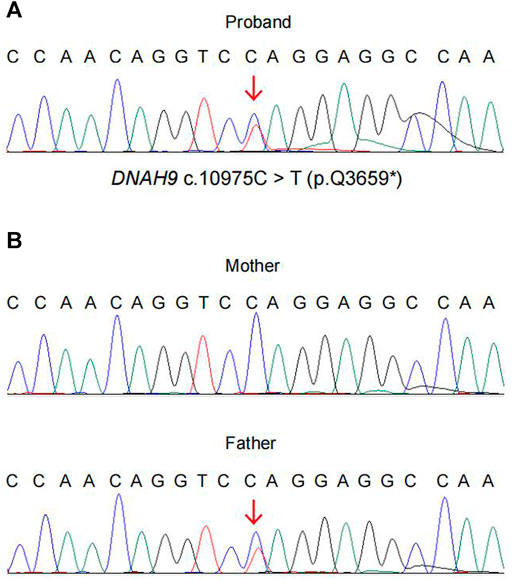
FIGURE 3. The evidence of the heterozygous nonsense mutation c.10975C> T (p.Q3659*) in the DNAH9 gene. WES and sanger sequencing verified the heterozygous mutation DNAH9 c.10975C> T in the proband (A) and her parents (B).
Effect of Mutations on Protein Structures
We used SWISS-MODEL web server (Waterhouse et al., 2018) to predict changes of DNAH9 protein structures when there were mutations in the sequence (Figure 4A). The deletion of 1–15 exons in DNAH9 gene will alter the 3D structure on β-sheet (Figure 4B), while the c.10975C>T (p.Q3659*) mutation will change the 3D structure from α-helix to termination (Figure 4C). The combination of these structural changes would alter the conformation of the DNAH9 protein and affect the protein stability and binding facility.
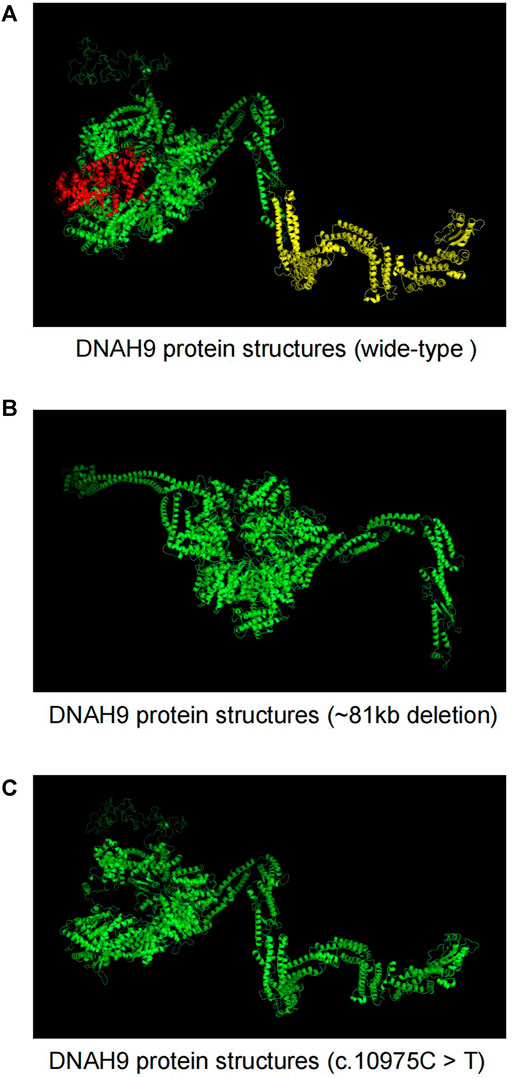
FIGURE 4. The Effects of DNAH9 mutations on protein structure. (A) The wide-type structure of DNAH9 protein predicted by SWISS-MODEL. The yellow and red parts were modelled using exon 1–15 in DNAH9 and amino acid residues from glutamine at position 3,659 to end. The mutant protein structures showed in (B) and (C) upon an 81 kb deletion at chr17p12 (11,486,795–11,568,385) or c.10975C> T mutation in DNAH9, respectively.
Discussion
Fetal CHD has become the most important birth defect type in China, accounting for the majority neonatal non-infectious diseases. However, the accuracy rate of prenatal diagnosis was only 36% by investigating 309 participants with CHD, including post-natal and fetal termination (Friedberg et al., 2009). The prenatal diagnosis rate of left ventricular outflow tract obstruction, transposition of great arteries and anomalous pulmonary venous drainage was the lowest. The traditional technology, ultrasound is the most effective non-invasive way of diagnosing CHD in fetal cases. However, because of the small meridian of the fetal heart, complex blood circulation, difficulties of capturing the fetal variable blood flow and the limitation of ultrasound itself, not all fetal CHD can be diagnosed by ultrasound in the prenatal setting. Although MRI can overcome shortcomings of the small field of vision and poor contrast of soft-tissue, the malformations of CHD patients are sometimes not limited to the heart. Additionally, many CHDs are accompanied by genetic changes, including chromosome aberration, single gene genetic defects, multi gene genetic defects and so on. Therefore, it is of great and urgent practical significance to make early and accurate prenatal genetic diagnosis of CHD in fetal cases by using next-generation sequencing technology.
