 Yazheng Di
Yazheng Di Jingying Wang
Jingying Wang Xiaoqian Liu
Xiaoqian Liu Tingshao Zhu
Tingshao Zhu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 20 December 2021
Sec. Computational Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.761141
This article is part of the Research Topic Machine Learning Used in Biomedical Computing and Intelligence Healthcare, Volume II View all 11 articles
Background: The application of polygenic risk scores (PRSs) in major depressive disorder (MDD) detection is constrained by its simplicity and uncertainty. One promising way to further extend its usability is fusion with other biomarkers. This study constructed an MDD biomarker by combining the PRS and voice features and evaluated their ability based on large clinical samples.
Methods: We collected genome-wide sequences and utterances edited from clinical interview speech records from 3,580 women with recurrent MDD and 4,016 healthy people. Then, we constructed PRS as a gene biomarker by p value-based clumping and thresholding and extracted voice features using the i-vector method. Using logistic regression, we compared the ability of gene or voice biomarkers with the ability of both in combination for MDD detection. We also tested more machine learning models to further improve the detection capability.
Results: With a p-value threshold of 0.005, the combined biomarker improved the area under the receiver operating characteristic curve (AUC) by 9.09% compared to that of genes only and 6.73% compared to that of voice only. Multilayer perceptron can further heighten the AUC by 3.6% compared to logistic regression, while support vector machine and random forests showed no better performance.
Conclusion: The addition of voice biomarkers to genes can effectively improve the ability to detect MDD. The combination of PRS and voice biomarkers in MDD detection is feasible. This study provides a foundation for exploring the clinical application of genetic and voice biomarkers in the diagnosis of MDD.
The deployment of bioinformatic evaluations in psychiatry would revolutionize the ability to diagnose, treat, and prevent major depressive disorder (MDD). MDD affects nearly 1 in 10 people (Kessler et al., 2003; Demyttenaere et al., 2004) and has lately been recognized as the world’s leading cause of disability (World Health Organization, 2017). However, only approximately half of the population suffering from MDD is currently identified and treated (Wells et al., 1989; Goldberg 1995). The difficulty in identifying MDD is one of the key barriers to the effective utilization of current medications. Diagnosis remains based on clinical interviews and mental status examination (Regier et al., 2013); screening instruments are hindered by poor specificity and sensitivity, and there are no reliable biomarkers. Furthermore, because MDD is a syndromic diagnosis, it possibly comprises several different diseases, each with its own set of symptoms and treatment response (Alexopoulos et al., 1997; Kendler et al., 2001, 2006; Kendler et al., 2013; Gustafsson et al., 2015; Masters et al., 2015; Peterson et al., 2018).
The study of constructing MDD biomarkers has shown two different orientations. On the one hand, researchers have been devoted to finding the biological basis of depression (Schneider and Prvulovic 2013), for example, genetic factors (23andMe Research Team et al., 2019; CONVERGE Consortium, 2015; eQTLGen et al., 2018), on which to build valid biomarkers. On the other hand, studies have started from behavioral indices that are easily accessible and nonintrusive, such as patient speech voice (Low et al., 2020). The studies focus on improving diagnostic accuracy by developing machine learning (ML) algorithms.
Researchers have spent decades looking for the genetic foundation for developing more accurate MDD diagnosis models (Reus et al., 2017; Mullins et al., 2019; Rantalainen et al., 2020). The results from genome-wide association studies (GWAS) (23andMe Research Team et al., 2019; CONVERGE Consortium, 2015; eQTLGen et al., 2018) suggested that MDD is polygenic, which means that hundreds of DNA variants impact its hereditary influences with very small effects. Polygenic risk scores (PRSs) provide an estimated risk for individuals suffering from MDD. PRS is calculated as a weighted sum of an individual’s risk alleles, where their weights are specified by loci and their assessed effects found by GWAS (Chatterjee et al., 2016). Advances in biotechnology have made sequencing technologies less expensive and the genetic screening of individuals easier. However, the utility of PRS in MDD prediction is currently constrained by its simplicity and uncertainty, which, to date, captures only part of the genetic contribution to MDD risk (Murray et al., 2021). Moreover, other non-genetic risk factors, such as lifestyles, also play important roles in MDD. As a result, extending the PRS models with other MDD biomarkers may be a more practical solution to addressing this problem (Torkamani et al., 2018).
Benefitting from the development of speech recognition technology, voice-based diagnostic models for depression have been validated and have achieved a high level of accuracy. Speech biomarkers can be used not only to identify depression (Low et al., 2020) but also to recognize the severity of depression (Shin et al., 2021) and predict depression-related symptoms (Zhang et al., 2020). One of the main obstacles hindering the application of voice biomarkers is its poor generalization ability, as traditional voice feature distribution can easily change due to different speech content and speakers (Wang et al., 2019). To address this issue, researchers developed the i-vector method, extracting the factors from voice features that are independent of speaker and channel variabilities (Dehak et al., 2011; Cummins et al., 2014). A study recognizing MDD in 1,808 clinical samples proved that voice i-vectors are effective and robust (Di et al., 2021). Therefore, combining technologies in speech recognition and integrating them into existing genetic models are likely to enable clinical diagnosis in general populations.
