 Dejian Yuan1,2†
Dejian Yuan1,2† Tizhen Yan1,2†Shiqiang Luo1,2Jun Huang1,2
Tizhen Yan1,2†Shiqiang Luo1,2Jun Huang1,2 Jianqiang Tan1,2Jianping Zhang3
Jianqiang Tan1,2Jianping Zhang3 Victor Wei Zhang4,5Yueyuan Lan3
Victor Wei Zhang4,5Yueyuan Lan3 Taobo Hu6Jing Guo7Mingwei Huang8Dingyuan Zeng9*
Taobo Hu6Jing Guo7Mingwei Huang8Dingyuan Zeng9*- 1Department of Medical Genetics, Liuzhou Municipal Maternity and Child Healthcare Hospital, Liuzhou, China
- 2Liuzhou Key Laboratory of Birth Defects Prevention and Control, Liuzhou Municipal Maternity and Child Healthcare Hospital, Liuzhou, China
- 3Department of Ophthalmology, Liuzhou Municipal Maternity and Child Healthcare Hospital, Liuzhou, China
- 4AmCare Genomics Laboratory, Guangzhou, China
- 5Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, United States
- 6Center of Breast Diseases, Peking University People’s Hospital, Beijing, China
- 7Reproductive Medical Center, The First Affiliated Hospital of Sun Yat-sen University, Guangzhou, China
- 8Aegicare (Sheznzhen) Technology Co., Ltd., Shenzhen, China
- 9Department of Gynecology, Liuzhou Municipal Maternity and Child Healthcare Hospital, Liuzhou, China
ARR3 has been associated with X-linked, female-limited, high myopia. However, using exome sequencing (ES), we identified the first high myopia case with hemizygous ARR3-related mutation in a male patient in a Southern Chinese family. This novel truncated mutation (ARR3: c.569C>G, p.S190*) co-segregated with the disease phenotype in affected family members and demonstrated that high myopia caused by ARR3 is not X-linked, female-limited, where a complicated X-linked inheritance pattern may exist. Thus, our case expanded the variant spectrum in ARR3 and provided additional information for genetic counseling, prenatal testing, and diagnosis. Moreover, we characterized the nonsense-mediated decay of the ARR3 mutant mRNA and discussed the possible underlying pathogenic mechanisms.
Introduction
Myopia occurs when the axial length of the eyes is too long, typically causing distant objects to be focused in the front of the retina (Young, 2009). High myopia (HM) is a common human genetic sensory defect characterized by reduced vision, retinal degeneration, and choroidal atrophy. Its global prevalence exceeds 2.9% (Saw, 2003; Holden et al., 2016). HM is defined by diopters (D) greater than −6.0 or an axial length greater than 26 mm (Young et al., 2007).
Both environmental and genetic factors can influence the severity of myopia (Saw et al., 1996; Morgan et al., 2012), with some family studies showing that genetic factors play a crucial role in the development of HM (Young et al., 2007; Cai et al., 2019). ARR3 (RefSeq NM_004312.3; see Materials and Methods, “Nomenclature”) gene is associated with X-linked, female-limited HM based on a study of three families where all affected patients were female and hemizygous male family members were asymptomatic (Xiao et al., 2016). Even though several male family members were hemizygous, they did not possess the phenotypic features of HM, thereby proving to be the second disease, after those associated with PCDH19, with this unusual inheritance pattern (Depienne et al., 2009).
In this study, a member of a Chinese family was referred for HM, and exome sequencing (ES) revealed that an ARR3 variant submitted to the LOVD (variant #0000660387) was responsible for the phenotype. Our identification of one male hemizygote with HM symptoms is in stark contrast with the previous report of the female-limited inheritance pattern of ARR3-related HM (Xiao et al., 2016), implying that ARR3 has more complicated hereditary patterns than initially thought.
Materials and Methods
Nomenclature
The nomenclature of arrestin protein and gene is very confusing. In fact, the nomenclature of arrestin should be as follows: arrestin-1 (historic names S-antigen, 48 kDa protein, and visual or rod arrestin), arrestin-2 (β-arrestin or β-arrestin-1), arrestin-3 (β-arrestin-2 or hTHY-ARRX), and arrestin-4 (cone or X-arrestin; for unclear reasons, its gene is called “arrestin-3” in the HUGO database) (Zhan et al., 2014). We would like to use ARR3 (OMIM: 301770) to avoid the confusion in the nomenclature.
