
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet., 03 December 2021
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.761264
 Gabriella Doddato1,2†Alessandra Fabbiani1,2,3†Chiara Fallerini1,2Mirella Bruttini1,2,3Theodora Hadjistilianou4Martino Landi5Caterina Coradeschi5
Gabriella Doddato1,2†Alessandra Fabbiani1,2,3†Chiara Fallerini1,2Mirella Bruttini1,2,3Theodora Hadjistilianou4Martino Landi5Caterina Coradeschi5 Salvatore Grosso6Barbara Tomasini5Maria Antonietta Mencarelli3
Salvatore Grosso6Barbara Tomasini5Maria Antonietta Mencarelli3 Alessandra Renieri1,2,3
Alessandra Renieri1,2,3 Francesca Ariani1,2,3*
Francesca Ariani1,2,3*Spondyloocular syndrome (SOS) is a skeletal disorder caused by pathogenic variants in XYLT2 gene encoding a xylotransferase involved in the biosynthesis of proteoglycans. This condition, with autosomal recessive inheritance, has a high phenotypic variability. It is characterized by bone abnormalities (osteoporosis, fractures), eye and cardiac defects, hearing impairment, and varying degrees of developmental delay. Until now only 20 mutated individuals have been reported worldwide. Here, we describe two siblings from consanguineous healthy parents in which a novel homozygous frameshift variant c.1586dup p(Thr530Hisfs*) in the XYLT2 gene was detected by exome sequencing (ES). The first patient (9 years) presented short stature with skeletal defects, long face, hearing loss and cataract. The second patient, evaluated at a few days of life, showed macrosomia, diffuse hypertrichosis on the back, overabundant skin in the retronucal area, flattened facial profile with drooping cheeks, elongated eyelid rims, wide and flattened nasal bridge and turned down corners of the mouth. During the prenatal period, high nuchal translucency and intestinal hyperechogenicity were observed at ultrasound. In conclusion, these two siblings with a novel pathogenic variant in XYLT2 further expand the clinical and mutational spectrum of SOS.
Spondyloocular syndrome (SOS) (OMIM #605822) is a very rare autosomal recessive skeletal disorder caused by pathogenic variants in XYLT2 gene located on chromosome 17q21 (Umair et al., 2018). The main features include generalized osteoporosis, multiple long bone and spinal compression fractures, platyspondyly, short stature, cataract, cardiopathy, sensorineural hearing loss and varying degree of intellectual disability (Alanay et al., 2006; Taylan et al., 2017).
The XYLT2 gene encodes a xylosyltransferase II, which catalyzes the initial phase of proteoglycans (PGs) assembly. PGs are found on the cell surface, in secretory granules and in the extracellular matrix. PGs are essential for many physiological processes, such as signal transduction, cellular homeostasis, membrane integrity, corepressor activity, morphogen gradient formation, lipid catabolism and scaffolding (MisMis et al., 2014). PGs consist of a core protein, which is linked to glycosaminoglycan (GAG) disaccharide chains. Assembly of GAGs on the core protein results in different groups of sulfated PGs such as chondroitin sulfate (CSPGs), heparan sulfate (HSPGs) and modified form of CSPGs, the dermatan sulfate (DSPGs) (Couchman and Pataki, 2012).
SOS was first clinically described in 2001 by Schmidt and others in a consanguineous family with six affected children. Clinical features of these patients included cataract, loss of vision due to retinal detachment, facial dysmorphisms and hypotonia, normal height with disproportionate short trunk, immobile spine with thoracic kyphosis and reduced lumbar lordosis (Schmidt et al., 2001). Alanay et al. reported another case with SOS in a Turkish family confirming that the syndrome was a distinct clinical entity (Alanay et al., 2006). In 2015, whole exome sequencing revealed homozygous mutations in the XYLT2 gene as the molecular basis of SOS (Munns et al., 2015). To date, only 20 individuals with XYLT2-deficiency have been described. We report on two additional siblings with a novel homozygous XYLT2 mutation. One of them represents the youngest patient with SOS ever described. Fetal abnormalities were documented pinpointing hints for the prenatal diagnosis.