To construct biomarkers for clinical disease detection, researchers have combined PRS with known risk factors (Hoang et al., 2021; Kapoor et al., 2021), neuroimaging, metabolites (Badhwar et al., 2020), or body indicators (Moldovan et al., 2021). However, to the best of our knowledge, there are no studies combining genes and voice in detecting MDD, which may be due to the difficulty in obtaining multiple types of samples of the same subject simultaneously. Evidence from clinical samples is needed to prove their ecological validity (Zhang et al., 2020; Murray et al., 2021). The combination and cross-validation of biological and behavioral biomarkers hold great promise to take us one step closer to the objective clinical diagnosis of MDD.
Here, based on a large sample of women with recurrent MDD diagnosed clinically, we used the PRS together with voice i-vectors to detect MDD. We examined whether their combination could surpass a single biomarker. We constructed models on different single nucleotide polymorphisms (SNPs) to examine their robustness. We also tested various ML models to find the better model.
We used a fivefold cross-validation design in this study. As shown in Figure 1, we split 80% of the samples into a training group and the rest into a test group. Firstly, we used voice data from the training samples to train the universal background model (UBM), and we used this UBM to extract i-vectors for each individual. Then, we used clumped SNP data from the training samples to train the PRS model, through which we calculated the PRS for each individual. Finally, we used the PRS and voice i-vectors from the training samples to train the ML models and used the same features from the test samples to validate the model performance. The details of each step are explained below.
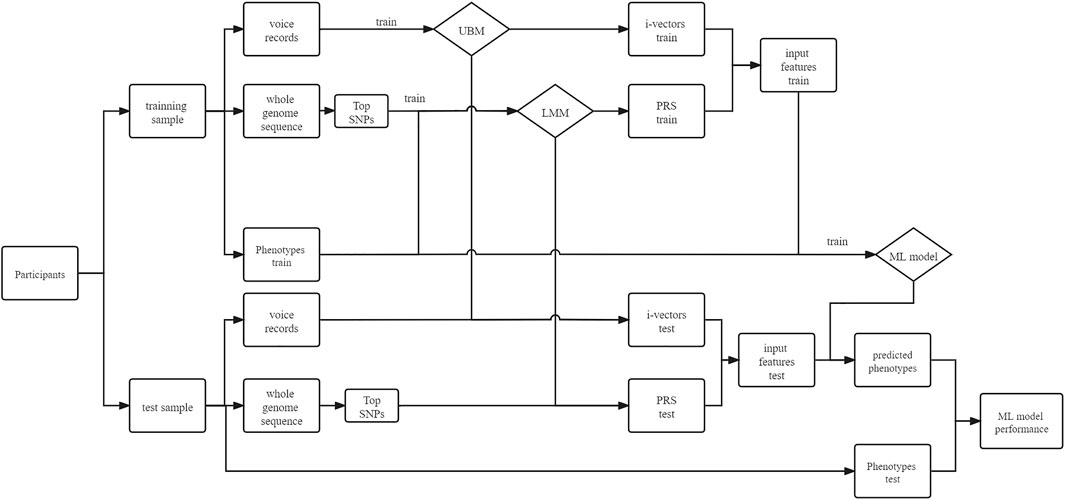
FIGURE 1. Fivefold cross-validation of voice–gene data. In each fold, the samples were split into a training group and a test group. Voice and genetic sequence data of the training group were used to train the universal background model (UBM) and linear mixed model (LMM) separately. Then, i-vectors for the training and test groups were extracted through the UBM, and the polygenic risk score (PRS) can be calculated through the LMM. The i-vectors and PRS will be concatenated as input features for a machine learning (ML) model.
The database used in this study was developed from the China, Oxford, and Virginia Commonwealth University Experimental Research on Genetic Epidemiology (CONVERGE). The CONVERGE study, designed for a genome-wide association of major depression disorders, recruited 11,670 Han Chinese women. There were 5,303 women with recurrent MDD aged between 30 and 60 years whose first episodes of MDD met the DSM-IV criteria (Association 1994). A total of 5,337 controls were recruited from patients undergoing minor surgical procedures at general hospitals or from local community centers. Only women were included in this study to minimize genetic heterogeneity because approximately 45% of the genetic liability to MDD is not shared between sexes (Kendler et al., 2007; Sullivan et al., 2000). The subject inclusion criteria and interview process were strictly controlled, as detailed in CONVERGE Consortium (2015).
The voice data of the patients were from the records during the semi-structured interview, which included assessments of psychopathology, demographic and personal characteristics, and psychosocial functioning. These voice data are characterized by a high degree of phonetic and content variety. A detailed description of the interview protocol is in Di et al. (2021).
DNA sequencing, variant calling, and the genotype likelihood calculation and imputation processes are described in CONVERGE Consortium, (2015). We used PLINK (Chang et al., 2015) to select SNPs with minor allele frequency (MAF) >0.5% and imputation quality INFO score >0.9 and clumped the SNP set using r2 = 0.5 with 50-kb windows. A total of 359,515 SNPs passed the filter.