Materials
Plasmid p3XFLAG-CMV-7.1 was a gift from Prof. Zuobin Zhu. The site-directed mutagenesis kit was purchased from Gene Company Limited, Shanghai, China. UPF1 small interfering RNA (UPF1 siRNA) was purchased from RiboBio, Guangdong, China. Monoclonal anti-GFP (B-2) antibody and monoclonal anti-UPF1 antibody were from ABclonal Technology, China. DMEM and fetal bovine serum (FBS) were from Hyclone, USA. DNA polymerase Gold premix was purchased from Tsingke Biotechnology, Beijing, China. The AxyPrepTM PCR Cleanup Kit was from Axygen Biosciences, USA. The TIANprep Mini Plasmid Kit was purchased from Tiangen Technology, Beijing, China. Liposomal Transfection Reagent and the BCA protein quantification kit were purchased from Yisheng Biotechnology, Shanghai, China. RNAiso Plus Total RNA Extraction Reagent, the PrimeScript 1st strand cDNA Synthesis Kit, and the PrimeScript RT Reagent Kit with gDNA Eraser were purchased from Thermo Scientific, Shanghai, China. The SYBR Green Real-Time PCR Master Mix was purchased from Toyobo Biotechnology, Shanghai, China.
Human Subjects
This study included a family comprising 14 individuals. Of the eight members who were affected, one patient was male. All patients had HM with refractive errors greater than −6.0 D. Participants were recruited at the Division of Medical Genetics at Liuzhou Municipal Maternity and Child Healthcare Hospital, Liuzhou, Guangxi, China. The study was approved by the Ethical Review Board of the Medical Faculty of the Liuzhou Municipal Maternity and Child Healthcare Hospital.
Exome Sequencing, Variant Calling, and Sanger Sequencing
Exomes sequencing (ES) was performed on the female proband and her parents to identify the causative mutation. The ES protocol achieved a mean coverage of 200x over 98% of the targeted regions. Data were filtered to generate cleanup reads by adapter trimming and reads with base quality < Q20. A minimum coverage of 20x was the standard (AmCare Genomics Laboratory, Guangdong, China) in the regions that contained 5,177 disease candidate genes. The SpeedSeq toolkit was used for the data analysis: 1) performing the human genome alignment with BWA-MEM, 2) marking duplicates using SAMBLASTER, 3) sorting and performing indexing of BAM files with Sambamba, and 4) executing variant calling using FreeBayes. SpeedSeq is an ultra-fast personal genome/exome analysis toolkit for next-generation sequencing (Chiang et al., 2015). It takes roughly 2 h to complete the analysis of one ES sample by using 7 cores 14 threads of the CPU. To quickly and accurately detect pathogenic variants, we used Exomiser to help narrow down the potential candidate variants. Variants were prioritized according to pathogenicity, quality, inheritance pattern, model organism phenotype data, and phenotypes known to be associated with disease genes in humans (Smedley et al., 2015). We removed variants with a minor allele frequency (MAF) < 0.01 in the 1000 Genomes Project, the Genome Aggregation Database (gnomAD), the Exome Aggregation Consortium (ExAC), and the Single Nucleotide Polymorphism Database (dbSNP). We used Exomiser to remove genes that are least to contribute to the phenotype with priorityScoreFilter < 0.501. Sanger sequencing was used to determine the segregation across all family members. The ARR3 c.569C>G (p.S190*) primers for PCR amplification were 5′-CCATAGGAAAACAGGCATCAGG-3′ (forward) and 5′-CCTTGTTGGTGCAGTTGTTG-3′ (reverse).
Plasmid Construction
The full-length ARR3 cDNA was synthesized and cloned into p3XFLAG-CMV-7.1 to get p3XFLAG-CMV-7.1-ARR3-wt. Subsequently, p3XFLAG-CMV-7.1-ARR3-mut was constructed from p3XFLAG-CMV-7.1-ARR3-wt using a site-directed mutagenesis kit.
Cell Culture and Transfection
Human retinal pigment epithelial cell line APRE19 was cultured in DMEM, with 10% FBS, 1% penicillin/streptomycin, and 5% of CO2 at 37°C. Transfection of p3XFLAG-CMV-7.1-wt/mut was performed with the liposomal transfection reagent according to the manufacturer’s instruction.
Translation Inhibition
At 40 h after transfection of p3XFLAG-CMV-7.1-wt/mut into APRE19 cells, 20 µg/ml of cycloheximide (CHX) was added to the medium, and translation inhibition was evaluated after 8 h of treatment.
RNA Interference
p3XFLAG-CMV-7.1-wt/mut was transfected into APRE19 cells, as mentioned above, together with si-RNA UPF1 (50 nmol/ml). Cells were collected for evaluation after 48 h.
Real-Time Quantitative PCR
For RT-PCR experiments, total RNA was extracted from APRE19 cells using an RNA kit and then reverse-transcribed into cDNAs. The real-time primer pairs upstream of the mutation site were 5′-ACAAGCTAGGGGACAATGCC-3 ′and 5′-AACCAGCCGCACATAGTCTC-3′, while the downstream mutation site primers were 5′-ACAAAGAGCTGCTGGGGATC-3 ′and 5′-CTAGCGGCCTCATGAGATGG-3′.