DNA samples were obtained at the Medical Genetics Unit of the Azienda Ospedaliera Universitaria Senese (A.O.U.S., Siena, Italy) upon the signature of informed consent for both diagnostic and research purposes. Genomic DNA was extracted from EDTA peripheral blood samples using MagCore HF16 (Diatech Lab Line, Jesi, Ancona, Italy).
Exome sequencing was performed on genomic DNA samples of the proband and both parents using the Life Technologies Ion Proton sequencer (Life Technologies, Carlsbad, CA, USA). This system enables >92% of bases covered ≥20×. Sample preparation and sequencing were performed with AmpliSeq Exome strategy, following the manufacturer’s protocol (Life Technologies). The library preparation was performed using the Ion AmpliSeq Exome Kit (Life Technologies), which allows us to target ∼33 Mb of coding exons plus 15 Mb of flanking regions for a total of ∼58 Mb, in total more than 97% of the coding regions, using 12 primer pools for highly specific enrichment of exons within the human genome. Taking advantage of a barcode system, three samples were loaded together in a single run and sequenced. Data analysis was performed with Torrent Suite Software v5.0.2 (Life Technologies). Using specific parameters, we were able to remove the adaptors’ contamination and low-quality sequences, so the total amount of clean data was mapped to the UCSC/hg19 reference genome. Indel and variant calls were made using GATK version 2.7 (Broad Institute, Cambridge, MA, USA) and then the variants were also annotated against external datasets, including 1000 genomes, ExAC, gnomAD database and dbSNP147.
The XYLT2 variant was confirmed by Sanger sequencing of exon eight including flanking intron sequences of the gene (NM_022167) in the probands and both parents.
DNA was amplified by PCR using specific primer pairs (forward 5′-ACAACAGCAGCAGGAAAAGC-3′; reverse 5′-CTTCAGACTGGGGCCTTGTT-3′), PCR products were sequenced employing ABIPRISM3130 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) and data were analyzed with Sequencher software V.4.9 (Gene Codes, Ann Arbor, USA).
The mutation is described according to Human Genome Variations Society (HGVS). Nucleotide numbers are derived from the cDNA sequence of XYLT2 (GenBank accession NM_022167).
Here, we present two SOS affected siblings from consanguineous healthy parents of Moroccan origin (Figure 1A).
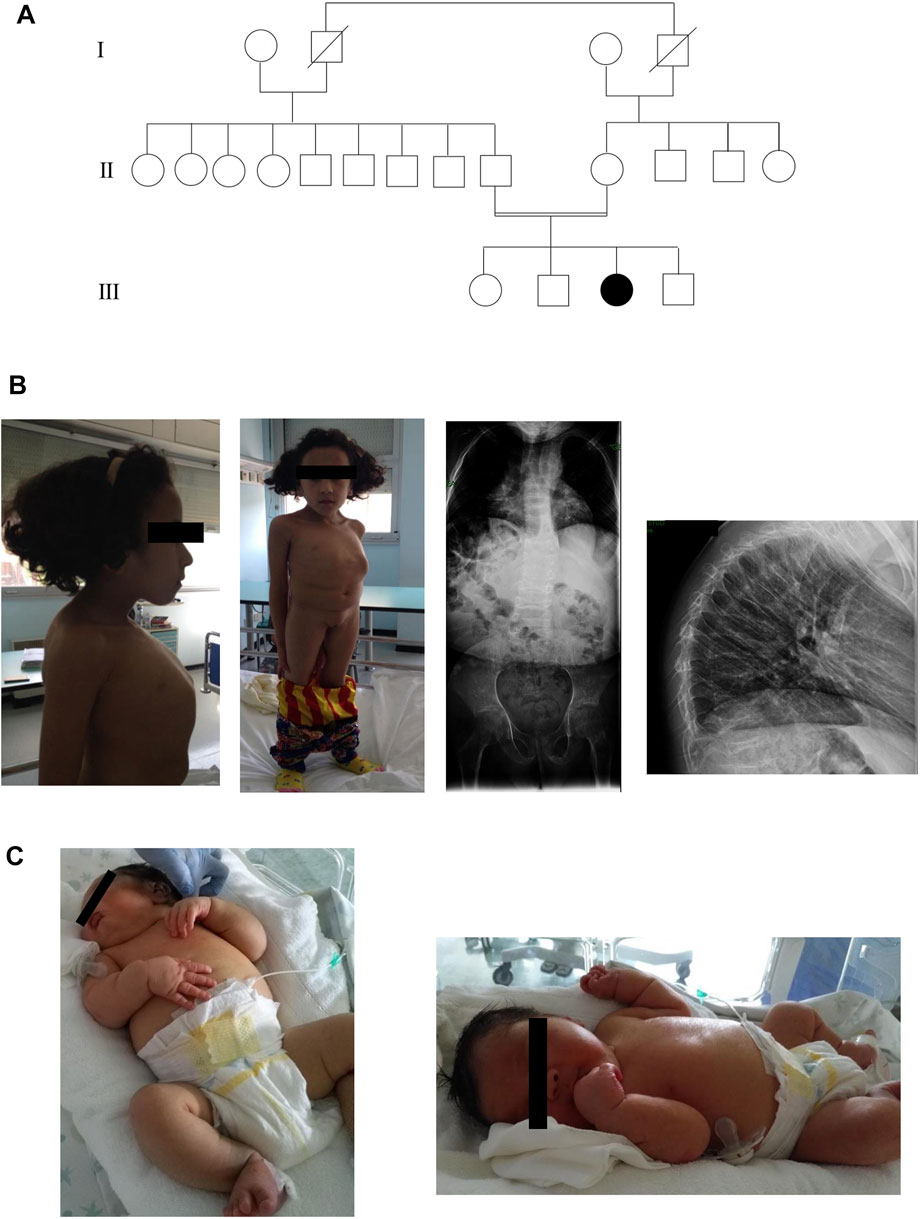
FIGURE 1. (A)Three generations of pedigree of family. (B) Anterior, lateral photographs and radiographs of sibling 1 that demonstrate platyspondyly with thinning of the vertebral bodies and secondary kyphosis. (C) Photographs of sibling 2 showing the main clinical features.