The utterances of participants were edited from recordings of the conversations between doctors and patients through the following steps. Firstly, voice segments from the participants were selected and labeled. Then, all the segments of one participant were combined into one utterance. Due to the variety of interviews, not all voice samples of participants had segments >2 s for the latter analysis. Thus, samples with both genetic data and enough voice data were passed to subsequent analysis, and the total number was 7,596 (3,580 cases and 4,016 controls). All utterances were downsampled to 8 kHz for subsequent processing.
We used the linear mixed model (Li and Zhu 2013) to calculate the PRS. The model can be written as follows:
Here,
We used the p value-based thresholding (P+T) method (Wray et al, 2007) to construct the PRS model. Usually, a lower threshold than genome-wide statistical significance can be applied to increase the overall predictability, generally at the sacrifice of generalizability (Murray et al., 2021). Different p-value thresholds (PTs) were tested, ranging from 5 × 10−8 to 5 × 10−3 (10−3 is a conservative significance threshold of p suggested by Euesden et al., 2015). The PRS model was trained using the FaST-LMM (Lippert et al., 2011) predictor, which efficiently reduced the computational time.
To assess how the confounders affect the model’s predictability and generalizability, we considered the following covariates: age, education, occupation, social class, marital status, height, weight, and 40 genetic principal components. We compared three different covariate use strategies. The first was a model ignoring the covariates (no-cov), the second was trained by and predicted on the genetic matrix along with covariates (all-cov), and the last was a model trained by a genetic matrix along with true covariates of the training samples, but made predictions on test samples whose covariate values were replaced with random numbers (random-cov).
The i-vector extraction process is shown in Figure 2. Firstly, mel frequency cepstral coefficients (MFCCs) were extracted with a window size of 25 ms, a window shift of 10 ms, a pre-emphasis filter with a coefficient of 0.97, and a sinusoidal lifter with a coefficient of 22. A filter bank with 23 filters was used, and 12 coefficients were extracted. Then, for the given voice features, we set the number of Gaussian mixtures as 256 to estimate the utterance-dependent Gaussian mixture model (GMM) parameters and adapted the UBM (Kenny et al., 2005), which represents the feature distribution of the acoustic space.
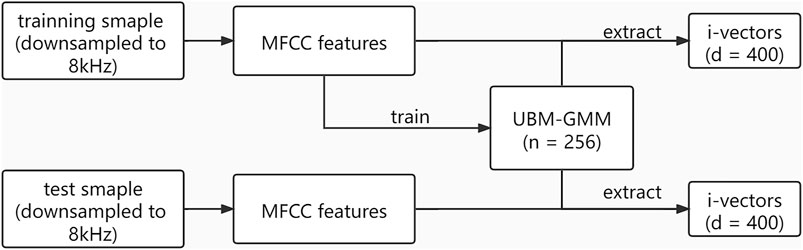
FIGURE 2. Process of i-vector extraction. UBM-GMM is a universal background model adapted by a Gaussian mixture model. n = 256 means there were 256 Gaussian mixture clusters. d = 400 means the dimension of i-vectors is 400.
The i-vectors are low-dimensional representations of the voice features based on factor analysis (Kenny et al., 2008), onto which the acoustic space is mapped via a linear transformation while keeping the majority of the variability inherent in the acoustic space. This approach has been widely used in speaker verification. The i-vector method (Dehak et al., 2011) can be expressed as follows:
where m is the mean supervector of the UBM. For the purpose of depression classification, it is expected that the UBM approximately models the phonetic variability of the acoustic space. M is the mean centered supervector of the speech utterance derived using the zeroth- and first-order Baum–Welch statistics. v is the i-vector, which captures variations in this structure caused by other factors, such as depression level, speaker identity, and channel effects (Cummins et al., 2014). We used the Kaldi speech recognition toolkit (Povey et al., 2011) and extracted the 400 dimensions of i-vectors.
We used a logistic regression (LR) classifier as a benchmark model, for which PRS, i-vectors, and both were used as input features. Then, we used random forest (RF), support vector machine (SVM), and multilayer perceptron (MLP) classifiers to test whether there was an improvement compared to the benchmark. We report the sensitivity, specificity, and area under the receiver operator characteristic curve (AUC) from the fivefold cross-validation. We used scikit-learn (Pedregosa et al., 2011) for the above process.
For the LR model, we also divided the test samples into the top 25%, middle 50%, and bottom 25% according to their PRS and calculated the accuracy on each stratification to test whether the accuracy of the biomarkers remains consistent across different genetic risk stratifications.
To check the contribution of voice and genes separately, we built three logistic regression models using a conditional forward step. Taking MDD as the dependent variable, voice i-vectors, PRS, and the combination of both were entered into the model separately as independent variables. Nagelkerke’s R2 (Nagelkerke, 1991) was utilized as an indicator of the contributing effect of the variables.