Statistical Analysis
Three independent experiments were conducted, and the data were presented as mean ± standard deviation (SD). Tukey’s or Dunnett’s test was used for one-way analysis of variance. GraphPad Prism 5 (GraphPad, Inc, San Diego, CA, United States) was used for statistical analysis. p < 0.05 was considered to be statistically significant.
Results
Clinical Features
All patients, including the only affected male family member, had a history of early-onset HM before the age of 7 years, without other ocular diseases or systemic comorbidities. The clinical description of family members is shown in Table 1. Affected members demonstrated peripapillary atrophy, tigroid appearance, and depigmentation changes by examination of fundus photographs (Figure 1). The only male patient in the family (II-1) stated that he had trouble seeing since childhood and began to wear glasses due to myopia when he was 20 years old. His spherical equivalent in the right eye (OD) is −12.00 D and that in the left (OS) is −11.00 D.
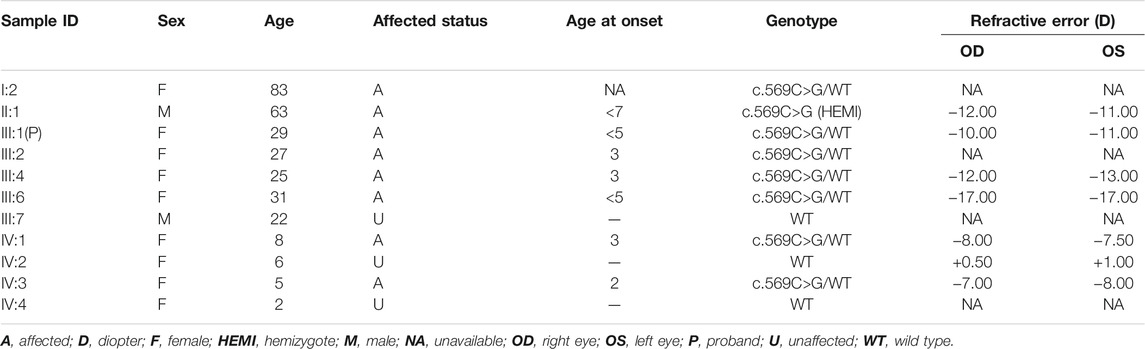
TABLE 1. Description of the clinical characteristics and the ARR3 genotypes of the family.
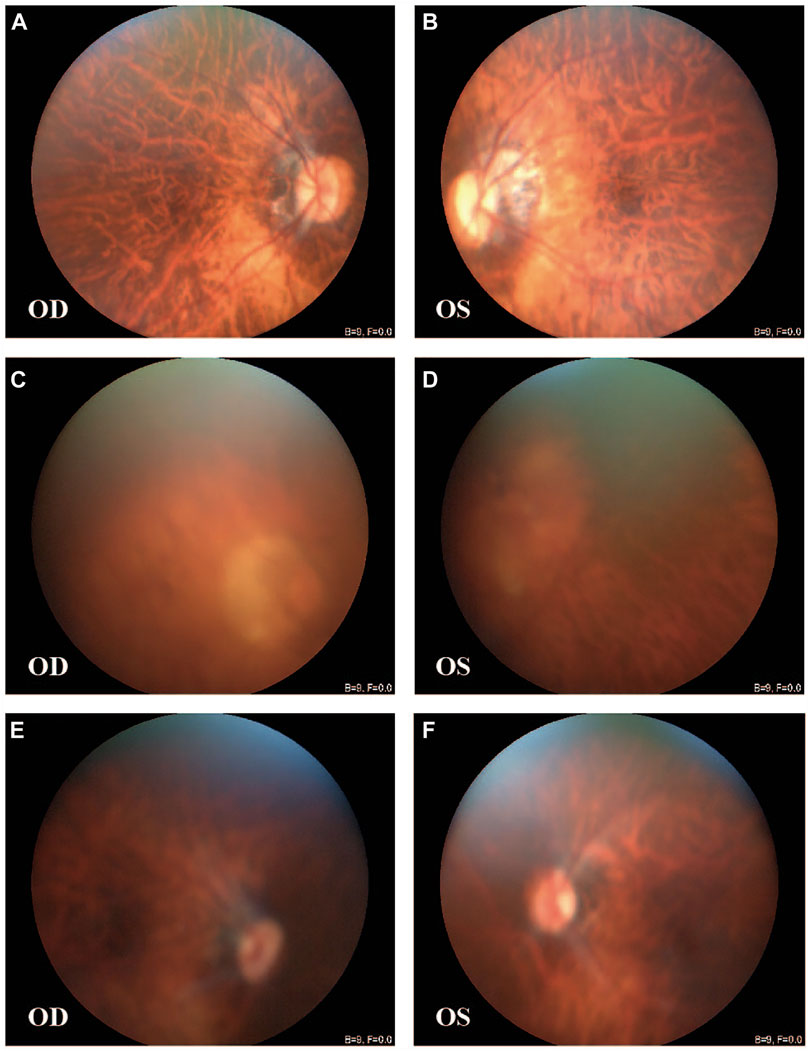
FIGURE 1. Fundus images of patients from the large family. [III-4: (A,B)] Peripapillary atrophy, tigroid appearance, and depigmentation were observed in both eyes. [II-1: (C,D); III-1: (E,F)] Tigroid appearance and depigmentation were observed. OD, right eye; OS, left eye.