Sibling 1 was evaluated for the first time in our Unit at the age of 9 years. We have little information about prenatal history. She was born from an uncomplicated pregnancy at full term. Her birth weight was 4,000 g (90° percentile). The baby showed hypovalid suction and poor growth. She had speech and psychomotor delay: she crawled at 5 years old, she was able to walk at 6 years old, she acquired sphincter control at 6 years old. She presented two fractures to the wrist and left foot after a fall. Physical examination revealed an occipital frontal circumference (OFC) of 51 cm (25th percentile), dental anomalies, short neck with pterygium, short stature with marked reduction in trunk length, severe kyphoscoliosis and barrel chest with increased intermammillary distance, in the absence of limb length reduction. In addition, she had long and oval face with flattened facial profile and blue sclerae, dorsal and pectoral hirsutism, generalized muscular hypotrophy. The instrumental examinations revealed platyspondyly with thinning of the vertebral bodies due to osteoporosis and thoracic deformity with secondary kyphosis and bilateral posterior subcapsular cataract and hearing loss (Figure 1B). At age of 13 years, biochemical analysis of patient’s blood revealed normal levels of calcium whereas serum 25-OH vitamin D level was decreased. Her ECG was normal, but she presented aneurysm of ascending aorta. The patient did not perform treatment with bisphosphonate.
Sibling 2, evaluated at a few days of life, was born at 36 weeks of gestational age with a caesarean section due by oligohydramnios, generalized fetal edema, hydrops and macrosomia. Moreover, during pregnancy, fetal malformations and maternal diabetes were observed. Infectious investigation of the mother and fetus was carried out and the serologies were negative.
An ultrasound performed at 15 + 3 weeks of gestation revealed an increased nuchal translucency (6.5 mm), hyperechoic intestine, oligohydramnios, cystic hygromas with difficulties in visualizing the other viscera and cerebral hemispheres, a discrepancy between fetal biometry and age of reported amenorrhea. The parents refused to perform prenatal invasive diagnosis. The auxological parameters at birth were length, 49 cm (75th percentile); weight, 3,990 g (>97° percentile); and OFC, 34 cm (75th percentile). Apgar score was 6 at the first minute, 9 at the fifth minute and 10 at the tenth minute. Echocardiography carried out immediately after the birth showed mild hypertrophy of the left ventricle, endowed with mild-to-moderate global contractile dysfunction. Furthermore, left bovine arch with origin of left common carotid artery from right brachiocephalic trunk, mitral-aortic discontinuity with subaortic conus and dysplastic aortic valve were displayed. During hospitalization, the patient kept hemodynamic stability.