The numbers of SNPs selected using different PTs are shown in Table 1. The SNPs and their estimated weights in previous GWAS (CONVERGE Consortium 2015) are provided in Supplementary Data Sheet S1. Figure 3 shows the detection ability of the PRS models with different PTs using different covariate use strategies. When
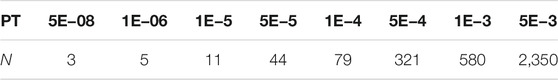
TABLE 1. Number of SNPs selected on different p-value thresholds (PTs)
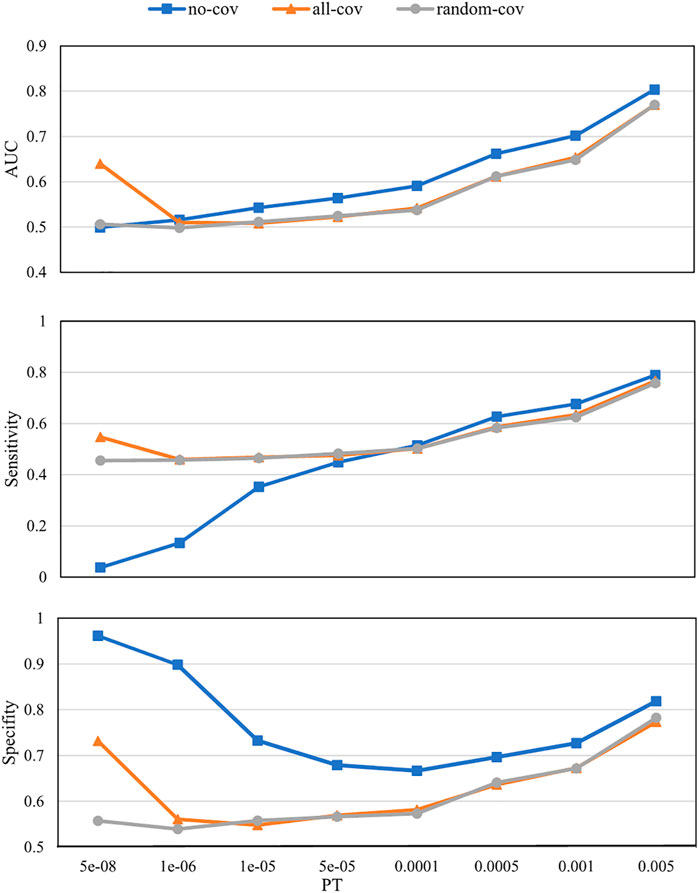
FIGURE 3. Polygenic risk score (PRS) model prediction results with different p-value thresholds (PTs) under different covariate use strategies. no-cov, no covariates were considered during the training and prediction processes; all-cov, all covariates were considered during the training and prediction processes; random-cov, the PRS model was trained with a sample genetic matrix along with covariates, but made predictions on samples whose covariates were replaced with random numbers. AUC, Area under the receiver operating characteristic curve.
Figure 4 shows the prediction results with different PTs using different biomarkers. The voice biomarkers achieved an AUC of 0.79. With the decrease in PT, the AUCs for genes only and the combined biomarkers both increased. Compared with genes only, the combined biomarkers always performed better. Compared with voice only, the combined biomarkers did not win until PT > 0.0005.
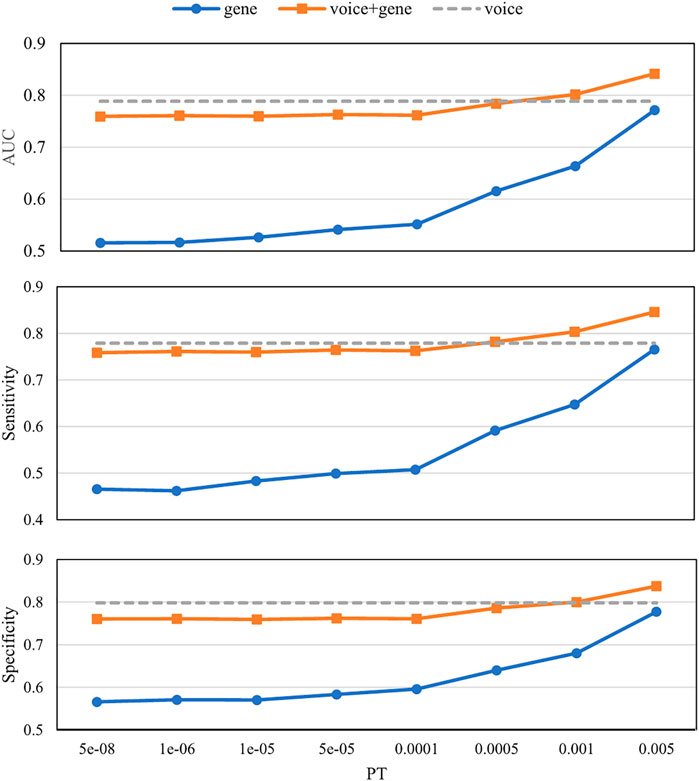
FIGURE 4. Prediction results with different p-value thresholds (PTs) using different biomarkers. The x-axis is the p-value threshold (PT) used in the gene model and the combined biomarkers. Voice biomarkers are not related to PT and are indicated by a dashed horizontal line. AUC, Area under the receiver operating characteristic curve.