Genetic Findings
Three family members (II-1, father; II-2, mother; and III-1, proband) were selected for ES to identify potential variants responsible for HM in this family. ES analysis revealed a novel heterozygous nonsense mutation in ARR3 (c.569C>G, p. S190*) in the proband (III-1), which was predicted to produce a shorter, truncated protein product. This mutation was verified by Sanger sequencing and co-segregated with all affected individuals (Figures 2A–D; Table 1). According to ACMG guidelines, the criteria of PM2 (absent from controls) and PP1_strong (greater than 5 affected segregations with HM) were met, and thus, the novel ARR3 mutation was classified as likely pathogenic.
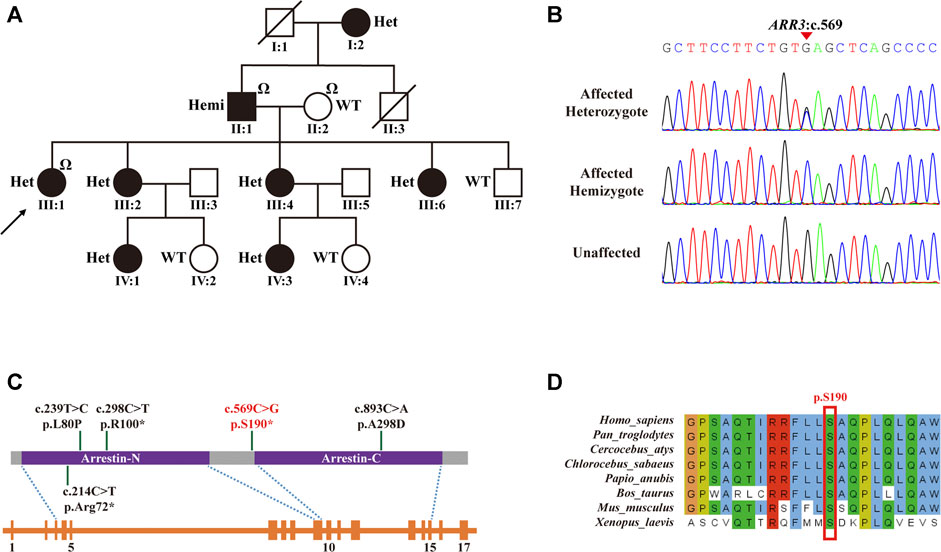
FIGURE 2. ARR3 mutation identified in myopia patients. (A) Pedigree for the family with the c.569C>G (p.S190*) mutation. Square = male, circle = female, dark symbol = affected, arrow = proband, slash = deceased member, “Ω” = samples used for exome sequencing, Het = heterozygote, Hemi = hemizygote, WT = wild type. (B) Sanger sequencing analysis for the mutation identified in the family with high myopia. (C) Top to bottom: schematic of the protein structure of ARR3, schematic of the gene structure of ARR3, and blocks are exons. The protein contains two important functional domains: an arrestin_N domain and an arrestin_C domain. (D) The nonsense ARR3 c.569C>G (p.S190*) mutation is highly conserved among different species.
Functional Consequences of the Identified ARR3 Mutation
The human ARR3 protein is composed of 388 amino acids and contains two arrestin-like domains in the N- and C-termini (Figure 2C). To date, there has been only one peer-reviewed report describing three pathogenic ARR3 variants, and no functional investigations of the underlying molecular mechanisms have been carried out (Xiao et al., 2016). In order to observe the effect of the identified variant on protein expression, the mutant and wild-type proteins were labeled with tag flags, transfected into the APRE19 human retinal pigment epithelial cell line, and cultured for 48 h. The mRNA expression of the mutant gene was significantly lower than that of the wild type (Figure 3A). Furthermore, ARR3 mutants had significantly lower protein expression than their wild-type counterparts (Figure 3B).