Previous studies have proved that CMA is an effective tool to detect fetal genomic imbalances, including abnormalities of chromosome number and copy number variations (Xia et al., 2018). However, the price of CMA is relatively expensive. Copy number variation sequencing (CNV-seq), is a technology by using next-generation sequencing technology, can detect CNVs with high resolution in the whole genome. Compared with CMA, it can increase different sequencing depth to obtain more accurate information, and has the advantages of more flexible, fast, accurate and low operation cost (Duan et al., 2013). Moreover, CNV-seq technology has been applied to the genetic diagnosis of fetal CHD, recently (Zhu et al., 2016). In our study, we found that the proband had a loss of heterozygosity of 81 kb at 17p12 (11,486,795–11,568,385) by high-resolution CNV-seq. This deletion was within DNAH9 gene. Then the parent’s samples were verified by CNV-seq, and found her mother was a carrier of this deletion.
The loss of heterozygosity in DNAH9 alone should not cause CHD. To search for other potential genetic defects, WES and sanger sequencing of the proband and the parents were further performed and found one heterozygous nonsense mutation of DNAH9 gene (c.10975C>T) in the proband inherited from her father. This mutation resulted in termination of translation at amino acid 3,659. With additional clinical evaluation, these two mutations meet ACMG criteria for classification as pathogenic of DNAH9 mutations in this case.
In summary, through the combined application of high-through sequencing technologies (CNV-seq and WES), we established a probable cause of the couple’s poor pregnancy outcomes. The proband had compound heterozygous mutations, the deletion of 81 kb at 17p12 (11,486,795–11,568,385) and DNAH9 c.10975C>T (p.Q3659*), inherited from each carrier parent. In addition, our results showed that the combination of CNV-seq and WES is an effective approach to prenatal diagnosis.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by the Institutional Ethics Committee of Shaoxing Maternity and Child Health Care Hospital Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
TZ and YZ conceived and designed the experiments. HP performed the experiments, HY, YH, XS and HZ contributed new materials, YY, JD and HD analyzed the data and wrote the paper. All authors read and improved the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (82071729), the China Postdoctoral Science Foundation (2020M681336), the Zhejiang Provincial Natural Science Foundation of China (LY19H040002), the Science Technology Department of Zhejiang Province, China (LGF21H040003, LGF21H040004, LGF19H040004, LGD20H040001) and the Health Commission of Zhejiang Province, China (2021KY375, 2021KY1156, 2019RC296, 2019KY717, 2021KY1154, 2021KY1157, 2019KY229, 2019KY230); the Science Technology Department of Shaoxing, China (2020A13032, 2020A13037, 2018C30042, 2018C30043, 2018C30044, 2018C30048, 2020A13034, 2020A13035).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ACMG, American College of Medical Genetics and Genomics; CHD, Congenital heart disease; CNV-seq, Copy number variation sequencing; DNAH9, dynein axonemal heavy chain 9; WES, Whole exome sequencing.
References
Aburawi, E. H., Aburawi, H. E., Bagnall, K. M., and Bhuiyan, Z. A. (2015). Molecular Insight into Heart Development and Congenital Heart Disease: An Update Review from the Arab Countries. Trends Cardiovasc. Med. 25, 291–301. doi:10.1016/j.tcm.2014.11.007
Charron, P., Arad, M., Arbustini, E., Basso, C., Bilinska, Z., Elliott, P., et al. (2010). European Society of Cardiology Working Group OnGenetic Counselling and Testing in Cardiomyopathies: a Position Statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 31, 2715–2726. doi:10.1093/eurheartj/ehq271
Chen, S., Huang, T., Zhou, Y., Han, Y., Xu, M., and Gu, J. (2017). AfterQC: Automatic Filtering, Trimming, Error Removing and Quality Control for Fastq Data. BMC Bioinformatics 18, 80. doi:10.1186/s12859-017-1469-3
Duan, J., Zhang, J.-G., Deng, H.-W., and Wang, Y.-P. (2013). Comparative Studies of Copy Number Variation Detection Methods for Next-Generation Sequencing Technologies. PLoS One 8, e59128. doi:10.1371/journal.pone.0059128