We examined how much gene and voice contributed to MDD using Nagelkerke’s R2. For voice only and gene only, Nagelkerke’s R2 values were 0.571 and 0.829, respectively. For the combined biomarkers, Nagelkerke’s R2 was 0.902. Details of the logistic regression models are in Supplementary Data Sheet S2.
Figure 5 shows the stratified accuracies of the different biomarkers in predicting MDD. The voice biomarker performed consistently in the three stratifications with different genetic risks, all at 0.79. However, the accuracy of genes varied considerably between the middle (0.64) and the two ends of the population (close to 0.9). The combined biomarker performed as well as the genes in the two ends and as well as the voice in the middle.
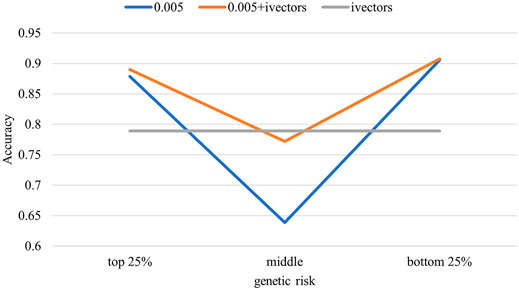
FIGURE 5. Stratified population accuracy using different biomarkers. The test samples were divided into three groups according to their predicted polygenic risk scores (PRSs). Accuracies were calculated for the three groups separately.
The classification results using LR, SVM, RF, and MLP are shown in Table 2. Two PTs (0.001 and 0.005) are presented here, while the results with more PTs are shown in Supplementary Data Sheet S3. The AUCs of LR were 0.79 and 0.83 at the two PTs. Taking LR as a benchmark, MLP achieved better results, with AUCs of 0.81 and 0.86 at the two PTs. The performance of SVM was close to that of LR, and that of RF was worse than that of LR.
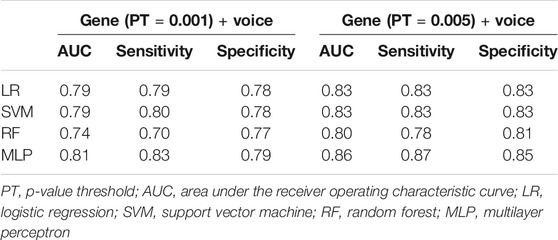
TABLE 2. Classification results using different machine learning (ML) models
This study combines the PRS and voice i-vectors to evaluate their ability to detect MDD. PRSs were calculated at different PTs. Using logistic regression, we compared the abilities of single biomarkers with the combined biomarker for MDD detection. We stratified the test group by genetic risk and examined whether the detection ability differed between stratifications. We also tested various ML models to find the best model.
A good PRS model would have high predictability, contributed mainly by capturing causal genetic variants instead of confounds. The estimated genetic fixed effect may be erroneously high for a linear mixed model if the confounding effects are not estimated. Thus, similar to our data from the same cohort, the no-cov PRS model always performed better than the all-cov PRS model (Figure 3). We believe that the results from the no-cov model are not capable of reflecting real situations because, in practical clinical applications, the distribution of covariates for a newly arrived patient is likely to be different from the distribution of the patients in our training set.
A comparison of PRS with the all-cov and random-cov models can demonstrate how the covariates affect the final prediction results in this study. When
The prediction results using different biomarkers demonstrated the ability of these biomarkers to detect MDD (Figure 4). The AUC of voice biomarkers was 0.79, which is consistent with our previous study on 1,808 clinical samples (Di et al., 2021). Since our previous study investigated the meaning of voice i-vectors, in this study, we attended to comparing its performance with the combination of PRS. Compared with genes only, the combined biomarkers can significantly improve the predictive ability at all PTs. Since the voice biomarker itself had an AUC of 0.79, only when the AUC of gene >0.65 (PT > 0.0005) can the combined biomarker perform better than voice only.
When only PRS was entered in the logistic model, it accounted for 82.9% of the variance in the dependent variable MDD (Nagelkerke’s R2). Combined with voice, the Nagelkerke’s R2 was 90.2%, indicating that the unique contribution of voice features was 8.7%. Furthermore, we illustrated how genes and voice work together to improve the predictive power by stratifying the test sample according to genetic risks and calculating the accuracies by stratifications. The results of the genes in identifying MDD for both high- and low-genetic-risk populations were consistent with the high accuracy (0.90). However, for the middle population, the accuracy of genes was poor (0.64), due mainly to the inability of genetic features to measure the effect of MDD-related environmental factors. Meanwhile, the accuracy of voice was consistent across the different genetic risk populations, suggesting that the predictive ability of voice was independent of genetic characteristics and that voice capture information was independent of genes. As a result, combining gene and voice biomarkers can effectively improve the detection ability of MDD.
We further explored whether different ML models can further improve the prediction of MDD. For the ML models, we tested the results using SVM, RF, and MLP and compared them with the results of LR. The results (Table 2 and Supplementary Data Sheet S3) showed that MLP could indeed further improve the prediction of the model, improving the AUC by 2.5% with PT = 0.001 and by 3.6% with PT = 0.005.