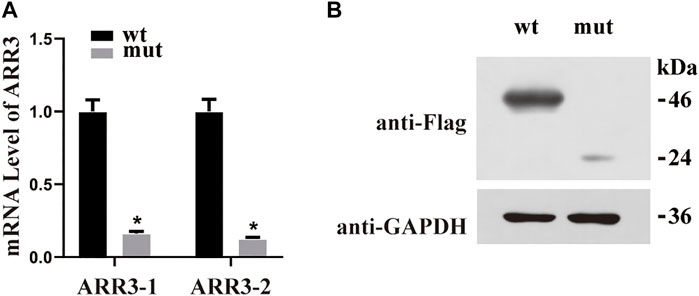
FIGURE 3. (A) Quantitative RT-PCR using GAPDH as the housekeeping gene demonstrated that the mRNA expression level was significantly decreased in mutant cases compared with wild-type controls. *p < 0.05 versus WT. (B) Western blot analysis of WT and S190* mutant ARR3 in whole-cell lysate.
Degradation of Nonsense ARR3 Mutant RNA via NMD
Nonsense-mediated decay (NMD) has been implicated in the degradation of mRNAs with premature stop codons and may be the reason that we observed a reduced level of mutant ARR3 mRNA compared to wild type. To test this hypothesis, we first expressed wild-type or mutant ARR3 in APRE19 cells and then treated the cells with cycloheximide (CHX). CHX is a potent NMD inhibitor, because NMD is translation-dependent and CHX inhibits translation (Noensie and Dietz, 2001). Our results showed that CHX treatment increased the mRNA level of mutant ARR3 but not that of wild-type ARR3 (Figure 4A), suggesting that CHX prevents mutant ARR3 mRNA from degradation through NMD.
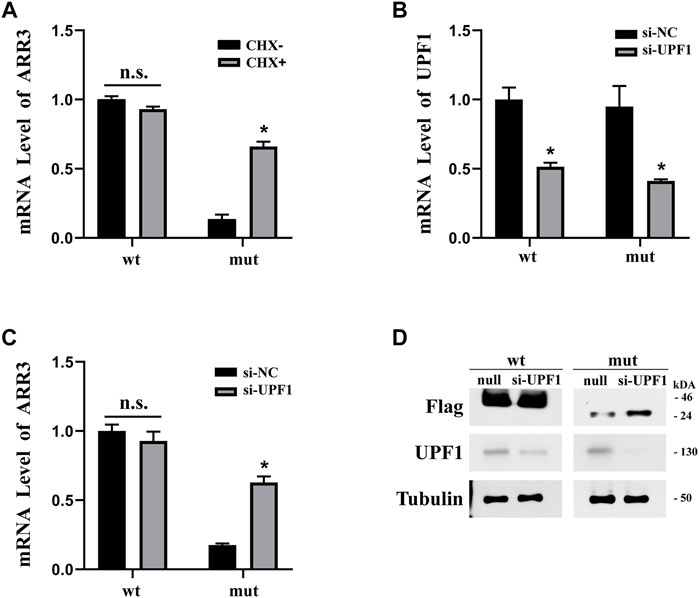
FIGURE 4. (A) CHX treatment increased the mRNA level of mutant ARR3 but not that of wild-type ARR3. (B) UPF1 siRNA treatment significantly decreased the mRNA level of UPF1. (C) UPF1 siRNA treatment significantly increased the mRNA level of mutant ARR3. (D) UPF1 siRNA treatment significantly increased the protein level of mutant ARR3 but not that of wild-type ARR3.
To directly test the role of NMD, we knocked down the expression level of UPF1, one critical component of the NMD pathway, and then evaluated its effect on the ARR3 mRNA level. During NMD, UPF1 is the central regulator of UPF proteins to recruit degradation enzymes for the decay of mRNAs with premature termination codons (PTCs) (Kervestin and Jacobson, 2012). As expected, transfection of UPF1 siRNA leads to efficient downregulation of UPF1 at both the mRNA level (Figure 4B) and the protein level (Figure 4D). However, knock-down of UPF1 expression only resulted in a significant increase of the mutant ARR3 mRNA and protein levels but not those of the wild-type ARR3 (Figures 4C,D). These data further supported our hypothesis that NMD contributed to the decreased expression of the mutant ARR3.
Discussion
In the present study, we reported an X-linked high myopia case and identified the associated genetic variant in a Southern Chinese family. As reported previously, this nonsense ARR3 mutation, c.569C>G (p.S190*), co-segregated within affected family members. However, our observation of HM symptoms in a male member was inconsistent with the established female-limited inheritance pattern.