At the physical examination, we found: severe facial dysmorphic features such as flattened profile with drooping cheeks, elongated eyelid rims, broad and flattened nasal root and turned down corners of the mouth, chin with horizontal crease, macrosomia, diffuse hypertrichosis on the back and overabundant skin in the retronucal area. Furthermore, the baby showed hepatomegaly and minimal bilateral subdiaphragmatic fluid flap. The child underwent brain ultrasonograpy and metabolic test with normal results. An opalescent appearance of the cornea bilaterally was observed. The ophthalmologist evaluation highlighted a more remarkable corneal leucoma on the right eye. Topical anti-edema therapy was then performed showing a progressive improvement of the corneal edema. The patient underwent brain MRI, eye sockets and spine which get normal results. Furthermore, the auditory brainstem response (ABR) test found severe deafness. In addition, the baby presented right cryptorchidism (Figure 1C). Biochemical analysis of patient’s blood revealed normal levels of calcium and 25-OH vitamin D.
Exome sequencing analysis was performed in sibling 1 and her parents. We obtained a mean depth of coverage of 91X. A total of 39,900 genetic variants on average for each sample was yielded. We filtered variants that were either loss-of-function (frameshift, nonsense and splicing variants) or missense variants (28,450 variants). Among them, variants with coverage ≥20X, variants CADD_phreed ≥25 (applied only to missense variants), frequency <1% or not reported were filtered. Considering the parental consanguinity, we decided to focus our attention on variants lying in disease genes consistent with autosomal recessive mode of inheritance (Figure 2). The analysis revealed a frameshift variant c.1586dup p.(Thr530Hisfs*16) in exon 8 of the XYLT2 gene on chromosome 17. Sanger sequencing confirmed the homozygous variant in the proband and the heterozygous variant in both parents. In sibling 2, Sanger sequencing revealed the same mutation in the XYLT2 gene in homozygous state (Figure 3A). This frameshift deletion has not been previously reported and it is absent in the ExAC or gnomAD database.

FIGURE 2. Workflow diagram of variants’ filtering in patient-parents trio exome.
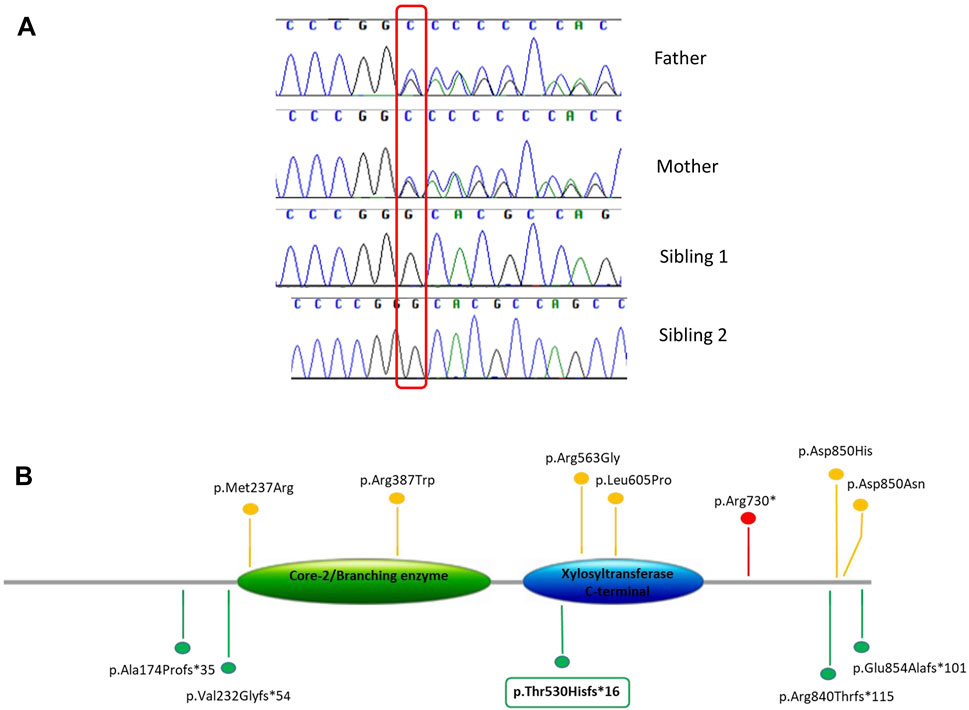
FIGURE 3. (A) Variant confirmation and segregation analysis by Sanger sequencing. (B) Schematic view of the XYLT2 protein (NM_022167), showing previously reported variants and the novel variant identified in this study (in box). Different types of variants are labeled with different colors.