There are several limitations in this research. To ensure homogeneity between subjects, this study selected women with recurrent MDD as cases, and 85% of the cases met the DSM-IV criteria for melancholia, which is a severe subtype of MDD (CONVERGE Consortium 2015). Thus, our samples represent the two poles of the distribution of depression severity in natural populations. Although our experiments effectively demonstrated that the combination of genes and voice could further improve their ability to identify MDD, experimental results based on a more general population are needed before clinical application.
This study combines the PRS and voice i-vectors to evaluate their ability to detect MDD. PRSs are calculated at different PTs. With the p-value threshold at 0.005, the combined biomarker improved the AUC by 9.09% compared to genes only and 6.73% compared to voice only. Genetic risk stratification analysis showed that the ability for MDD detection of voice is genetically independent. Multilayer perceptron further improved the AUC by 3.6% compared to logistic regression. The combination of PRS and voice biomarkers in MDD detection is feasible. This study provides a foundation for exploring the clinical application of genetic and voice biomarkers in the diagnosis of MDD (Wray et al., 2018).
Publicly available datasets were analyzed in this study. The data can be found here: https://www.ebi.ac.uk/ena/browser/view/PRJNA289433?show=related-records.
The studies involving human participants were reviewed and approved by Ethical Review Board of Oxford University (Oxford Tropical Research Ethics Committee). The patients/participants provided written informed consent to participate in this study.
All authors contributed to the conception and design of the study. XL and JW organized the database. YD performed the statistical analysis. YD wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
This research is funded by the Key Research Program of the Chinese Academy of Sciences (ZDRW-XH-2019-4).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.761141/full#supplementary-material
Alexopoulos, G. S., Meyers, B. S., Young, R. C., Campbell, S., Silbersweig, D., and Charlson, M. (1997). 'Vascular Depression' Hypothesis. Arch. Gen. Psychiatry 54 (10), 915–922. doi:10.1001/archpsyc.1997.01830220033006
Association, A. P. (1994). Diagnostic and Statistical Manual of Mental Disorders. Washington, D.C: American Psychiatric Association.
Badhwar, A., McFall, G. P., Sapkota, S., Black, S. E., Chertkow, H., Duchesne, S., et al. (2020). A Multiomics Approach to Heterogeneity in Alzheimer's Disease: Focused Review and Roadmap. Brain 143 (May), 1315–1331. doi:10.1093/brain/awz384
Chang, C. C., Chow, C. C., Tellier, L. C., Vattikuti, S., Purcell, S. M., and Lee, J. J. (2015). Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets. GigaSci 4 (1), 7. doi:10.1186/s13742-015-0047-8
Chatterjee, N., Shi, J., and García-Closas, M. (2016). Developing and Evaluating Polygenic Risk Prediction Models for Stratified Disease Prevention. Nat. Rev. Genet. 17 (7), 392–406. doi:10.1038/nrg.2016.27
CONVERGE consortium (2015). Sparse Whole-Genome Sequencing Identifies Two Loci for Major Depressive Disorder. Nature 523 (7562), 588–591. doi:10.1038/nature14659
Cummins, N., Epps, J., Sethu, V., and Krajewski, J. (2014).Variability Compensation in Small Data: Oversampled Extraction of I-Vectors for the Classification of Depressed Speech, Proceeding of the 2014 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP), May 2014, Florence, Italy. IEEE, 970–974. doi:10.1109/ICASSP.2014.6853741
Dehak, N., Kenny, P. J., Dehak, R., Dumouchel, P., and Ouellet, P. (2011). Front-End Factor Analysis for Speaker Verification. IEEE Trans. Audio Speech Lang. Process. 19 (4), 788–798. doi:10.1109/tasl.2010.2064307
Demyttenaere, K., Bruffaerts, R., Posada-Villa, J., Gasquet, I., Kovess, V., Lepine, J. P., et al. (2004). Prevalence, Severity, and Unmet Need for Treatment of Mental Disorders in the World Health Organization World Mental Health Surveys. Jama 291 (21), 2581–2590. doi:10.1001/jama.291.21.2581
Di, Y., Wang, J., Li, W., and Zhu, T. (2021). Using I-Vectors from Voice Features to Identify Major Depressive Disorder. J. Affective Disord. 288 (June), 161–166. doi:10.1016/j.jad.2021.04.004
Euesden, J., Lewis, C. M., and O’Reilly, P. F. (2015). PRSice: Polygenic Risk Score Software. Bioinformatics 31 (9), 1466–1468. doi:10.1093/bioinformatics/btu848
Goldberg, D. (1995). Epidemiology of Mental Disorders in Primary Care Settings. Epidemiologic Rev. 17 (1), 182–190. doi:10.1093/oxfordjournals.epirev.a036174
Gustafsson, H., Nordstrom, A., and Nordstrom, P. (2015). Depression and Subsequent Risk of Parkinson Disease: A Nationwide Cohort Study. Neurology 84 (24), 2422–2429. doi:10.1212/wnl.0000000000001684