Arrestin is an important family of proteins that can desensitize G-protein–coupled receptors (GPCRs). Rhodopsin belongs to a class of GPCRs that can sense external light signals and transmit these to cells that produce vision. In mammals, there are two types of visual arrestins: arrestin-1 (encoded by ARR1, also called S-antigen or 48K protein) (Wacker et al., 1977; Kuhn et al., 1984; Pfister et al., 1985; Vishnivetskiy et al., 2018) and arrestin-4 (encoded by ARR3, also called cone arrestin or X-arrestin) (Murakami et al., 1993; Craft et al., 1994). Both arrestins are required for normal inactivation of phototransduction in mouse cones, where it was proposed that arrestin-4 has undergone evolutionary specialization and arrestin-1 maintains the basic function (Nikonov et al., 2008; Deming et al., 2015).
As shown in Figure 2C, of the four previously described pathogenic ARR3 mutations, two were missense mutations (p.L80P and p. A298D) and two were nonsense mutations (p.R100* and p. R72*), the latter of which would lead to truncated protein translation, similar to our reported c.569C>G (p.S190*). All four mutations could cause functional impairment of arrestin-4 because both the C- and N-terminal domains play a critical role in binding to the receptor molecule (Gurevich and Benovic, 1993; Hirsch et al., 1999). In addition, alignment of multiple arrestin-4 amino acid sequences has shown that the serine at codon 190 is highly conserved (Figure 2D), where evolutionary conservation indicates functional importance.
The pathogenicity of the ARR3 mutations and their association with HM were described for the first time in 2016, where an X-linked, female-limited pattern of inheritance was proposed based on the observation that none of the hemizygous male family members were diagnosed with HM (Xiao et al., 2016). However, in our study, the proband’s father was affected with HM and possessed this particular pathogenic ARR3 mutation. This suggested that the hereditary pattern of the ARR3 gene may not be X-linked, female-limited. HM is a complex, heterogeneous disease which is influenced by environmental and genetic factors. Occurring in females, an X chromosome is compacted during X-chromosome inactivation, which results in the random silencing of one of the X chromosomes (Galupa and Heard, 2018). X-chromosome inactivation could, however, not explain the phenotypes observed in this study since a nonsense mutation was identified in an affected male hemizygote, while unaffected male hemizygotes carried a missense mutation in other families. So we speculate that the mechanism of loss of function caused by null variants may be more detrimental for the ARR3 gene. Both Arr1 and Arr3 genes are expressed in mouse cone cells. If only one of the two genes is knocked out, the recovery time will slow down after being stimulated by saturated strong light, but knockout of both genes will greatly increase the delay in recovery time, which indicates that there may be functional compensation between Arr1 and Arr3 in mice (Nikonov et al., 2008). So it becomes reasonable to question whether there is also a complementary mechanism in the human retina which leads to the occurrence and absence of diseases in different hemizygous individuals? Further proof is needed in the future. PCDH19-related epilepsies illustrate an unusual X-linked inheritance pattern (Dibbens et al., 2008). The female heterozygote of the PCDH19 gene located on the X chromosome is usually patient, but the hemizygous male is usually not affected (Dibbens et al., 2008). Some studies believe that this is a kind of “cellular interference” caused by the cell adhesion affinities of PCDH19 protein (Pederick et al., 2018). Without skewed X chromosome inactivation, the brains of Pcdh19+/−female mice showed different cell populations that can adhere improperly to the other. Cells containing mutant Pcdh19 changed the original adhesive condition to another. The presence of two distinctive adhesive properties led to adhesive errors of cells on the developing brain. On the contrary, nerve cells of Pcdh19+/+ (or Pcdh19−/−) mice sorted suitably since all cells showed the same adhesive condition. Moreover, arrestin appears to have the function of adhesion (Cleghorn et al., 2015). Perhaps there exist a “cellular interference”–like mechanism and other unknown inheritance patterns with incomplete penetrance in ARR3, resulting in both normal and affected hemizygous males. This is the goal of our research in the future.
One key limitation of the investigations and discussions mentioned above is the small number of ARR3-related HM patients available for further evaluation. Future identification and characterization of more HM-related ARR3 mutations and their corresponding inheritance patterns will clarify this genotype–phenotype relationship. Only after that can we start to devise plans for precision medicine accordingly.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://db.cngb.org/search/project/CNP0002221/
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethical Review Board of the Medical Faculty of the Liuzhou Municipal Maternity and Child Healthcare Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
DY and TY designed the experiment, analyzed the data, and drafted the manuscript; SL, JH, and JT conducted cell culture and cell transfection and molecular biology experiments including Western blot and real-time PCR; JG, JZ, and YL enrolled clinical data of the family and enrolled the samples and confirmed the diagnosis; and TH, VZ, MH, and DZ revised the manuscript. All authors approved the final manuscript.