The variant has been submitted to the LOVD database (https://www.LOVD.nl) with the following ID: 0000272447.
In the present study, we described two additional patients with SOS and reported one novel homozygous variant, a frameshift deletion in the XYLT2 gene c.1586dup p.(Thr530Hisfs*16). The variant was identified by exome sequencing and confirmed by Sanger sequencing in the two siblings while parents were heterozygous carriers, in accordance with an autosomal recessive inheritance pattern. To date, only 20 patients with XYLT2 mutations have been reported worldwide. These two siblings with a novel pathogenic variant further expand the clinical and mutational spectrum of SOS.
The XYLT2 gene, located on chromosome 17q21.3–q22, encodes an 865 amino acid protein that transport xylose molecules from the nucleoside diphosphate donor (UDP-xylose) to targeted serine molecules of the core protein during the synthesis of proteoglycans. The XYLT2 is composed of four domains: an N terminus domain, a xylosyltransferase terminal domain (catalytic domain), a core2/I-branching enzyme domain, and a C-terminus domain (Umair et al., 2018). The identified variant is predicted to truncate the C-terminus but this has not been shown experimentally and a haploinsufficiency effect could not be ruled out. To date, only 11 homozygous variants including a nonsense, six missense, one frameshift duplication and three frameshift deletions in the XYLT2 gene have been reported (Umair et al., 2018; Taylan et al., 2017; Munns et al., 2015; Guleray et al., 2019; Kausar et al., 2019; Taylan et al., 2016) (Figure 3B). The phenotypic severity of SOS is suggested to be associated with the mutation type, localization and domain, but there is no agreement on this point and no obvious genotype phenotype correlation can be made (Alanay et al., 2006; Taylan et al., 2017; Umair et al., 2018). Missense variants are not always correlated with a milder phenotype and different truncating variants are not associated with the phenotypic spectrum depending on the localization. Indeed, our patients show a severe phenotype even if the variant truncates only the C-terminus. There is high phenotypic variability and other factors in the genome could modulate the clinical spectrum.
Concerning the phenotype, consistent findings include skeletal dysplasia with short stature (13/22), low weight (9/22), multiple fractures (19/22), kyphosis (15/22), and facial dysmorphisms (14/22). Other, more frequent features were ocular problems (21/22) and hearing loss (14/22). Dental problems (3/22), cardiovascular defects (7/22) and neurodevelopmental delay (10/22) are variably present (Umair et al., 2018; Taylan et al., 2017; Munns et al., 2015; Guleray et al., 2019; Kausar et al., 2019; Taylan et al., 2016) (Table 1). In some patients, the treatment with bisphosphonate appears to have a positive effect on the clinical course, but the exact mechanism between bisphosphonate treatment and proteoglycan biosynthesis has not been established (Munns et al., 2015; Guleray et al., 2019).