Hoang, T., Nguyen Ngoc, Q., Lee, J., Lee, E. K., Hwangbo, Y., and Kim, J. (2021). Evaluation of Modifiable Factors and Polygenic Risk Score in Thyroid Cancer. Endocrine-Related Cancer 28 (7), 481–494. doi:10.1530/ERC-21-0078
Howard, D. M., Adams, M. J., Adams, M. J., Clarke, T.-K., Hafferty, J. D., Gibson, J., et al. (2019). Genome-Wide Meta-Analysis of Depression Identifies 102 Independent Variants and Highlights the Importance of the Prefrontal Brain Regions. Nat. Neurosci. 22 (3), 343–352. doi:10.1038/s41593-018-0326-7
Kapoor, P. M., Middha, P., Mavaddat, N., Choudhury, P. P., Wilcox, A. N., Lindstrom, S., et al. (2021). Combined Associations of a Polygenic Risk Score and Classical Risk Factors with Breast Cancer Risk. Jnci-Journal Natl. Cancer Inst. 113 (3), 329–337. doi:10.1093/jnci/djaa056
Kendler, K. S., Aggen, S. H., and Neale, M. C. (2013). Evidence for Multiple Genetic Factors Underlying DSM-IV Criteria for Major Depression. JAMA Psychiatry 70 (6), 599–607. doi:10.1001/jamapsychiatry.2013.751
Kendler, K. S., Gardner, C. O., Neale, M. C., and Prescott, C. A. (2001). Genetic Risk Factors for Major Depression in Men and Women: Similar or Different Heritabilities and Same or Partly Distinct Genes. Psychol. Med. 31 (4), 605–616. doi:10.1017/s0033291701003907
Kendler, K. S., Gatz, M., Gardner, C. O., and Pedersen, N. L. (2006). A Swedish National Twin Study of Lifetime Major Depression. Am. J. Psychiatry 163 (1), 109–114. doi:10.1176/appi.ajp.163.1.109
Kendler, K. S., Gatz, M., Gardner, C. O., and Pedersen, N. L. (2007). Clinical Indices of Familial Depression in the Swedish Twin Registry. Acta Psychiatr. Scand. 115 (3), 214–220. doi:10.1111/j.1600-0447.2006.00863.x
Kenny, P., Boulianne, G., and Dumouchel, P. (2005). Eigenvoice Modeling with Sparse Training Data. IEEE Trans. Speech Audio Process. 13 (3), 345–354. doi:10.1109/TSA.2004.840940
Kenny, P., Ouellet, P., Dehak, N., Gupta, V., and Dumouchel, P. (2008). A Study of Interspeaker Variability in Speaker Verification. IEEE Trans. Audio Speech Lang. Process. 16 (5), 980–988. doi:10.1109/TASL.2008.925147
Kessler, R. C., Berglund, P., Demler, O., Jin, R., Koretz, D., Merikangas, K. R., et al. (2003). The Epidemiology of Major Depressive Disorder. JAMA 289 (23), 3095–3105. doi:10.1001/jama.289.23.3095
Li, G., and Zhu, H. (2013). Genetic Studies: The Linear Mixed Models in Genome-wide Association Studies. Open Bioinformatics J. 7 (1), 27–33. doi:10.2174/1875036201307010027
Lippert, C., Listgarten, J., Liu, Y., Kadie, C. M., Davidson, R. I., and Heckerman, D. (2011). FaST Linear Mixed Models for Genome-wide Association Studies. Nat. Methods 8 (10), 833–835. doi:10.1038/nmeth.1681
Low, D. M., Bentley, K. H., and Ghosh, S. S. (2020). Automated Assessment of Psychiatric Disorders Using Speech: A Systematic Review. Laryngoscope Invest. Otolaryngol. 5 (1), 96–116. doi:10.1002/lio2.354
Masters, M. C., Morris, J. C., and Roe, C. M. (2015). "Noncognitive" Symptoms of Early Alzheimer Disease: A Longitudinal Analysis. Neurology 84 (6), 617–622. doi:10.1212/wnl.0000000000001238
Moldovan, A., Waldman, Y. Y., Brandes, N., and Linial, M. (2021). Body Mass Index and Birth Weight Improve Polygenic Risk Score for Type 2 Diabetes. J. Personalized Med. 11 (6), 582. doi:10.3390/jpm11060582
Mullins, N., Bigdeli, T. B., Børglum, A. D., Coleman, J. R. I., Demontis, D., Mehta, D., et al. (2019). GWAS of Suicide Attempt in Psychiatric Disorders and Association with Major Depression Polygenic Risk Scores. Am. J. Psychiatry 176 (8), 651–660. doi:10.1176/appi.ajp.2019.18080957
Murray, G. K., Lin, T., Austin, J., McGrath, J. J., Hickie, I. B., and Wray, N. R. (2021). Could Polygenic Risk Scores Be Useful in Psychiatry. JAMA Psychiatry 78 (2), 210. doi:10.1001/jamapsychiatry.2020.3042
Nagelkerke, N. J. D. (1991). A Note on a General Definition of the Coefficient of Determination. Biometrika 78 (3), 691–692. doi:10.1093/biomet/78.3.691
Pedregosa, F., Varoquaux, G., Gramfort, A., Michel, V., Thirion, B., Grisel, O., et al. (2011). Scikit-Learn: Machine Learning in Python. J. Machine Learn. Res. 12, 2825–2830. doi:10.5555/1953048.2078195
Peterson, R. E., Cai, N., Dahl, A. W., Bigdeli, T. B., Edwards, A. C., Webb, B. T., et al. (2018). Molecular Genetic Analysis Subdivided by Adversity Exposure Suggests Etiologic Heterogeneity in Major Depression. Am. J. Psychiatry 175 (6), 545. doi:10.1176/appi.ajp.2017.17060621
Povey, D., Ghoshal, A., Boulianne, G., Burget, L., Glembek, O., Goel, N., et al. (2011).The Kaldi Speech Recognition Toolkit, IEEE 2011 Workshop on Automatic Speech Recognition and Understanding. IEEE Signal Processing Society.