Funding
This work was supported by the Construction Project of the Ability and Condition in Science and Technology Innovation of Liuzhou (2014G020404 and 2018AF10501); the Science and Technology Development Projects of Local Government guided by the Central Government (ZY18164008); the Health Department Research Fund of Guangxi Zhuang Autonomous Region, Guangxi, People’s Republic of China (Z20190789); the Scientific Research and Technology Development Project of Liuzhou (2018DB20501); the Key Research and Development Program of Guangxi (Guike AB18126056); and the Key Research and Development Program of Liuzhou (2018BJ10301).
Conflict of Interest
Author VZ is employed by the company AmCare Genomics Laboratory. Author MH is employed by the company Aegicare (Sheznzhen) Technology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank all the members in the family of the patient for their interest and cooperation.
References
Cai, X.-B., Zheng, Y.-H., Chen, D.-F., Zhou, F.-Y., Xia, L.-Q., Wen, X.-R., et al. (2019). Expanding the Phenotypic and Genotypic Landscape of Nonsyndromic High Myopia: A Cross-Sectional Study in 731 Chinese Patients. Invest. Ophthalmol. Vis. Sci. 60, 4052–4062. doi:10.1167/iovs.19-27921
Chiang, C., Layer, R. M., Faust, G. G., Lindberg, M. R., Rose, D. B., Garrison, E. P., et al. (2015). SpeedSeq: Ultra-Fast Personal Genome Analysis and Interpretation. Nat. Methods 12, 966–968. doi:10.1038/nmeth.3505
Cleghorn, W. M., Branch, K. M., Kook, S., Arnette, C., Bulus, N., Zent, R., et al. (2015). Arrestins Regulate Cell Spreading and Motility via Focal Adhesion Dynamics. MBoC 26, 622–635. doi:10.1091/mbc.e14-02-0740
Craft, C. M., Whitmore, D. H., and Wiechmann, A. F. (1994). Cone Arrestin Identified by Targeting Expression of a Functional Family. J. Biol. Chem. 269, 4613–4619. doi:10.1016/s0021-9258(17)41820-5
Deming, J. D., Pak, J. S., Shin, J.-a., Brown, B. M., Kim, M. K., Aung, M. H., et al. (2015). Arrestin 1 and Cone Arrestin 4 Have Unique Roles in Visual Function in an All-Cone Mouse Retina. Invest. Ophthalmol. Vis. Sci. 56, 7618–7628. doi:10.1167/iovs.15-17832
Depienne, C., Bouteiller, D., Keren, B., Cheuret, E., Poirier, K., Trouillard, O., et al. (2009). Sporadic Infantile Epileptic Encephalopathy Caused by Mutations in PCDH19 Resembles Dravet Syndrome but Mainly Affects Females. Plos Genet. 5, e1000381. doi:10.1371/journal.pgen.1000381
Dibbens, L. M., Tarpey, P. S., Hynes, K., Bayly, M. A., Scheffer, I. E., Smith, R., et al. (2008). X-Linked Protocadherin 19 Mutations Cause Female-Limited Epilepsy and Cognitive Impairment. Nat. Genet. 40, 776–781. doi:10.1038/ng.149
Galupa, R., and Heard, E. (2018). X-Chromosome Inactivation: A Crossroads Between Chromosome Architecture and Gene Regulation. Annu. Rev. Genet. 52, 535–566. doi:10.1146/annurev-genet-120116-024611
Gurevich, V. V., and Benovic, J. L. (1993). Visual Arrestin Interaction with Rhodopsin. Sequential Multisite Binding Ensures Strict Selectivity toward Light-Activated Phosphorylated Rhodopsin. J. Biol. Chem. 268, 11628–11638. doi:10.1016/s0021-9258(19)50248-4
Hirsch, J. A., Schubert, C., Gurevich, V. V., and Sigler, P. B. (1999). A Model for Arrestin's Regulation: The 2.8 Å Crystal Structure of Visual Arrestin. Cell 97, 257–269. doi:10.1016/s0092-8674(00)80735-7
Holden, B. A., Fricke, T. R., Wilson, D. A., Jong, M., Naidoo, K. S., Sankaridurg, P., et al. (2016). Global Prevalence of Myopia and High Myopia and Temporal Trends from 2000 through 2050. Ophthalmology 123, 1036–1042. doi:10.1016/j.ophtha.2016.01.006
Kervestin, S., and Jacobson, A. (2012). NMD: a Multifaceted Response to Premature Translational Termination. Nat. Rev. Mol. Cel. Biol. 13, 700–712. doi:10.1038/nrm3454