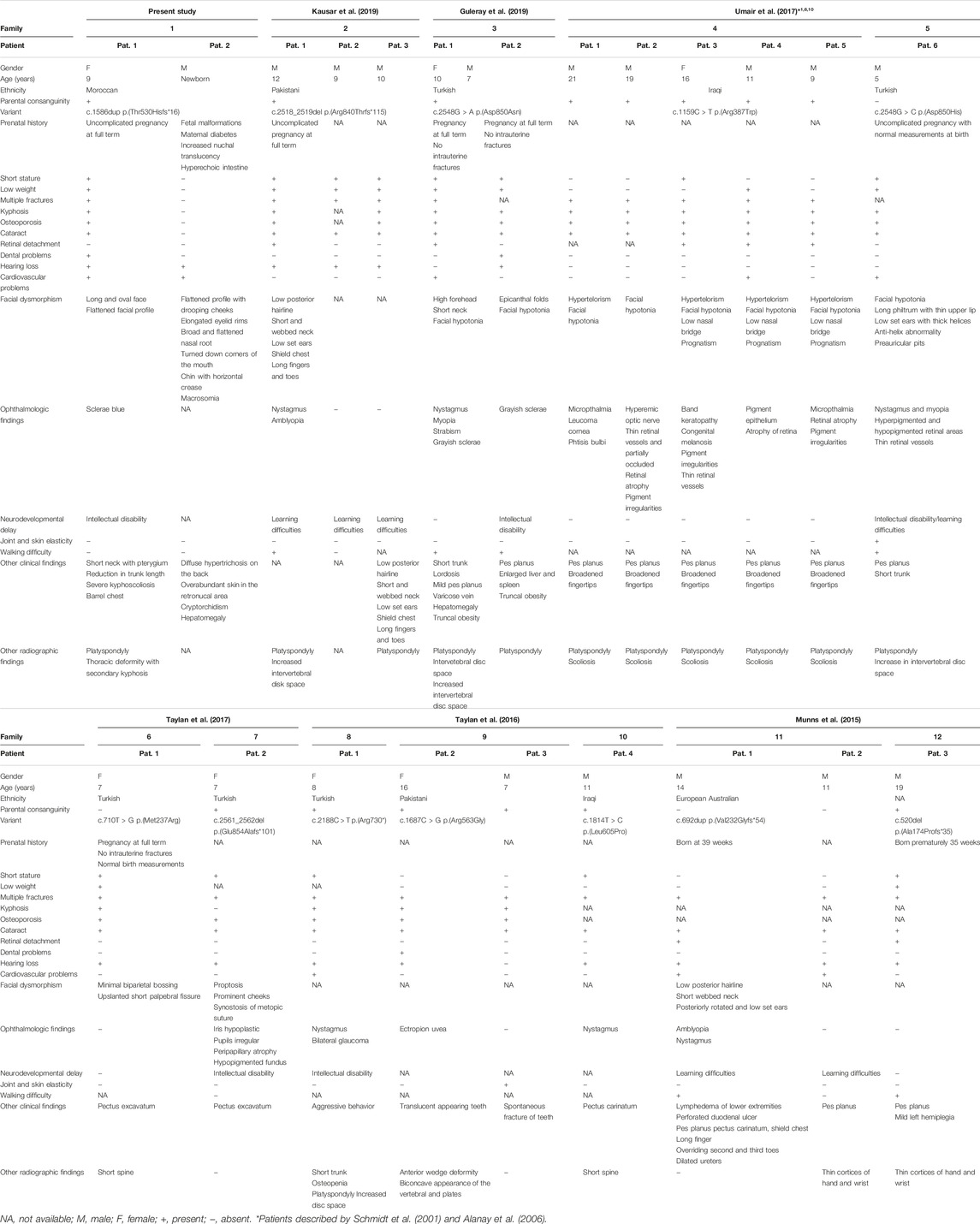
TABLE 1. Clinical features of the present case compared with the previously reported patients with XYLT2 variants
We described the youngest patient affected by SOS with important hints for prenatal diagnosis: increased nuchal translucency, hyperechoic intestine, oligohydramnios and cystic hygromas. The phenotype of the newborn includes macrosomia, diffuse hypertrichosis on the back and overabundant skin in the retronucal area. He also displayed severe facial dysmorphic features. Moreover, minor cardiac abnormalities, ocular alteration and hearing loss were in place.
In conclusion, we have reported a novel frameshift deletion in two additional patients with SOS. The variant is a truncating deletion that disrupts the C-terminus domain of the protein. These findings extend the mutation spectrum of XYLT2-related disorders and help proper genotype–phenotype correlation.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Written informed consent was obtained from both parents for the publication of any potentially identifiable images or data included in this article.
GD, AR, and FA have made substantial contributions to conceptions and design and have been involved in drafting the paper. AF and MM conducted the genetic counseling to the family and drafted the article. CF and MB have performed the experiments and data acquisition. TH, ML, CC, SG, and BT made ophthalmologic, neonatal, and pediatric clinical evaluation, respectively. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank the family for participating in the study. Two of the authors of this publication are members of the European Reference Network for rare malformation syndromes and rare intellectual and neurodevelopmental disorders, ERN-ITHACA.