Rantalainen, V., Binder, E. B., Lahti‐Pulkkinen, M., Czamara, D., Laivuori, H., Villa, P. M., et al. (2020). Polygenic Prediction of the Risk of Perinatal Depressive Symptoms. Depress. Anxiety 37 (9), 862–875. doi:10.1002/da.23066
Regier, D. A., Narrow, W. E., Clarke, D. E., Kraemer, H. C., Kuramoto, S. J., Kuhl, E. A., et al. (2013). DSM-5 Field Trials in the United States and Canada, Part II: Test-Retest Reliability of Selected Categorical Diagnoses. Am. J. Psychiatry 170 (1), 59–70. doi:10.1176/appi.ajp.2012.12070999
Reus, L. M., Shen, X., Gibson, J., Wigmore, E., Ligthart, L., Adams, M. J., et al. (2017). Association of Polygenic Risk for Major Psychiatric Illness with Subcortical Volumes and White Matter Integrity in UK Biobank. Sci. Rep. 7 (1), 42140. doi:10.1038/srep42140
the Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium Ripke, S., Mattheisen, M., Trzaskowski, M., Byrne, E. M., Abdellaoui, A., Adams, M. J., et al. (2018). Genome-Wide Association Analyses Identify 44 Risk Variants and Refine the Genetic Architecture of Major Depression. Nat. Genet. 50 (5), 668–681. doi:10.1038/s41588-018-0090-3
Schneider, B., and Prvulovic, D. (2013). Novel Biomarkers in Major Depression. Curr. Opin. Psychiatry 26 (1), 47–53. doi:10.1097/YCO.0b013e32835a5947
Shin, D., Cho, W. I., Park, C. H. K., Rhee, S. J., Kim, M. J., Lee, H., et al. (2021). Detection of Minor and Major Depression through Voice as a Biomarker Using Machine Learning. J. Clin. Med. 10 (14), 3046. doi:10.3390/jcm10143046
Sullivan, P. F., Neale, M. C., and Kendler, K. S. (2000). Genetic Epidemiology of Major Depression: Review and Meta-Analysis. Am. J. Psychiatry 157 (10), 1552–1562. doi:10.1176/appi.ajp.157.10.1552
Torkamani, A., Wineinger, N. E., and Topol, E. J. (2018). The Personal and Clinical Utility of Polygenic Risk Scores. Nat. Rev. Genet. 19 (9), 581–590. doi:10.1038/s41576-018-0018-x
Wang, J., Zhang, L., Liu, T., Pan, W., Hu, B., and Zhu, T. (2019). Acoustic Differences between Healthy and Depressed People: A Cross-Situation Study. BMC Psychiatry 19 (1), 300. doi:10.1186/s12888-019-2300-7
Wells, K. B., Hays, R. D., Burnam, M. A., Rogers, W., Greenfield, S., and Ware, J. E. (1989). Detection of Depressive Disorder for Patients Receiving Prepaid or Fee-For-Service Care. Jama 262 (23), 3298–3302. doi:10.1001/jama.1989.03430230083030
World Health Organization, (2017). Depression and Other Common Mental Disorders: Global Health Estimates. Available at: https://www.who.int/publications/i/item/depression-global-health-estimates (Accessed December 6, 2021).
Wray, N. R., Goddard, M. E., and Visscher, P. M. (2007). Prediction of Individual Genetic Risk to Disease from Genome-wide Association Studies. Genome Res. 17 (10), 1520–1528. doi:10.1101/gr.6665407
Keywords: biomarkers, polygenic risk score (PRS), computer technology, major depressive disorder (MDD), voice biomarkers, depression
Citation: Di Y, Wang J, Liu X and Zhu T (2021) Combining Polygenic Risk Score and Voice Features to Detect Major Depressive Disorders. Front. Genet. 12:761141. doi: 10.3389/fgene.2021.761141
Received: 19 August 2021; Accepted: 12 November 2021;
Published: 20 December 2021.
Edited by:
Honghao Gao, Shanghai University, ChinaReviewed by:
Bingxin Zhao, Purdue University, United StatesCopyright © 2021 Di, Wang, Liu and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tingshao Zhu, dHN6aHVAcHN5Y2guYWMuY24mI3gwMjAwYTs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.