Kühn, H., Hall, S. W., and Wilden, U. (1984). Light-induced Binding of 48-kDa Protein to Photoreceptor Membranes Is Highly Enhanced by Phosphorylation of Rhodopsin. FEBS Lett. 176, 473–478. doi:10.1016/0014-5793(84)81221-1
Morgan, I. G., Ohno-Matsui, K., and Saw, S.-M. (2012). Myopia. Lancet 379, 1739–1748. doi:10.1016/s0140-6736(12)60272-4
Murakami, A., Yajima, T., Sakuma, H., Mclaren, M. J., and Inana, G. (1993). X-Arrestin: a New Retinal Arrestin Mapping to the X Chromosome. FEBS Lett. 334, 203–209. doi:10.1016/0014-5793(93)81712-9
Nikonov, S. S., Brown, B. M., Davis, J. A., Zuniga, F. I., Bragin, A., Pugh, E. N., et al. (2008). Mouse Cones Require an Arrestin for Normal Inactivation of Phototransduction. Neuron 59, 462–474. doi:10.1016/j.neuron.2008.06.011
Noensie, E. N., and Dietz, H. C. (2001). A Strategy for Disease Gene Identification through Nonsense-Mediated mRNA Decay Inhibition. Nat. Biotechnol. 19, 434–439. doi:10.1038/88099
Pederick, D. T., Richards, K. L., Piltz, S. G., Kumar, R., Mincheva-Tasheva, S., Mandelstam, S. A., et al. (2018). Abnormal Cell Sorting Underlies the Unique X-Linked Inheritance of PCDH19 Epilepsy. Neuron 97, 59–66. doi:10.1016/j.neuron.2017.12.005
Pfister, C., Chabre, M., Plouet, J., Tuyen, V. V., De Kozak, Y., Faure, J. P., et al. (1985). Retinal S Antigen Identified as the 48K Protein Regulating Light-Dependent Phosphodiesterase in Rods. Science 228, 891–893. doi:10.1126/science.2988124
Saw, S.-M., Katz, J., Schein, O. D., Chew, S.-J., and Chan, T.-K. (1996). Epidemiology of Myopia. Epidemiol. Rev. 18, 175–187. doi:10.1093/oxfordjournals.epirev.a017924
Saw, S. m. (2003). A Synopsis of the Prevalence Rates and Environmental Risk Factors for Myopia. Clin. Exp. Optom. 86, 289–294. doi:10.1111/j.1444-0938.2003.tb03124.x
Smedley, D., Jacobsen, J. O. B., Jäger, M., Köhler, S., Holtgrewe, M., Schubach, M., et al. (2015). Next-generation Diagnostics and Disease-Gene Discovery with the Exomiser. Nat. Protoc. 10, 2004–2015. doi:10.1038/nprot.2015.124
Vishnivetskiy, S. A., Sullivan, L. S., Bowne, S. J., Daiger, S. P., Gurevich, E. V., and Gurevich, V. V. (2018). Molecular Defects of the Disease-Causing Human Arrestin-1 C147F Mutant. Invest. Ophthalmol. Vis. Sci. 59, 13–20. doi:10.1167/iovs.17-22180
Wacker, W. B., Donoso, L. A., Kalsow, C. M., Yankeelov, J. A., and Organisciak, D. T. (1977). Experimental Allergic Uveitis. Isolation, Characterization, and Localization of a Soluble Uveitopathogenic Antigen from Bovine Retina. J. Immunol. 119, 1949–1958.
Xiao, X., Li, S., Jia, X., Guo, X., and Zhang, Q. (2016). X-Linked Heterozygous Mutations in ARR3 Cause Female-Limited Early Onset High Myopia. Mol. Vis. 22, 1257–1266. doi:10.3389/fcell.2021.645501
Young, T. L., Metlapally, R., and Shay, A. E. (2007). Complex Trait Genetics of Refractive Error. Arch. Ophthalmol. 125, 38–48. doi:10.1001/archopht.125.1.38
Young, T. L. (2009). Molecular Genetics of Human Myopia: an Update. Optom. Vis. Sci. 86, E8–E22. doi:10.1097/opx.0b013e3181940655
Keywords: ARR3, NMD, high myopia, cone arrestin, X-linked
Citation: Yuan D, Yan T, Luo S, Huang J, Tan J, Zhang J, Zhang VW, Lan Y, Hu T, Guo J, Huang M and Zeng D (2021) Identification and Functional Characterization of a Novel Nonsense Variant in ARR3 in a Southern Chinese Family With High Myopia. Front. Genet. 12:765503. doi: 10.3389/fgene.2021.765503
Received: 27 August 2021; Accepted: 10 November 2021;
Published: 13 December 2021.
Edited by:
Lawrence Todd Reiter, University of Tennessee Health Science Center (UTHSC), United StatesReviewed by:
Li Zhuo, Central South University, ChinaShi Song Rong, Massachusetts Eye and Ear Infirmary and Harvard Medical School, United States
Copyright © 2021 Yuan, Yan, Luo, Huang, Tan, Zhang, Zhang, Lan, Hu, Guo, Huang and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dingyuan Zeng, bHpmeXpkeUAxNjMuY29t
†These authors have contributed equally to this work