Alanay, Y., Superti-Furga, A., Karel, F., and Tunçbilek, E. (2006). Spondylo-ocular Syndrome: a New Entity Involving the Eye and Spine. Am. J. Med. Genet. 140A140 (6), 652–656. doi:10.1002/ajmg.a.31119
Couchman, J. R., and Pataki, C. A. (2012). An Introduction to Proteoglycans and Their Localization. J. Histochem. Cytochem. 60 (12), 885–897. doi:10.1369/0022155412464638
Guleray, N., Simsek Kiper, P. O., Utine, G. E., Boduroglu, K., and Alikasifoglu, M. (2019). Intrafamilial Variability of XYLT2-Related Spondyloocular Syndrome. Eur. J. Med. Genet. 62 (11), 103585. doi:10.1016/j.ejmg.2018.11.019
Kausar, M., Chew, E. G. Y., Ullah, H., Anees, M., Khor, C. C., Foo, J. N., et al. (2019). A Novel Homozygous Frameshift Variant in XYLT2 Causes Spondyloocular Syndrome in a Consanguineous Pakistani Family. Front. Genet. 10, 144. doi:10.3389/fgene.2019.00144
MisMis, E. K., Liem, K. F., Kong, Y., Schwartz, N. B., Domowicz, M., and WeatherbeeWeatherbee, S. D. (2014). Forward Genetics Defines Xylt1 as a Key, Conserved Regulator of Early Chondrocyte Maturation and Skeletal Length. Dev. Biol. 385 (1), 67–82. doi:10.1016/j.ydbio.2013.10.014
Munns, C. F., Fahiminiya, S., Poudel, N., Munteanu, M. C., Majewski, J., Sillence, D. O., et al. (2015). Homozygosity for Frameshift Mutations in XYLT2 Result in a Spondylo-Ocular Syndrome with Bone Fragility, Cataracts, and Hearing Defects. Am. J. Hum. Genet. 96, 971–978. doi:10.1016/j.ajhg.2015.04.017
Schmidt, H., Rudolph, G., Hergersberg, M., Schneider, K., Moradi, S., and Meitinger, T. (2001). Retinal Detachment and Cataract, Facial Dysmorphism, Generalized Osteoporosis, Immobile Spine and Platyspondyly in a Consanguinous kindred - a Possible New Syndrome. Clin. Genet. 59 (2), 99–105. doi:10.1034/j.1399-0004.2001.590206.x
Taylan, F., Costantini, A., Coles, N., Pekkinen, M., Héon, E., Şıklar, Z., et al. (2016). Spondyloocular Syndrome: Novel Mutations in XYLT2 Gene and Expansion of the Phenotypic Spectrum. J. Bone Miner Res. 31 (8), 1577–1585. doi:10.1002/jbmr.2834
Taylan, F., Yavaş Abalı, Z., Jäntti, N., Güneş, N., Darendeliler, F., Baş, F., et al. (2017). Two Novel Mutations in XYLT2 Cause Spondyloocular Syndrome. Am. J. Med. Genet. 173, 3195–3200. doi:10.1002/ajmg.a.38470
Keywords: spondyloocular syndrome (SOS), xylosyltransferase II, exome sequencing (ES), skeletal dysplasia, XYLT2
Citation: Doddato G, Fabbiani A, Fallerini C, Bruttini M, Hadjistilianou T, Landi M, Coradeschi C, Grosso S, Tomasini B, Mencarelli MA, Renieri A and Ariani F (2021) Spondyloocular Syndrome: A Novel XYLT2 Variant with Description of the Neonatal Phenotype. Front. Genet. 12:761264. doi: 10.3389/fgene.2021.761264
Received: 19 August 2021; Accepted: 26 October 2021;
Published: 03 December 2021.
Edited by:
Sadaf Naz, University of the Punjab, PakistanReviewed by:
Khushnooda Ramzan, King Faisal Specialist Hospital and Research Centre, Saudi ArabiaCopyright © 2021 Doddato, Fabbiani, Fallerini, Bruttini, Hadjistilianou, Landi, Coradeschi, Grosso, Tomasini, Mencarelli, Renieri and Ariani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca Ariani, ZnJhbmNlc2NhLmFyaWFuaUB1bmlzaS5pdA==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.