Egbe, A., Ho, D., Lee, S., Uppu, S., and Srivastava, S. (2014). Prevalence of Congenital Anomalies in Newborns with Congenital Heart Disease Diagnosis. Ann. Pediatr. Card. 7, 86–91. doi:10.4103/0974-2069.132474
Fassad, M. R., Shoemark, A., Legendre, M., Hirst, R. A., Koll, F., Le Borgne, P., et al. (2018). Mutations in Outer Dynein Arm Heavy Chain DNAH9 Cause Motile Cilia Defects and Situs Inversus. Am. J. Hum. Genet. 103, 984–994. doi:10.1016/j.ajhg.2018.10.016
Friedberg, M. K., Silverman, N. H., Moon-Grady, A. J., Tong, E., Nourse, J., Sorenson, B., et al. (2009). Prenatal Detection of Congenital Heart Disease. J. Pediatr. 155, 26–31.e21. doi:10.1016/j.jpeds.2009.01.050
Hoffman, J. I. E., Kaplan, S., and Liberthson, R. R. (2004). Prevalence of Congenital Heart Disease. Am. Heart J. 147, 425–439. doi:10.1016/j.ahj.2003.05.003
Kalisch-Smith, J. I., Ved, N., and Sparrow, D. B. (2020). Environmental Risk Factors for Congenital Heart Disease. Cold Spring Harb Perspect. Biol. 12, 1. doi:10.1101/cshperspect.a037234
Lambrechts, D., Devriendt, K., Driscoll, D. A., Goldmuntz, E., Gewillig, M., Vlietinck, R., et al. (2005). Low Expression VEGF Haplotype Increases the Risk for Tetralogy of Fallot: a Family Based Association Study. J. Med. Genet. 42, 519–522. doi:10.1136/jmg.2004.026443
Li, H., and Durbin, R. (2010). Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 26, 589–595. doi:10.1093/bioinformatics/btp698
Loges, N. T., Antony, D., Maver, A., Deardorff, M. A., Güleç, E. Y., Gezdirici, A., et al. (2018). Recessive DNAH9 Loss-Of-Function Mutations Cause Laterality Defects and Subtle Respiratory Ciliary-Beating Defects. Am. J. Hum. Genet. 103, 995–1008. doi:10.1016/j.ajhg.2018.10.020
Mckenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: A MapReduce Framework for Analyzing Next-Generation DNA Sequencing Data. Genome Res. 20, 1297–1303. doi:10.1101/gr.107524.110
Pediatric Cardiac Genomics Consortium Gelb, B., Brueckner, M., Chung, W., Goldmuntz, E., Kaltman, J., et al. (2013). The Congenital Heart Disease Genetic Network Study: Rationale, Design, and Early Results. Circ. Res. 112, 698–706. doi:10.1161/CIRCRESAHA.111.300297
Rehm, H. L. (2013). Disease-Targeted Sequencing: A Cornerstone in the Clinic. Nat. Rev. Genet. 14, 295–300. doi:10.1038/nrg3463
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Van Der Bom, T., Zomer, A. C., Zwinderman, A. H., Meijboom, F. J., Bouma, B. J., and Mulder, B. J. M. (2011). The Changing Epidemiology of Congenital Heart Disease. Nat. Rev. Cardiol. 8, 50–60. doi:10.1038/nrcardio.2010.166
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., et al. (2018). SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 46, W296–W303. doi:10.1093/nar/gky427
Winston, J. B., Schulkey, C. E., Chen, I.-B. D., Regmi, S. D., Efimova, M., Erlich, J. M., et al. (2012). Complex Trait Analysis of Ventricular Septal Defects Caused by Nkx2-5 Mutation. Circ. Cardiovasc. Genet. 5, 293–300. doi:10.1161/circgenetics.111.961136
Xia, Y., Yang, Y., Huang, S., Wu, Y., Li, P., and Zhuang, J. (2018). Clinical Application of Chromosomal Microarray Analysis for the Prenatal Diagnosis of Chromosomal Abnormalities and Copy Number Variations in Fetuses with Congenital Heart Disease. Prenatal Diagn. 38, 406–413. doi:10.1002/pd.5249
Keywords: congenital heart disease, DNAH9 gene, copy number variation sequencing, whole exome sequencing, 3D structure
Citation: Zhang T, Yuan H, Zhu H, Ying Y, Ding J, Ding H, Shi X, He Y, Pan H and Zhong Y (2022) Fetal Congenital Heart Disease Caused by Compound Heterozygous Mutations in the DNAH9 Gene: A Case Report. Front. Genet. 12:771756. doi: 10.3389/fgene.2021.771756
Received: 07 September 2021; Accepted: 20 December 2021;
Published: 18 January 2022.
Edited by:
Emiliano González Vioque, University Clinical Hospital of Santiago, SpainReviewed by:
Gillian Blue, The Children’s Hospital at Westmead, AustraliaLauren Jeffries, Yale University, United States
Copyright © 2022 Zhang, Yuan, Zhu, Ying, Ding, Ding, Shi, He, Pan and Zhong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haitao Pan, cGFuaGFpdGFvQHN4ZmJ5LmNvbQ==; Yongxing Zhong, emhvbmd5b25neGluZ0BzeGZieS5jb20=
†These authors have contributed equally to this work